
Cyclopropanation of 5-(1-Bromo-2-phenyl-vinyl)-3-methyl-4-nitro-isoxazoles under Phase Transfer Catalysis (PTC) Conditions


Abstract
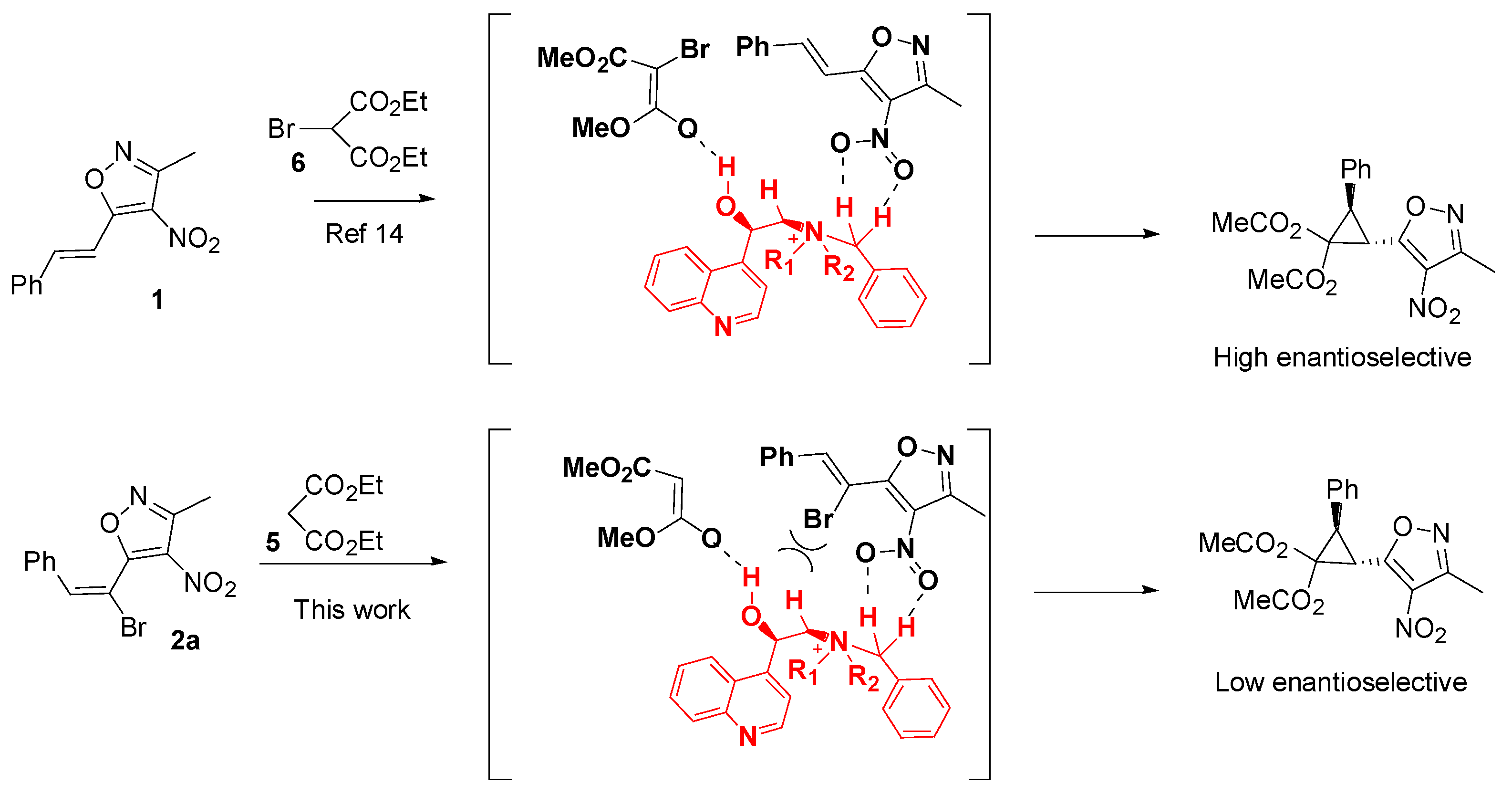
:1. Introduction
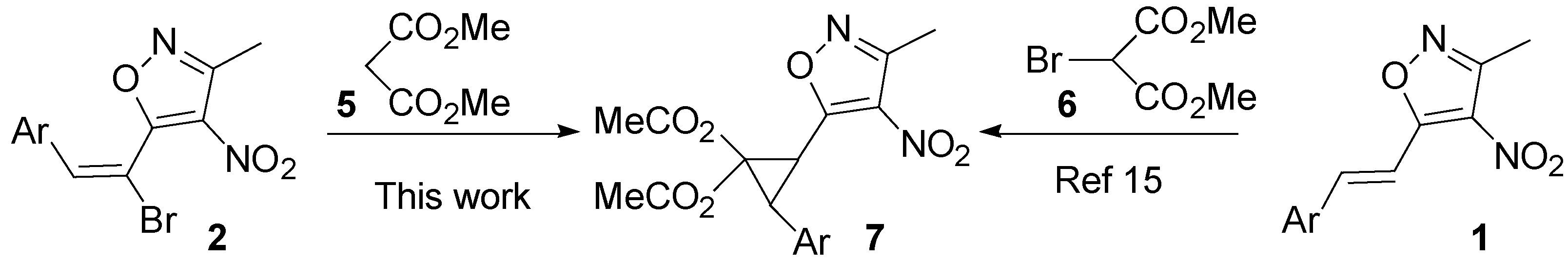
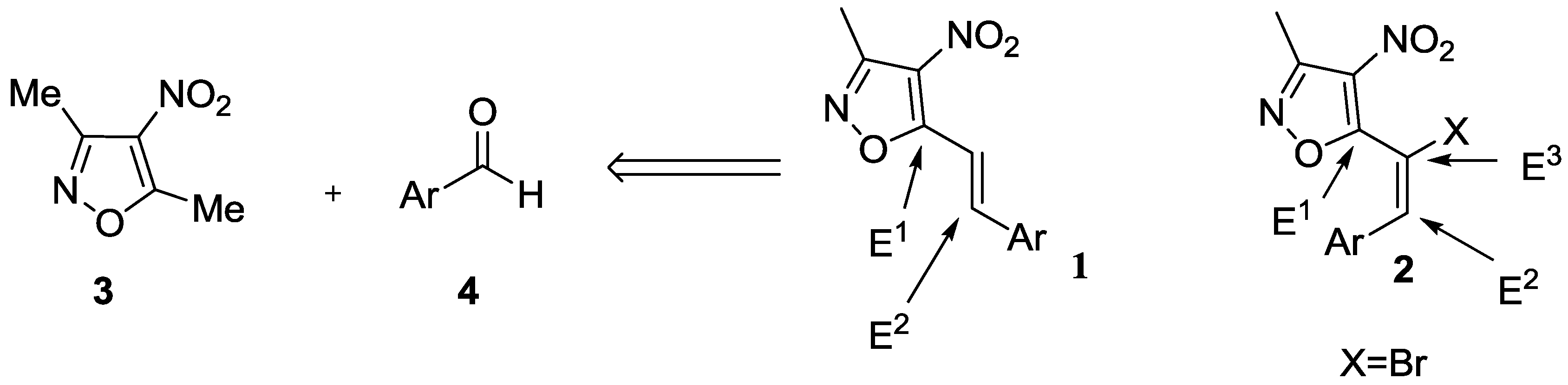
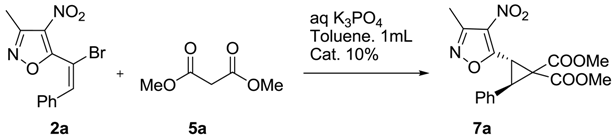
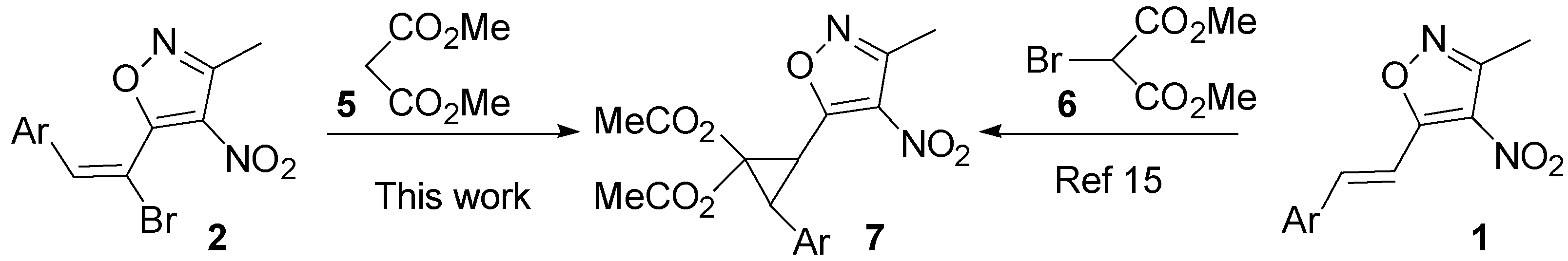
2. Results and Discussion

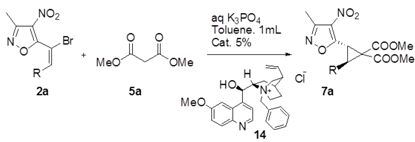
| Entry [a] | Catalyst | Time/h | Conv. % of 2a [b] | ee% of 7a [c] |
|---|---|---|---|---|
| 1 | 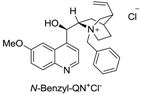 | 48 | >90% | −42 |
| 2 | 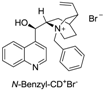 | 48 | <60% | −20 |
| 3 |  | 24 | >95% | +36 |
| 4 |  | 24 | >90% | +10 |
| 5 |  | 12 | >90% | +4 |
| 6 |  | 48 | >95% | −40 |
| 7 | 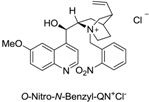 | 48 | >95% | −26 |
| 8 |  | 48 | >95% | −24 |
| 9 |  | 48 | >95% | −29 |
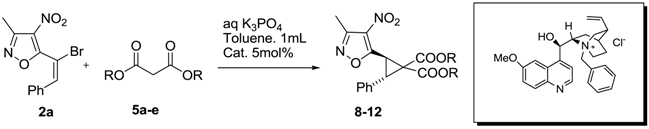
{kind=link}
{kind=link}
{kind=link}
| Entry | R | Product | Time/h | Conv.of 2a [b] | ee% [c] |
|---|---|---|---|---|---|
| 1 | Me | 8 | 48 | >95% | −42 |
| 2 | Et | 9 | 48 | >95% | −40 |
