
One-Pot Synthesis of (+)-Nootkatone via Dark Singlet Oxygenation of Valencene: The Triple Role of the Amphiphilic Molybdate Catalyst

 , ,
, , 
Abstract
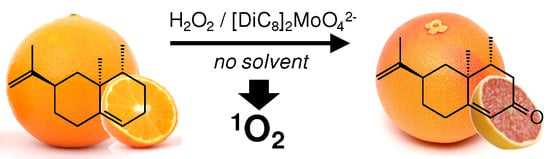
:
1. Introduction
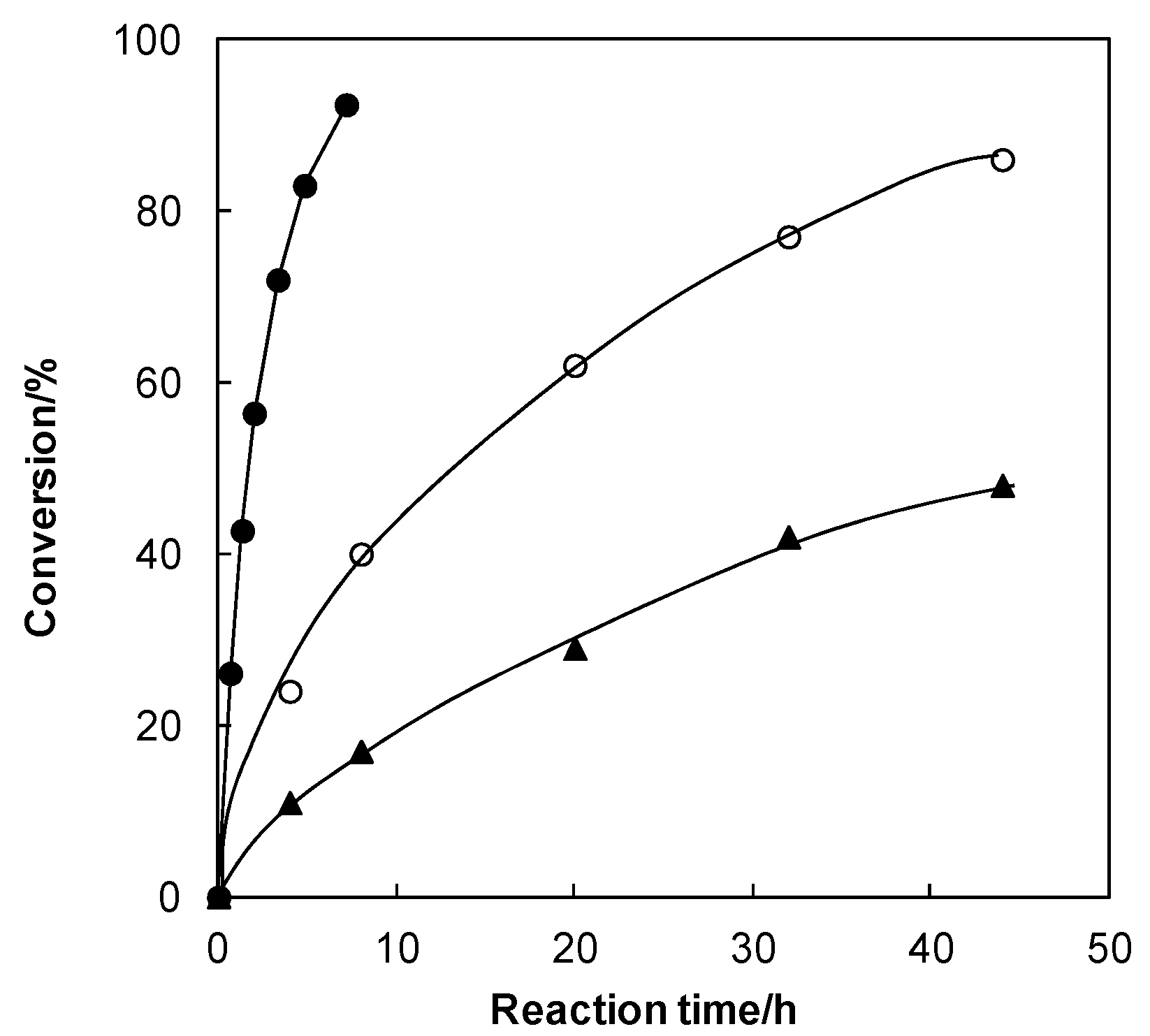
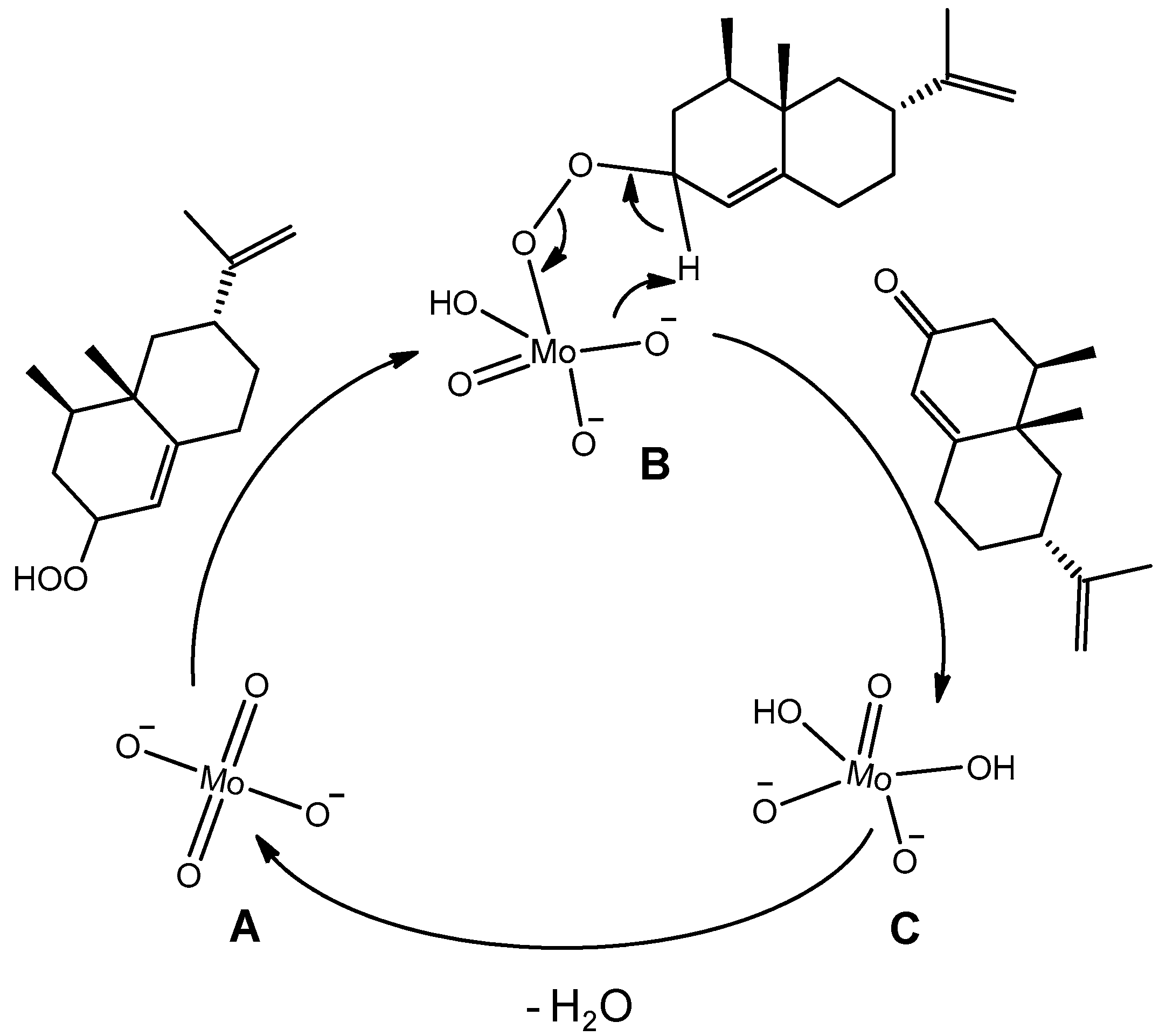
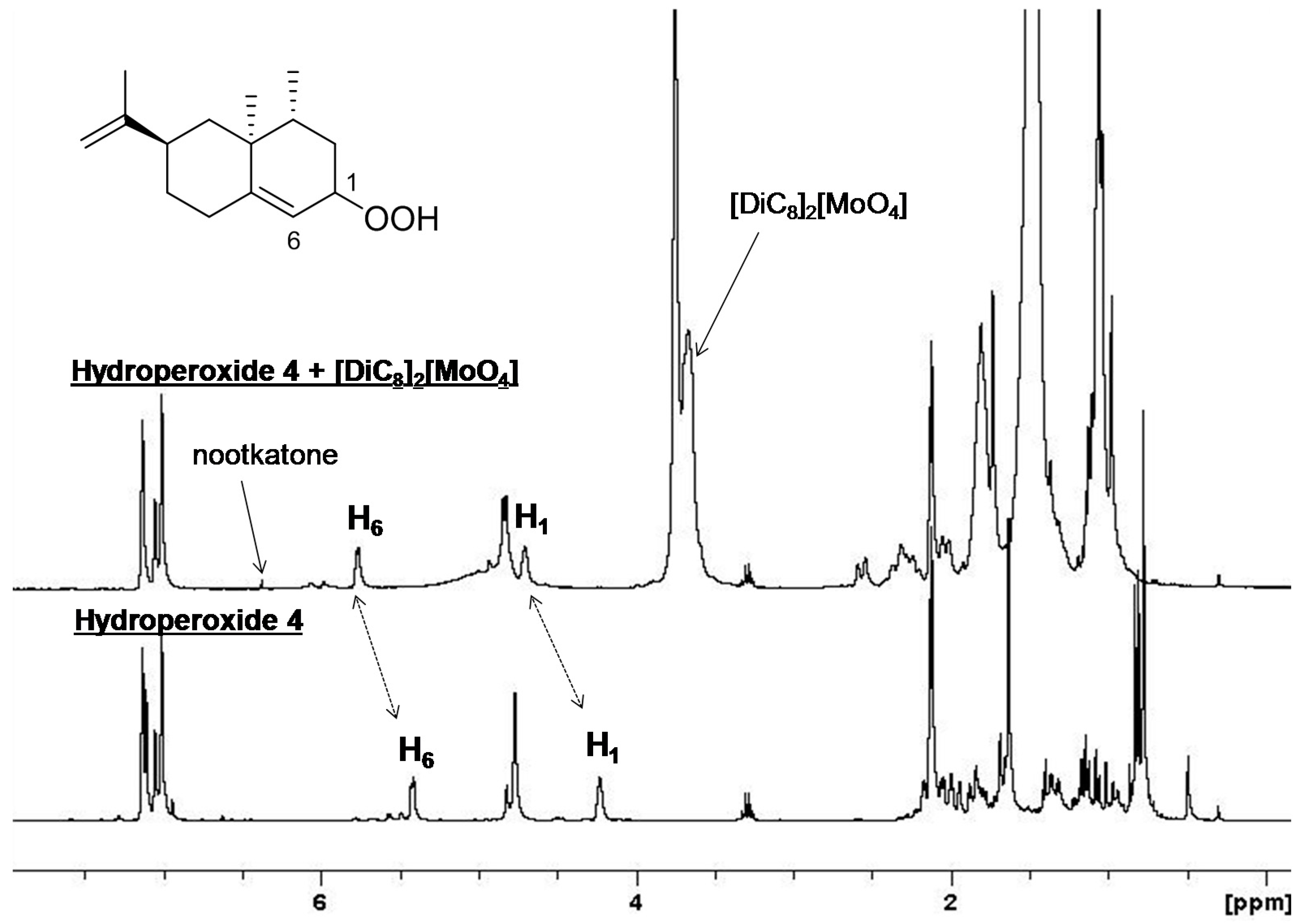
