
Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides


Abstract
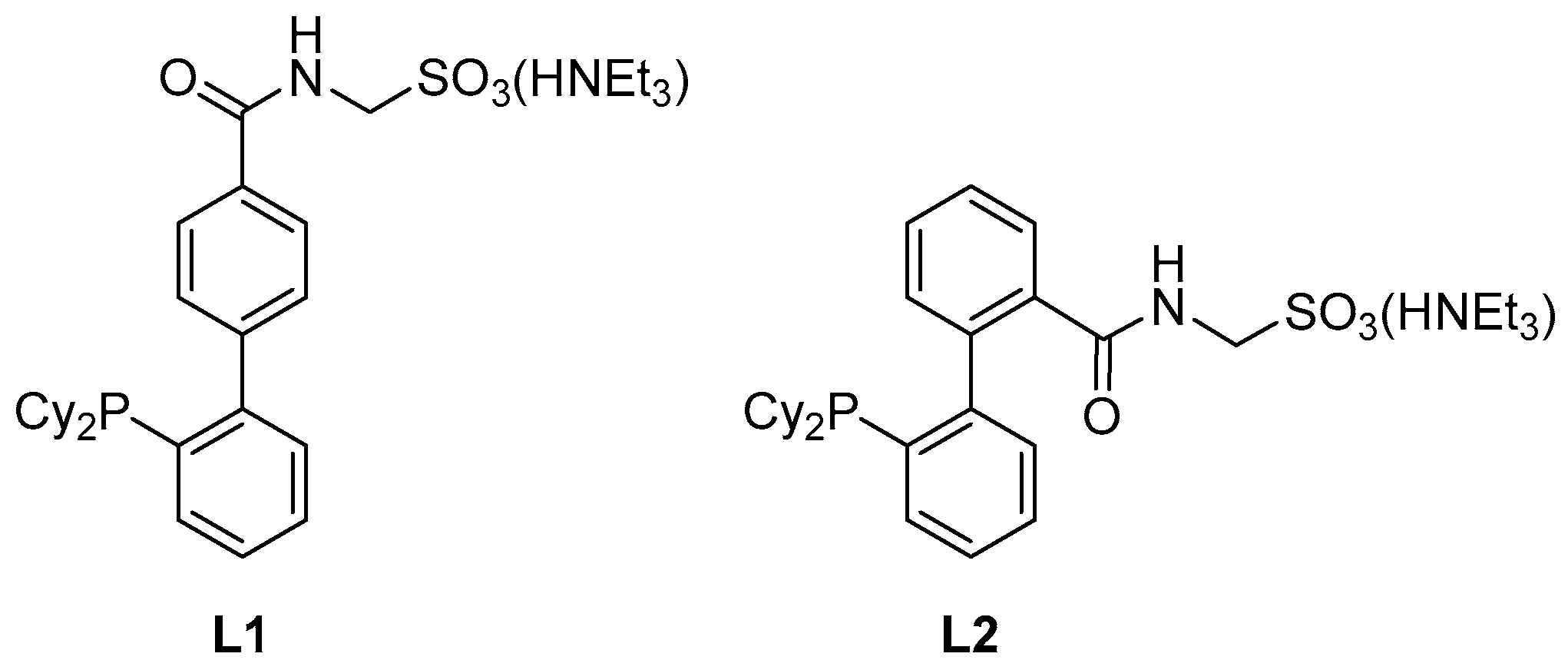
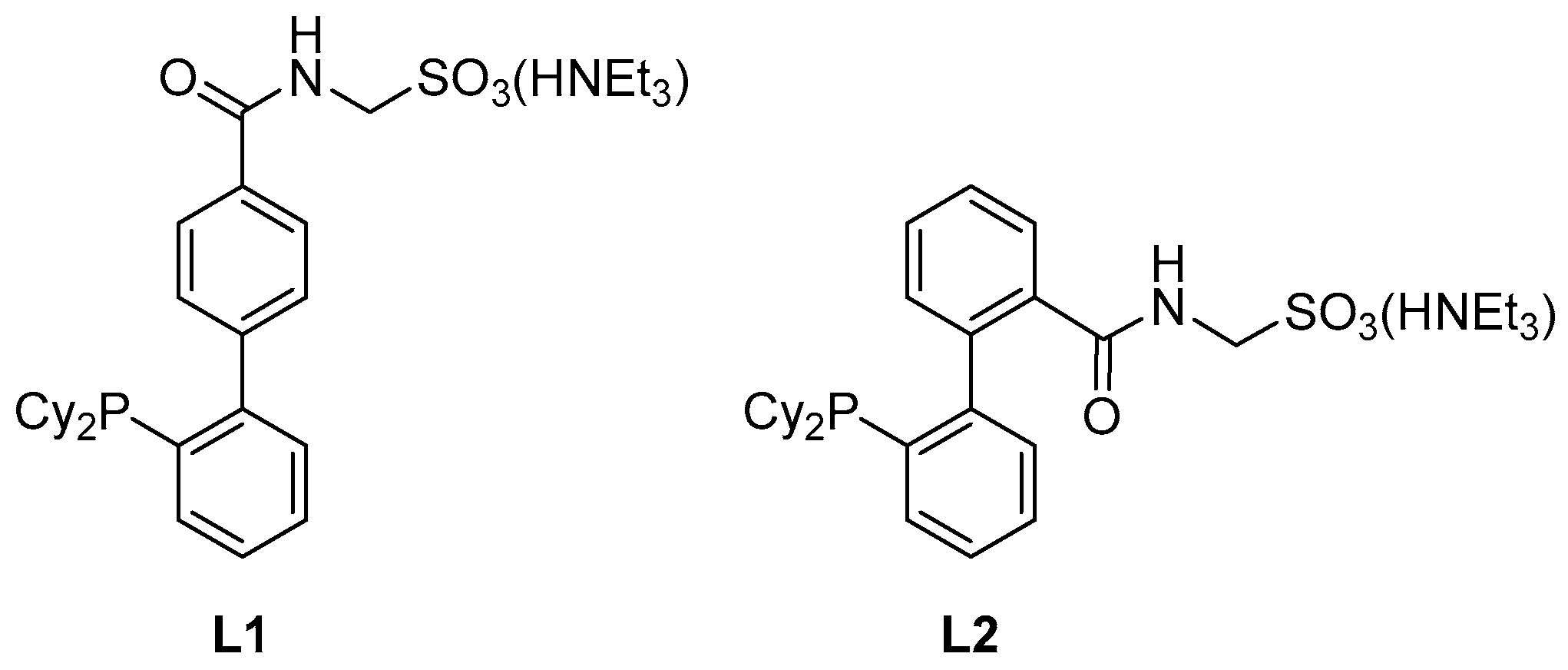
:1. Introduction
2. Results and Discussion
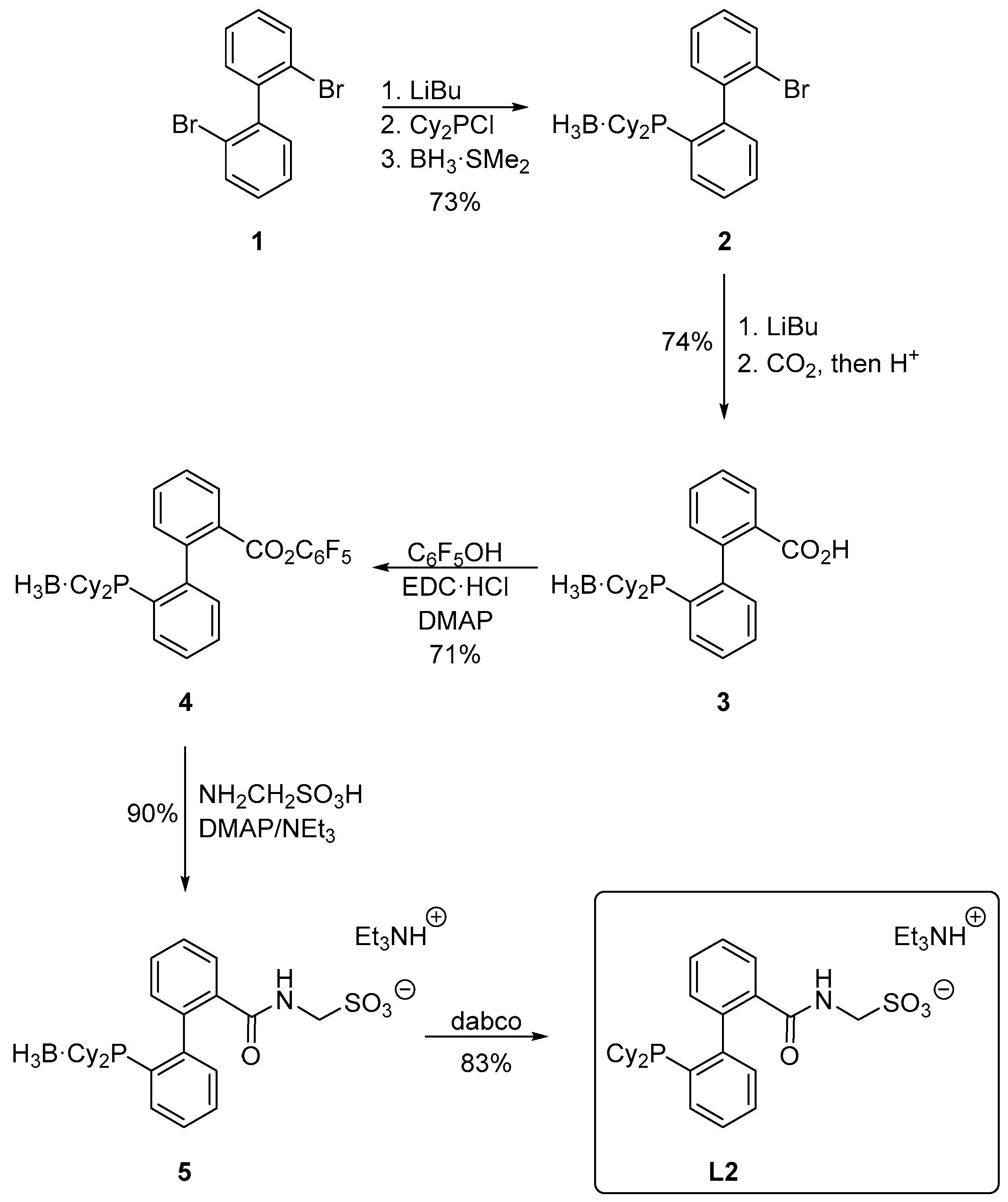
2.1. The Synthesis of L2
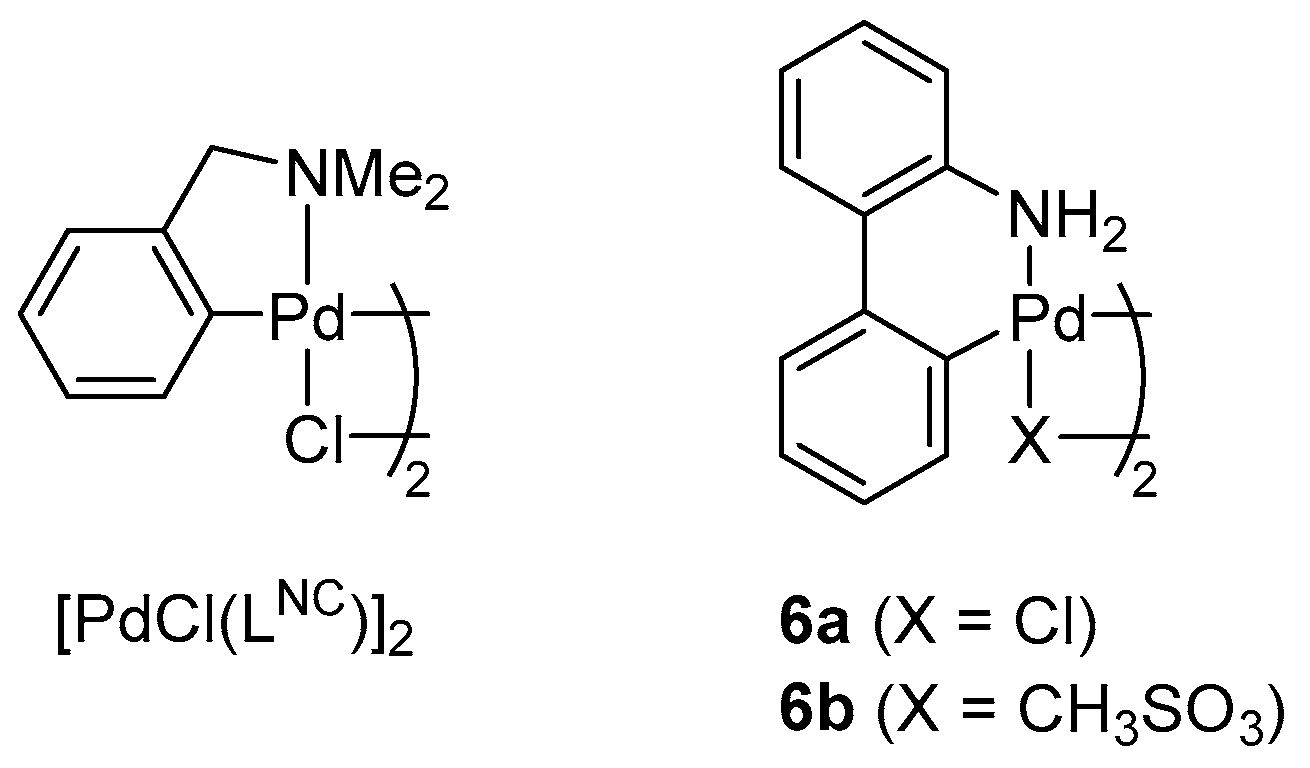
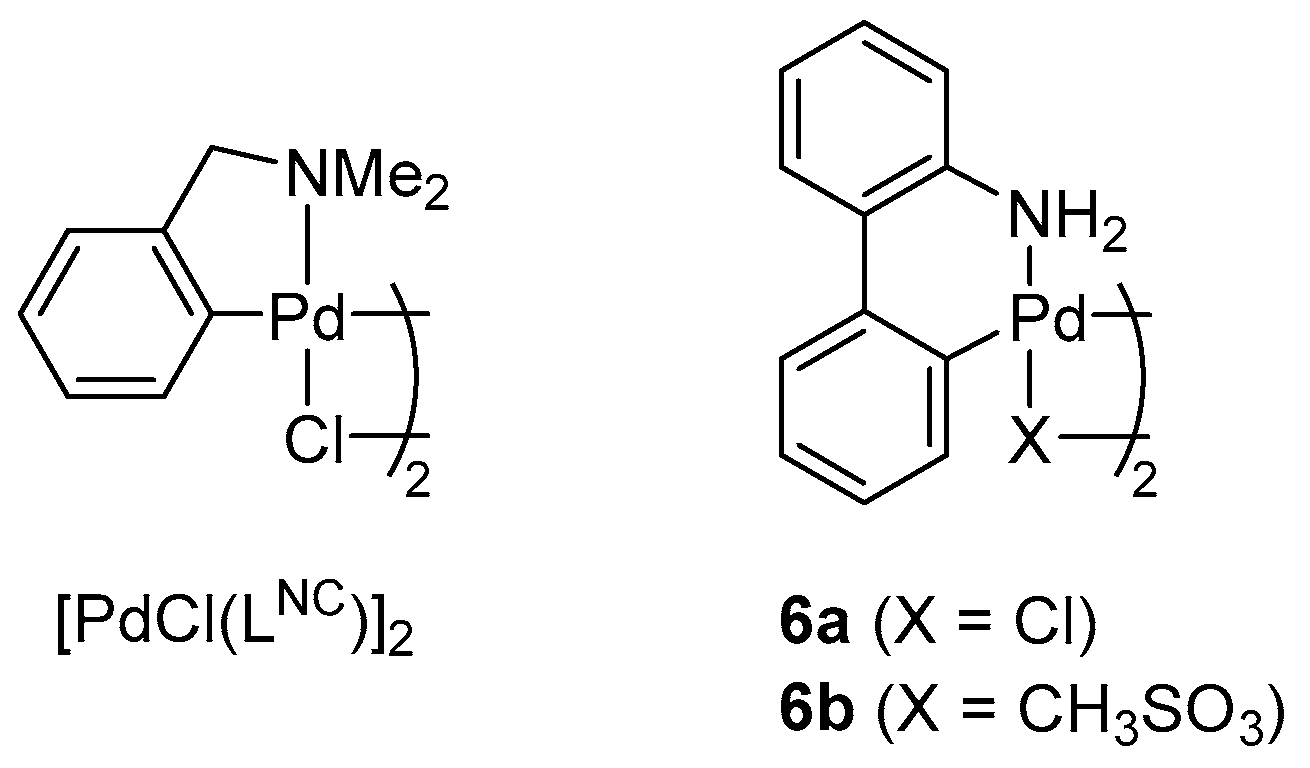