| 3 | iPr | 10 | 72 | <70% | −26 |
| 4 | Bn | 11 | 72 | >95% | −30 |
| 5 | Ph | 12 | 72 | >95% | 0 |
| Entry | R | Prod. | Time/h | Yield[%] [b] | ee[%] [c] | Isomer |
|---|---|---|---|---|---|---|
| 1 | C6H5 | 7a | 48 | 97 | 42 | (−) |
| 2 | 4-OCH3- C6H4 | 7b | 48 | 97 | 45 | (−) |
| 3 | 2,3-Cl2- C6H3 | 7c | 48 | 95 | 40 | (−) |
| 4 | 2-Cl- C6H4 | 7d | 48 | 94 | 48 | (−) |
| 5 | 2-thienyl | 7e | 48 | 95 | 49 | (−) |
| 6 | 2-OMe- C6H4 | 7f | 48 | 97 | 56 | (−) |
| 7 | 2-Br- C6H4 | 7g | 48 | 94 | 54 | (−) |
| 8 | 2,4-OMe- C6H3 | 7h | 48 | 91 | 58 | (−) |
| 9 | n-heptyl | 7i | 48 | 96 | 46 | (−) |
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
References
- Adamo, M.F.A.; Duffy, E.F.; Konda, V.R.; Murphy, F. An improved synthesis of 3-methyl-4-nitro-5-heteroarylethenylisoxazoles. Heterocycles 2007, 71, 1173–1181. [Google Scholar] [CrossRef]
- Adamo, M.F.A.; Duffy, E.F. Multicomponent synthesis of 3-heteroarylpropionic acids. Org. Lett. 2006, 8, 5157–5159. [Google Scholar] [CrossRef] [PubMed]