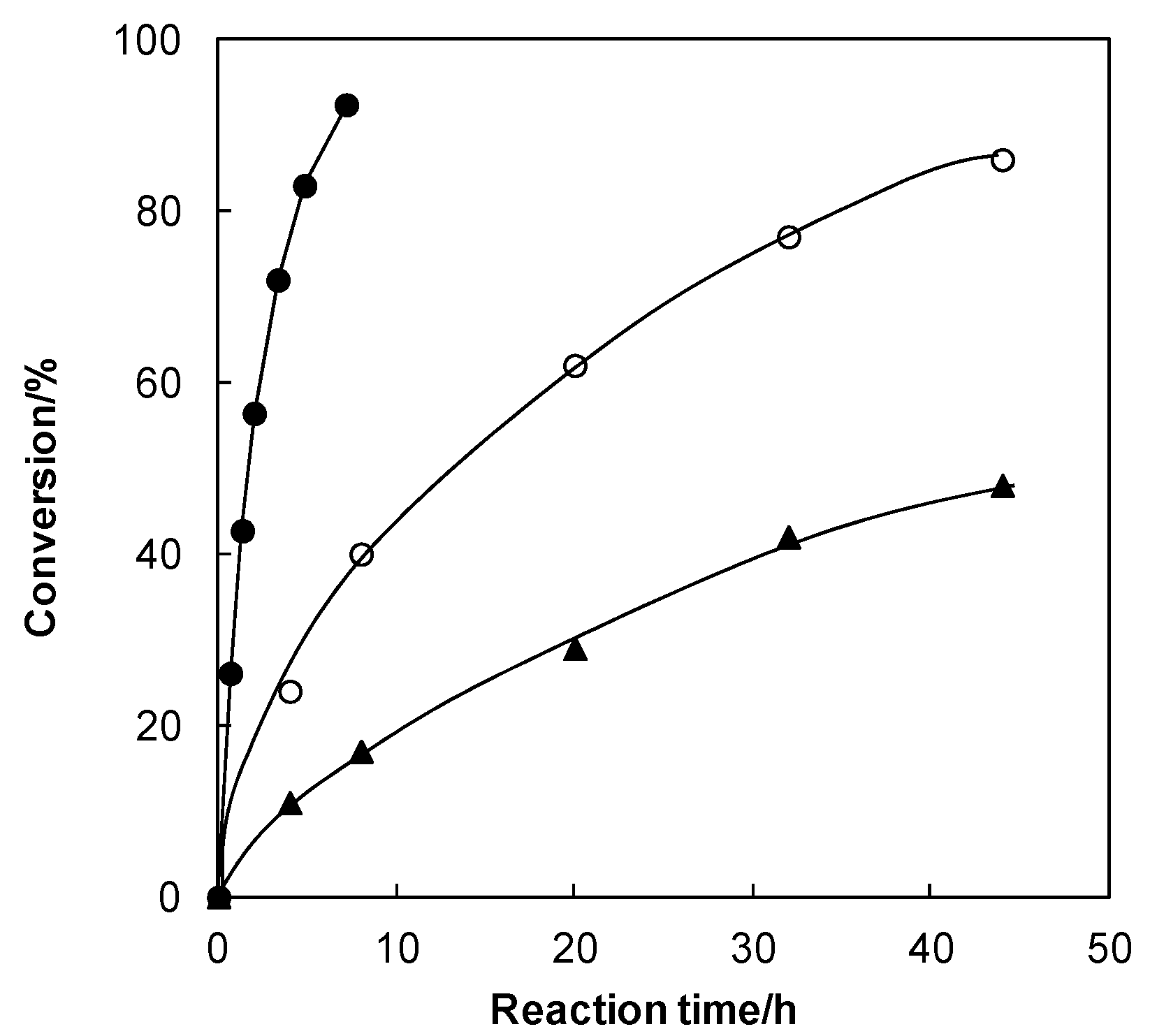
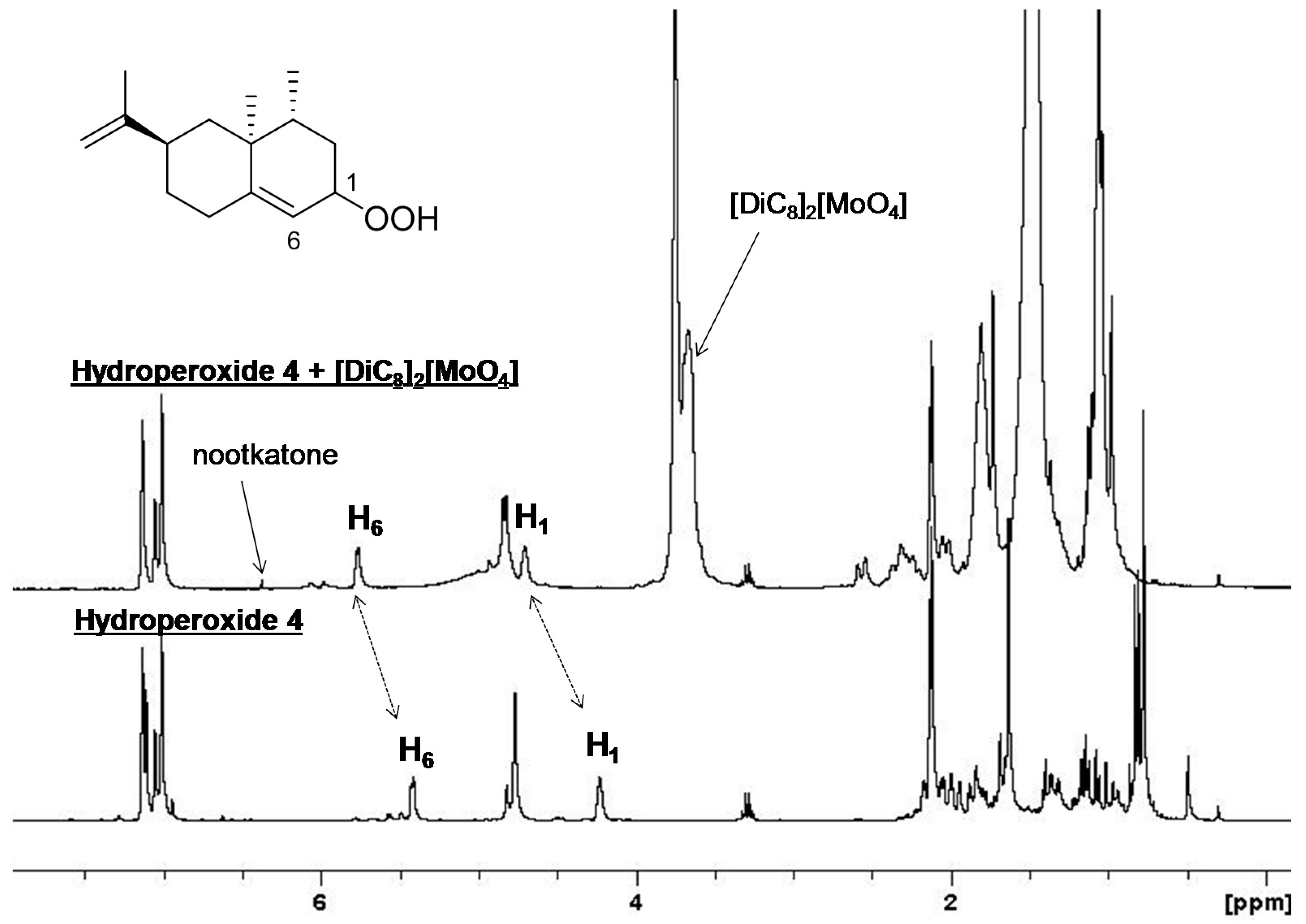
2. Results and Discussion
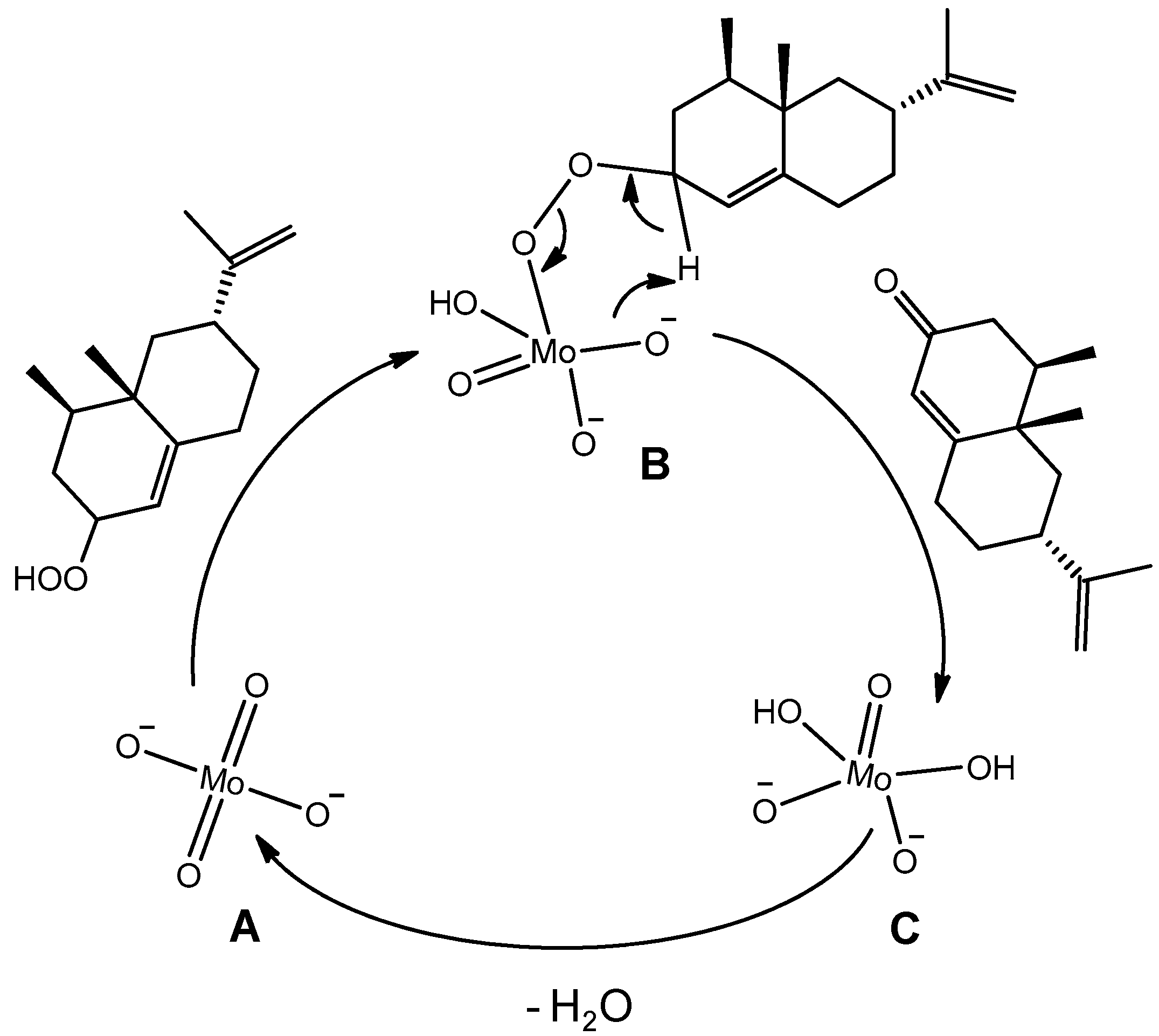
- the dark singlet oxygenation (i.e., the chemical oxidation versus the photooxidation [53]) of valencene can be efficiently performed in the absence of organic solvent thanks to the surface activity of the catalyst, which lowered the interfacial tension between the organic phase, i.e., the substrate, and the aqueous H2O2, thus generating nanodroplets and a higher interfacial area in the solvent-less medium;
- [DiC8]2[MoO4] can play a dual catalytic role in Step 1 (see above) and Step 3 of the process;
- it is a “balanced catalytic surfactant” that can thus provide a three-phase-microemulsion system (i.e., a microemulsion phase in equilibrium with both water and solvent excess phases) in the presence of water and an appropriate solvent, allowing for easy product isolation and catalyst recycling via simple phase separation [52].