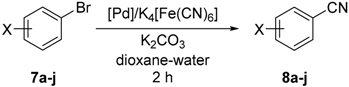
2.2. Pd-Catalyzed Cyanation of Aryl Bromides
3. Experimental
3.1. Materials and Methods
3.2. Preparation of 2′-(Dicyclohexylphosphino)-2-bromo[1,1′-biphenyl]–borane (1:1) (Compound 2)
3.3. Preparation of 2′-(Dicyclohexylphosphino)[1,1′-biphenyl]-2-carboxylic acid–borane (1:1) (Compound 3)
3.4. Synthesis of 2′-(Dicyclohexylphosphino)[1,1′-biphenyl]-2-carboxylic acid–borane (1:1), Pentafluorophenyl Ester (Compound 4)
3.5. Preparation of 2′-(Dicyclohexylphosphino)-2-{[(sulfonatomethyl)amino]carbonyl}[1,1′-biphenyl]–borane (1:1), Triethylammonium Salt (Compound 5)
3.6. Preparation of 2′-(Dicyclohexylphosphino)-2-{[(sulfonatomethyl)amino]carbonyl}[1,1′-biphenyl], Triethylammonium Salt (Ligand L2)
3.7. Pd-Catalyzed Cyanation of Aryl Bromides. General Procedure for Screening Experiments
3.8. Pd-Catalyzed Cyanation of Aryl Bromides. General Procedure for Preparative Experiments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Meijere, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004.