- Adamo, M.F.A.; Suresh, S.; Piras, L. Reaction of 5-(1-bromo-2-arylvinyl)-3-methyl-4-nitroisoxazoles and 1,3-dicarbonyl compounds. Tetrahedron 2009, 65, 5402–5408. [Google Scholar] [CrossRef]
- Adamo, M.F.A.; Chimichi, S.; De Sio, F.; Donati, D.; Sarti-Fantoni, P. The reactivity of 3-methyl-4-nitro-5-styrylisoxazole with some bis-enolisable ketones. Tetrahedron Lett. 2002, 43, 4157–4160. [Google Scholar] [CrossRef]
- Adamo, M.F.A.; Donati, D.; Duffy, E.F.; Sarti-Fantoni, P. Multicomponent synthesis of spiroisoxazolines. J. Org. Chem. 2005, 70, 8395. [Google Scholar] [CrossRef] [PubMed]
- Adamo, M.F.A.; Duffy, E.F.; Donati, D.; Sarti-Fantoni, P. Modular synthesis of isoxazolepyridones and pyrazolepyridones. Tetrahedron 2007, 63, 2047–2052. [Google Scholar] [CrossRef]
- Adamo, M.F.A.; Konda, V.R. Multicomponent synthesis of 3-indolepropionic acids. Org. Lett. 2007, 9, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Fini, F.; Nagabelli, M.; Adamo, M.F.A. Development of a Mild Procedure for the Addition of Bisulfite to Electrophilic Olefins. Adv. Synth. Catal. 2010, 352, 3163–3168. [Google Scholar] [CrossRef]