3. Materials and Methods
3.1. General Information
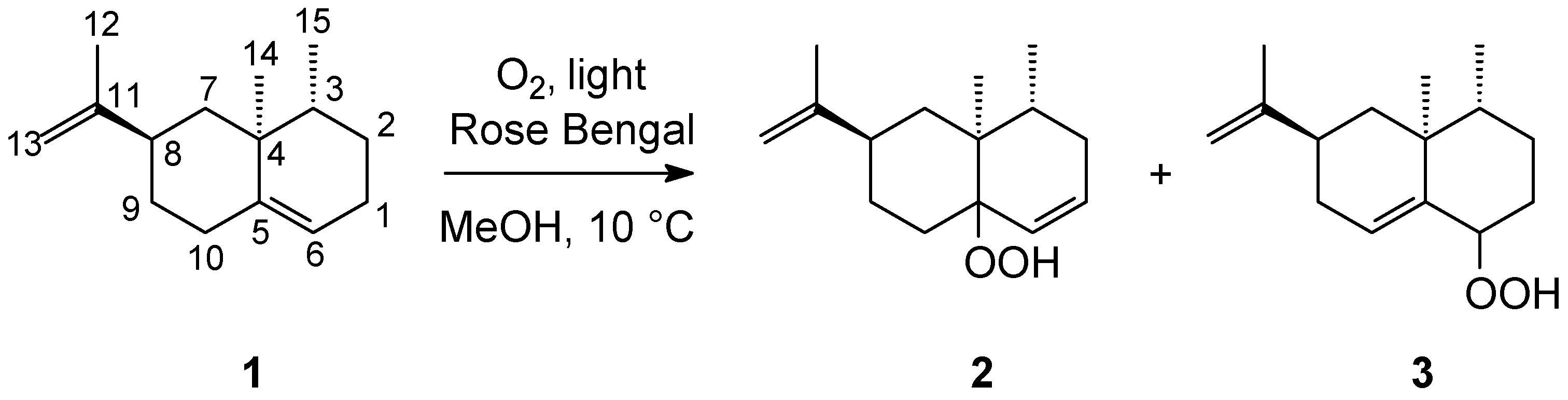
3.2. Photooxidation of (+)-Valencene 1 in Methanol with Rose Bengal as a Photosensitizer
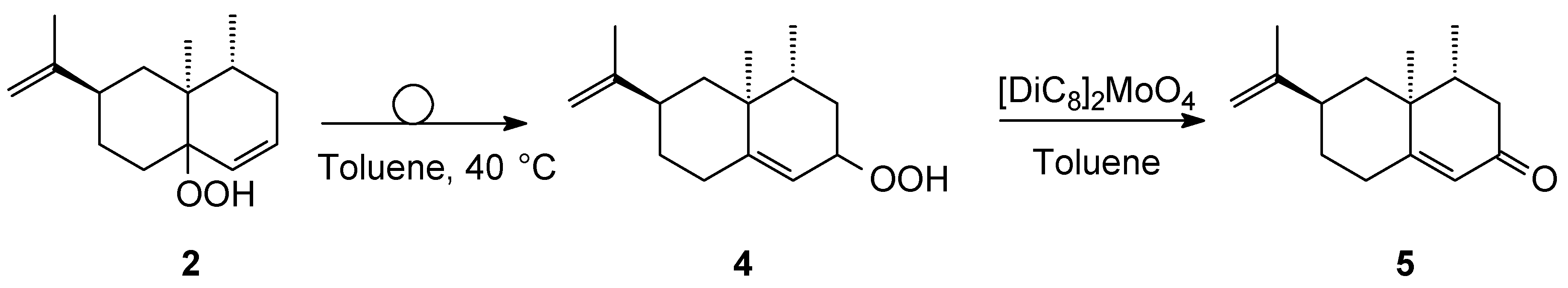
3.3. Conversion of Hydroperoxide 2 into Hydroperoxide 4 via Schenck Rearrangement and Further Catalytic Conversion into (+)-Nootkatone 5
3.4. Direct Synthesis of (+)-Nootkatone 5 in Toluene by Photooxidation of (+)-Valencene 1 in the Presence of [DiC8]2MoO4
3.5. Dark Singlet Oxygenation of (+)-Valencene 1 to (+)-Nootkatone 5
3.6. Preparative Scale Synthesis of (+)-Nootkatone 5
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Erdtman, H.; Hirose, Y. The chemistry of the natural order cupressales. Xlvi. The structure of nootkatone. Acta Chem. Scand. 1962, 16, 1311–1314. [Google Scholar] [CrossRef]
- MacLeod, W.D., Jr.; Buigues, N.M. Sesquiterpenes. I. Nootkatone, a new grapefruit flavor constituent. J. Food Sci. 1964, 29, 565–568. [Google Scholar] [CrossRef]
- Haring, H.G.; Rijkens, F.; Boelens, H.; Van der Gen, A. Olfactory studies on enantiomeric eremophilane sesquiterpenoids. J. Agric. Food Chem. 1972, 20, 1018–1021. [Google Scholar] [CrossRef]
- EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Safety and efficacy of secondary alicyclic saturated and unsaturated alcohols, ketones, ketals and esters with ketals containing alicyclic alcohols or ketones and esters containing secondary alicyclic alcohols from chemical group 8 when used as flavourings for all animal species. EFSA J. 2016, 14, 44–75. [Google Scholar]
- Zhu, B.C.R.; Henderson, G.; Sauer, A.M.; Yu, Y.; Crowe, W.; Laine, R.A. Structure-activity of valencenoid derivatives and their repellence to the formosan subterranean termite. J. Chem. Ecol. 2003, 29, 2695–2701. [Google Scholar] [CrossRef] [PubMed]
- Panella, N.A.; Dolan, M.C.; Karchesy, J.J.; Xiong, Y.; Peralta-Cruz, J.; Khasawneh, M.; Montenieri, J.A.; Maupin, G.O. Use of novel compounds for pest control: Insecticidal and acaricidal activity of essential oil components from heartwood of alaska yellow cedar. J. Med. Entomol. 2005, 42, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Leitereg, T.J. Synthesis of sesquiterpenoids related to nootkatone. Structure determination by nmr using tris(dipivalomethanato)europium. Tetrahedron Lett. 1972, 13, 2617–2620. [Google Scholar] [CrossRef]
- Pesaro, M.; Bozzato, G.; Schudel, P. The total synthesis of racemic nootkatone. Chem. Commun. 1968, 1152–1154. [Google Scholar] [CrossRef]
- Odom, H.C.; Pinder, A.R. Total synthesis of (±)-nootkatone. Chem. Commun. 1969, 26–27. [Google Scholar] [CrossRef]
- Marshall, J.A.; Ruden, R.A. Stereoselective total synthesis of racemic nootkatone. J. Org. Chem. 1971, 36, 594–596. [Google Scholar] [CrossRef]
- McGuire, H.M.; Odom, H.C., Jr.; Pinder, A.R. Further synthetic studies in the nootkatane sesquiterpene group. New total synthesis of (±)-valencene and (±)-nootkatone. J. Chem. Soc. Perkin Trans. 1 1974, 1879–1883. [Google Scholar] [CrossRef]
- Dastur, K.P. Stereoselective approach to eremophilane sesquiterpenes. Synthesis of (±)-nootkatone and (±)-α-vetivone. J. Am. Chem. Soc. 1974, 96, 2605–2608. [Google Scholar] [CrossRef]
- Naf, F.; Decorzant, R.; Thommen, W. A novel entry to the eremophilane and valencane sesquiterpenes via a stereoselective intramolecular diels-alder reaction. Helv. Chim. Acta 1979, 62, 114–118. [Google Scholar] [CrossRef]