- Molnár, A. (Ed.) Palladium-Catalyzed Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2013.
- Colacot, T.J. (Ed.) New Trends in Cross-Coupling: Theory and Applications; The Royal Society of Chemistry: Cambridge, UK, 2015.
- Anbarasan, P.; Schareina, T.; Beller, M. Recent developments and perspectives in palladium-catalyzed cyanation of aryl halides: Synthesis of benzonitriles. Chem. Soc. Rev. 2011, 40, 5049–5067. [Google Scholar] [CrossRef] [PubMed]
- Vafaeezadeh, M.; Hashemi, M.M.; Karbalaie-Reza, M. The possibilities of palladium-catalyzed aromatic cyanation in aqueous media. Inorg. Chem. Commun. 2016, 72, 86–90. [Google Scholar] [CrossRef]
- Schareina, T.; Zapf, A.; Beller, M. Potassium hexacyanoferrate(II)—A new cyanating agent for the palladium-catalyzed cyanation of aryl halides. Chem. Commun. 2004, 35, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Yeung, P.Y.; So, C.M.; Lau, C.P.; Kwong, F.Y. A mild and efficient palladium-catalyzed cyanation of aryl mesylates in water or tBuOH/Water. Angew. Chem. Int. Ed. 2010, 49, 8918–8922. [Google Scholar] [CrossRef] [PubMed]
- Hajipour, A.R.; Karami, K.; Pirisedigh, A. Palladium-catalyzed cyanation reaction of aryl halides using K4[Fe(CN)6] as non-toxic source of cyanide under microwave irradiation. Appl. Organomet. Chem. 2010, 24, 454–457. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Hu, T.; Zhang, Y.; Xu, K.; Yu, Y.; Huang, J. Highly efficient Pd-catalyzed cyanation of aryl chlorides and arenesulfonates with potassium ferrocyanide in Aqueous Media. Catal. Lett. 2010, 139, 56–60. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, H.; Zhang, L.; Zhou, Y.; Li, C.; Jiang, H.; Chen, K.; Liu, H. An expedient Pd/DBU mediated cyanation of aryl/heteroaryl bromides with K4[Fe(CN)6]. Chem. Commun. 2012, 43, 2909–2911. [Google Scholar] [CrossRef] [PubMed]
- Senecal, T.D.; Shu, E.; Buchwald, S.L. A general, practical palladium-catalyzed cyanation of (hetero)aryl chlorides and bromides. Angew. Chem. Int. Ed. 2013, 52, 10035–10039. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Feng, X.; Liu, H.; Yu, X.; Yamamoto, Y.; Bao, M. Efficient palladium-catalyzed cyanation of aryl/heteroaryl bromides with K4[Fe(CN)6] in t-BuOH–H2O using tris(2-morpholinophenyl)phosphine as a ligand. RSC Adv. 2013, 3, 20379–20384. [Google Scholar] [CrossRef]