- Hiyama, T.; Shinoda, M.; Nozaki, H. A new stereoselective synthesis of (±)-nootkatone by means of cyclopentenone annulation. Tetrahedron Lett. 1979, 20, 3529–3532. [Google Scholar] [CrossRef]
- Majetich, G.; Behnke, M.; Hull, K. A stereoselective synthesis of (±)-nootkatone and (±)-valencene via an intramolecular sakurai reaction. J. Org. Chem. 1985, 50, 3615–3618. [Google Scholar] [CrossRef]
- Handore, K.L.; Seetharamsingh, B.; Reddy, D.S. Ready access to functionally embellished cis-hydrindanes and cis-decalins: Protecting group-free total syntheses of (±)-nootkatone and (±)-noreremophilane. J. Org. Chem. 2013, 78, 8149–8154. [Google Scholar] [CrossRef] [PubMed]
- Yanami, T.; Miyashita, M.; Yoshikoshi, A. Stereocontrolled synthesis of (+)-nootkatone from (−)-β-pinene. J. Chem. Soc. Chem. Commun. 1979, 525–527. [Google Scholar] [CrossRef]
- Yanami, T.; Miyashita, M.; Yoshikoshi, A. Synthetic study of (+)-nootkatone from (−)-β-pinene. J. Org. Chem. 1980, 45, 607–612. [Google Scholar] [CrossRef]
- Sauer, A.M.; Crowe, W.E.; Henderson, G.; Laine, R.A. An efficient and economic asymmetric synthesis of (+)-nootkatone, tetrahydronootkatone, and derivatives. Org. Lett. 2009, 11, 3530–3533. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Greenhagen, B. Plant Sesquiterpene Synthase Cdna, Production with Recombinant Organisms of the Enzyme, and Its Use for Production of Sesquiterpenes. U.S. Patent 20060218661A1, 28 September 2006. [Google Scholar]
- Hunter, G.L.K.; Brogden, W.B., Jr. Conversion of valencene to nootkatone. J. Food Sci. 1965, 30, 876–878. [Google Scholar] [CrossRef]
- Shaffer, G.W.; Eschinasi, E.H.; Purzycki, K.L.; Doerr, A.B. Oxidations of valencene. J. Org. Chem. 1975, 40, 2181–2185. [Google Scholar] [CrossRef]
- Wilson, C.W., III; Shaw, P.E. Synthesis of nootkatone from valencene. J. Agric. Food Chem. 1978, 26, 1430–1432. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Clark, J.H. The allylic oxidation of unsaturated steroids by tert-butyl hydroperoxide using surface functionalised silica supported metal catalysts. Green Chem. 2002, 4, 352–356. [Google Scholar] [CrossRef]
- Silvestre, S.M.; Salvador, J.A.R. Allylic and benzylic oxidation reactions with sodium chlorite. Tetrahedron 2007, 63, 2439–2445. [Google Scholar] [CrossRef]
- Garcia-Cabeza, A.L.; Marin-Barrios, R.; Moreno-Dorado, F.J.; Ortega, M.J.; Massanet, G.M.; Guerra, F.M. Allylic oxidation of alkenes catalyzed by a copper-aluminum mixed oxide. Org. Lett. 2014, 16, 1598–1601. [Google Scholar] [CrossRef] [PubMed]
- Girhard, M.; Machida, K.; Itoh, M.; Schmid, R.D.; Arisawa, A.; Urlacher, V.B. Regioselective biooxidation of (+)-valencene by recombinant E. coli expressing CYP109B1 from Bacillus subtilis in a two-liquid-phase system. Microb. Cell Fact. 2009, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, H.; Itoh, K.-i.; Taniai, T.; Kitanaka, S.; Takagi, Y.; Chai, W.; Horiuchi, C.A. Biotransformation of valencene by cultured cells of Gynostemma pentaphyllum. J. Mol. Catal. B Enzym. 2005, 32, 103–106. [Google Scholar] [CrossRef]
- Furusawa, M.; Hashimoto, T.; Noma, Y.; Asakawa, Y. Highly efficient production of nootkatone, the grapefruit aroma from valencene, by biotransformation. Chem. Pharm. Bull. 2005, 53, 1513–1514. [Google Scholar] [CrossRef] [PubMed]