- Schulz, J.; Císařová, I.; Štěpnička, P. Phosphinoferrocene amidosulfonates: Synthesis, palladium complexes, and catalytic use in Pd-Catalyzed cyanation of aryl bromides in an aqueous reaction medium. Organometallics 2012, 31, 729–738. [Google Scholar] [CrossRef]
- Škoch, K.; Císařová, I.; Štěpnička, P. Phosphinoferrocene Ureas: Synthesis, structural characterization, and catalytic use in palladium-catalyzed cyanation of aryl bromides. Organometallics 2015, 34, 1942–1956. [Google Scholar] [CrossRef]
- Martin, R.; Buchwald, S.L. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Císařová, I.; Štěpnička, P. Synthesis of an amidosulfonate-tagged biphenyl phosphine and its application in the Suzuki-Miyaura reaction affording biphenyl-substituted amino acids in water. J. Organomet. Chem. 2015, 796, 65–72. [Google Scholar] [CrossRef]
- Anderson, K.W.; Buchwald, S.L. General catalysts for the Suzuki–Miyaura and Sonogashira coupling reactions of aryl chlorides and for the coupling of challenging substrate combinations in water. Angew. Chem. Int. Ed. 2005, 44, 6173–6177. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Štěpnička, P. Phosphino-carboxamides: The inconspicuous gems. Chem. Soc. Rev. 2012, 41, 4273–4305. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.M.; Faure, B.; Maffei, M. Phosphane–boranes: Synthesis, characterization and synthetic applications. Coord. Chem. Rev. 1998, 178–180, 665–698. [Google Scholar] [CrossRef]
- In the IR spectrum of neat ethyl acetate, the C=O stretching band occurs at 1743 cm−1. Data from National Institute of Advanced Industrial Science and Technology. Spectral Database for Organic Compounds: SDBS. Available online: http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 22 September 2016).
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 2nd ed.; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Kinzel, T.; Zhang, Y.; Buchwald, S.L. A new palladium precatalyst allows for the fast Suzuki-Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J. Am. Chem. Soc. 2010, 132, 14073–14075. [Google Scholar] [CrossRef] [PubMed]