- Krügener, S.; Krings, U.; Zorn, H.; Berger, R.G. A dioxygenase of Pleurotus sapidus transforms (+)-valencene regio-specifically to (+)-nootkatone via a stereo-specific allylic hydroperoxidation. Bioresour. Technol. 2010, 101, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Rickert, A.; Krombach, V.; Hamers, O.; Zorn, H.; Maison, W. Enzymatic allylic oxidations with a lyophilisate of the edible fungus Pleurotus sapidus. Green Chem. 2012, 14, 639–644. [Google Scholar] [CrossRef]
- Kaspera, R.; Krings, U.; Nanzad, T.; Berger, R.G. Bioconversion of (+)-valencene in submerged cultures of the ascomycete Chaetomium globosum. Appl. Microbiol. Biotechnol. 2005, 67, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Sowden, R.J.; Yasmin, S.; Rees, N.H.; Bell, S.G.; Wong, L.-L. Biotransformation of the sesquiterpene (+)-valencene by cytochrome p450cam and p450BM-3. Org. Biomol. Chem. 2005, 3, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Cankar, K.; van Houwelingen, A.; Bosch, D.; Sonke, T.; Bouwmeester, H.; Beekwilder, J. A chicory cytochrome p450 mono-oxygenase cyp71av8 for the oxidation of (+)-valencene. FEBS Lett. 2011, 585, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Fraatz, M.A.; Berger, R.G.; Zorn, H. Nootkatone—A biotechnological challenge. Appl. Microbiol. Biotechnol. 2009, 83, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Tatsumi, A.; Terada, M.; Hirai, N. Process for Producing Organic Compounds by Catalysis of Imide Compounds. EP1164131A1, 19 December 2001. [Google Scholar]
- Huang, R.; Christenson, P.A.; Labuda, I.M. Process for the Preparation of Nootkatone by Laccase Catalysis. U.S. Patent 6200786B1, 13 March 2001. [Google Scholar]
- Davies, A.G.; Davison, I.G.E. Rearrangements of allylic hydroperoxides derived from (+)-valencene. J. Chem. Soc. Perkin Trans. 2 1989, 825–830. [Google Scholar] [CrossRef]
- Ohloff, G. Singlet oxygen: A reagent in organic synthesis. Pure Appl. Chem. 1975, 43, 481–502. [Google Scholar] [CrossRef]
- Davies, A.G. The schenck rearrangement of allylic hydroperoxides. J. Chem. Res. 2009, 2009, 533–544. [Google Scholar] [CrossRef]
- Neuenschwander, U.; Jensen, K.F. Olefin autoxidation in flow. Ind. Eng. Chem. Res. 2014, 53, 601–608. [Google Scholar] [CrossRef]
- Avila, D.V.; Davies, A.G.; Davison, I.G.E. Stereoselectivity in the formation and allylic rearrangement of 8a-methyl- and 8a-ethyl-1,2,3,4,4a,7,8,8a-octahydronaphthalenyl hydroperoxides. J. Chem. Soc. Perkin Trans. 2 1988, 1847–1852. [Google Scholar] [CrossRef]
- Ponce, M.A.; Ramirez, J.A.; Galagovsky, L.R.; Gros, E.G.; Erra-Balsells, R. Singlet-oxygen ene reaction with 3[small beta]-substituted stigmastanes. An alternative pathway for the classical schenck rearrangement. J. Chem. Soc. Perkin Trans. 2 2000, 2351–2358. [Google Scholar] [CrossRef]
- Porter, N.A.; Mills, K.A.; Caldwell, S.E.; Dubay, G.R. The mechanism of the [3,2] allylperoxyl rearrangement: A radical-dioxygen pair reaction that proceeds with stereochemical memory. J. Am. Chem. Soc. 1994, 116, 6697–6705. [Google Scholar] [CrossRef]