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Carole, W.A.; Colacot, T.J. Understanding palladium acetate from a user perspective. Chem. Eur. J. 2016, 22, 7686–7695. [Google Scholar] [CrossRef] [PubMed]
- Klinkenberg, J.L.; Hartwig, J.F. Reductive elimination from arylpalladium cyanide complexes. J. Am. Chem. Soc. 2012, 134, 5758–5761. [Google Scholar] [CrossRef] [PubMed]
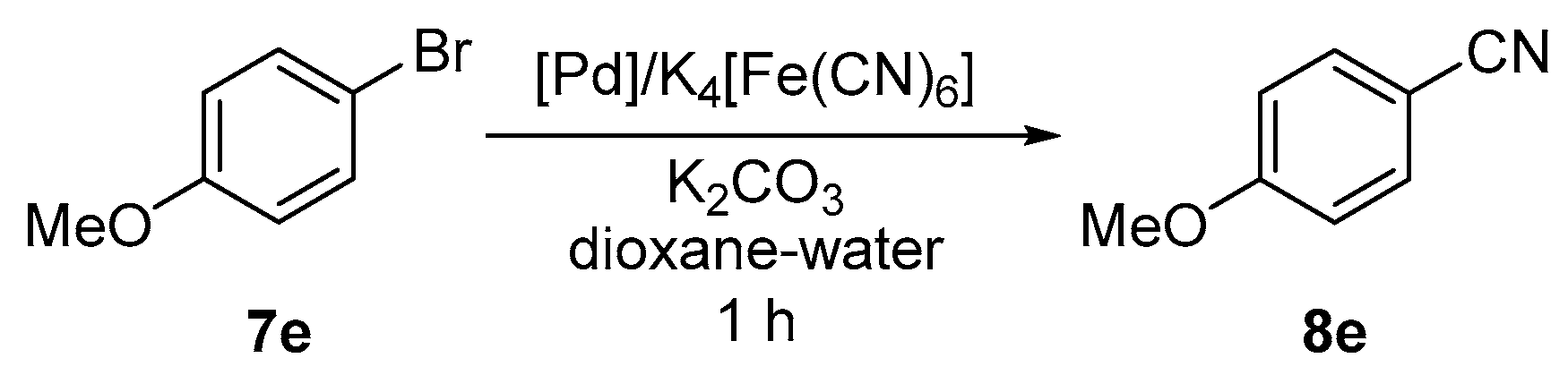

{kind=link}
{kind=link}
{kind=link}
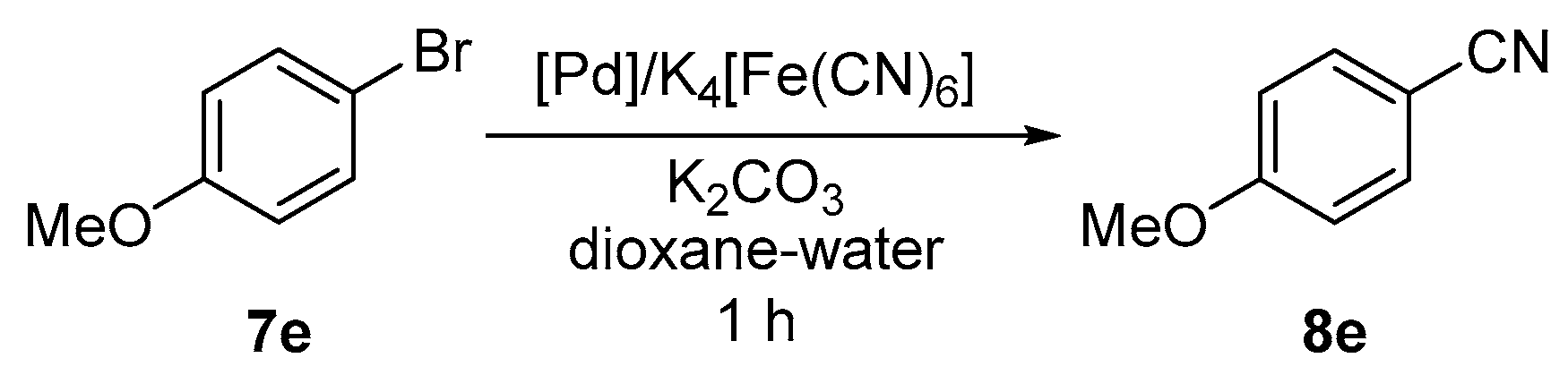{kind=link}
| Entry | Pd Source | L:Pd Ratio | Ligand L1 | Ligand L2 |
|---|---|---|---|---|
| 1 | Pd(OAc)2 | 1:1 | 0 | 33 |
| 2 | Pd(OAc)2 | 2:1 | 96 (6 b) | 16 |
| 3 | [PdCl2(cod)] | 1:1 | 100 | 30 |
| 4 | [PdCl2(cod)] | 2:1 | 20 | 14 |
| 5 | K2[PdCl4] | 1:1 | 0 | 0 |
| 6 | K2[PdCl4] | 2:1 | 2 | 0 |
| 7 | 6a | 1:1 | 63 | 18 |
| 8 | 6b | 1:1 | 70 | 26 |
| 9 | [PdCl(LNC)]2 | 1:1 | 66 | 29 |
| 10 | [(η3-C3H5)PdCl]2 | 1:1 | 0 | 3 |
 | |||||
|---|---|---|---|---|---|
| Substituent | Product | Conversion (Yield) (%) | Substituent | Product | Conversion after 2 h (24 h) (%) |
| 2-Me | 8a | 100 (70) | 4-NO2 | 8f | <5 [<5] |
| 3-Me | 8b | 100 (84) | 4-Cl | 8g | <5 [10] |
| 4-Me | 8c | 100 (87) | 4-CF3 | 8h | <5 [<5] |
| 4-t-Bu | 8d | 100 (87) | 4-CHO | 8i | 0 [0] |
| 4-MeO | 8e | 100 (82) | 4-CO2H b | 8j | <5 [<5] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, J.; Horký, F.; Štěpnička, P. Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides. Catalysts 2016, 6, 182. https://doi.org/10.3390/catal6120182
Schulz J, Horký F, Štěpnička P. Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides. Catalysts. 2016; 6(12):182. https://doi.org/10.3390/catal6120182
Chicago/Turabian StyleSchulz, Jiří, Filip Horký, and Petr Štěpnička. 2016. "Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides" Catalysts 6, no. 12: 182. https://doi.org/10.3390/catal6120182
APA StyleSchulz, J., Horký, F., & Štěpnička, P. (2016). Different Performance of Two Isomeric Phosphinobiphenyl Amidosulfonates in Pd-Catalyzed Cyanation of Aryl Bromides. Catalysts, 6(12), 182. https://doi.org/10.3390/catal6120182