- Brill, W.F. The isolation and rearrangement of pure acyclic allylic hydroperoxides. J. Am. Chem. Soc. 1965, 87, 3286–3287. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Davies, A.G.; Davison, I.G.E.; Maccoll, A.; Mruzek, M.H. The mechanisms of the rearrangements of allylic hydroperoxides: 5[small alpha]-hydroperoxy-3[small beta]-hydroxycholest-6-ene and 7[small alpha]-hydroperoxy-3[small beta]-hydroxycholest-5-ene. J. Chem. Soc. Perkin Trans. 2 1989, 815–824. [Google Scholar] [CrossRef]
- Partenheimer, W. The effect of zirconium in metal/bromide catalysts during the autoxidation of p-xylene: Part i. Activation and changes in benzaldehyde intermediate formation. J. Mol. Catal. A Chem. 2003, 206, 105–119. [Google Scholar] [CrossRef]
- Landheer, I.; Ginsburg, D. Propellanes—LIII. Tetrahedron 1981, 37, 143–150. [Google Scholar] [CrossRef]
- Schenck, G.O.; Neumuller, O.A.; Eisfeld, W. Photosensitive autoxidation of steroids. Ii. New type of allyl rearrangement of steroid hydroperoxides. Angew. Chem. 1958, 70, 595. [Google Scholar] [CrossRef]
- Sato, K.; Aoki, M.; Takagi, J.; Zimmermann, K.; Noyori, R. A practical method for alcohol oxidation with aqueous hydrogen peroxide under organic solvent- and halide-free conditions. Bull. Chem. Soc. Jpn. 1999, 72, 2287–2306. [Google Scholar] [CrossRef]
- Nardello, V.; Marko, J.; Vermeersch, G.; Aubry, J.M. 90mo nmr and kinetic studies of peroxomolybdic intermediates involved in the catalytic disproportionation of hydrogen peroxide by molybdate ions. Inorg. Chem. 1995, 34, 4950–4957. [Google Scholar] [CrossRef]
- Nardello-Rataj, V.; Caron, L.; Borde, C.; Aubry, J.-M. Oxidation in three-liquid-phase microemulsion systems using “balanced catalytic surfactants”. J. Am. Chem. Soc. 2008, 130, 14914–14915. [Google Scholar] [CrossRef] [PubMed]
- Alsters, P.L.; Jary, W.; Nardello-Rataj, V.; Aubry, J.-M. “Dark” singlet oxygenation of β-citronellol: A key step in the manufacture of rose oxide. Org. Process. Res. Dev. 2010, 14, 259–262. [Google Scholar] [CrossRef]
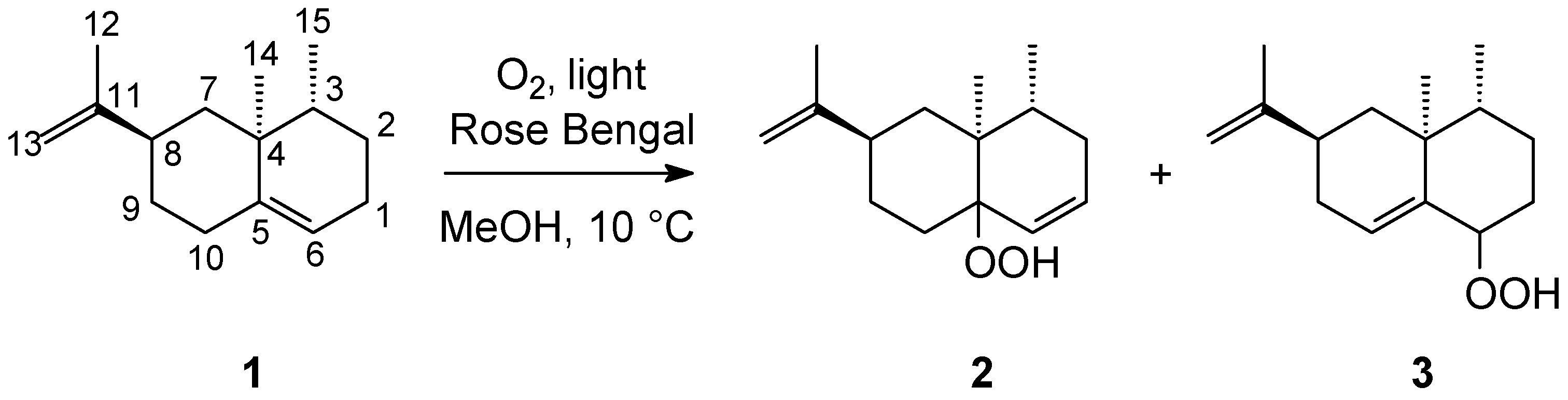
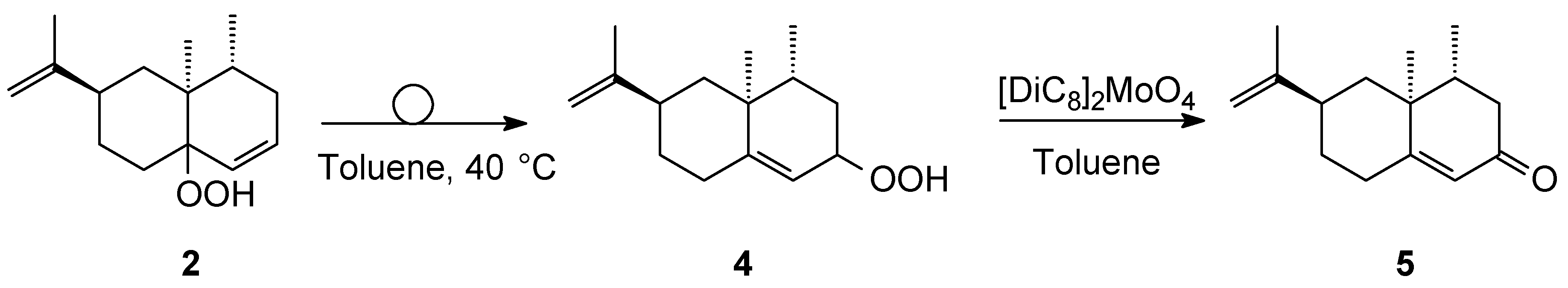


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvents | Conversion of 2 into 4 (%) |
|---|---|
| MeOH | <5 |
| Acetone | 55 |
| Acetonitrile | 66 |
| Chloroform | 89 |
| Toluene | >95 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, B.; Lebeuf, R.; Delbaere, S.; Alsters, P.L.; Nardello-Rataj, V. One-Pot Synthesis of (+)-Nootkatone via Dark Singlet Oxygenation of Valencene: The Triple Role of the Amphiphilic Molybdate Catalyst. Catalysts 2016, 6, 184. https://doi.org/10.3390/catal6120184
Hong B, Lebeuf R, Delbaere S, Alsters PL, Nardello-Rataj V. One-Pot Synthesis of (+)-Nootkatone via Dark Singlet Oxygenation of Valencene: The Triple Role of the Amphiphilic Molybdate Catalyst. Catalysts. 2016; 6(12):184. https://doi.org/10.3390/catal6120184
Chicago/Turabian StyleHong, Bing, Raphaël Lebeuf, Stéphanie Delbaere, Paul L. Alsters, and Véronique Nardello-Rataj. 2016. "One-Pot Synthesis of (+)-Nootkatone via Dark Singlet Oxygenation of Valencene: The Triple Role of the Amphiphilic Molybdate Catalyst" Catalysts 6, no. 12: 184. https://doi.org/10.3390/catal6120184
APA StyleHong, B., Lebeuf, R., Delbaere, S., Alsters, P. L., & Nardello-Rataj, V. (2016). One-Pot Synthesis of (+)-Nootkatone via Dark Singlet Oxygenation of Valencene: The Triple Role of the Amphiphilic Molybdate Catalyst. Catalysts, 6(12), 184. https://doi.org/10.3390/catal6120184