- Baschieri, A.; Bernardi, L.; Ricci, A.; Suresh, S.; Adamo, M.F.A. Catalytic Asymmetric Conjugate Addition of Nitroalkanes to 4-Nitro-5-styrylisoxazoles. Angew. Chem. Int. Ed. 2009, 48, 9342–9345. [Google Scholar]
- Wells, R.; Moccia, M.; Adamo, M.F.A. The preparation of 3-methyl-4-nitro-5-(2-alkylethenyl)isoxazoles. Tetrahedron Lett. 2014, 55, 803–805. [Google Scholar] [CrossRef]
- Chimichi, S.; De Sio, F.; Donati, D.; Fina, G.; Pepino, R.; Sarti-Fantoni, P. The preparation of coumaric acids via styrylisoxazoles. Heterocycles 1983, 20, 263–267. [Google Scholar] [CrossRef]
- Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Trifluoromethylation of Aromatic Isoxazoles: Regio- and Diastereoselective route to 5-Trifluoromethyl-2-isoxazolines. Angew. Chem. Int. Ed. 2011, 50, 7803–7806. [Google Scholar] [CrossRef]
- Kawai, H.; Sugita, Y.; Tokunaga, E.; Sato, H.; Shiro, M.; Shibata, N. Diastereoselective Additive Trifluoromethylation/Halogenation of Isoxazole Triflones: Synthesis of All-Carbon-Functionalized Trifluoromethyl Isoxazoline Triflones. Chemistry Open. 2014, 3, 14–18. [Google Scholar] [PubMed]
- Wang, X.; Tokunaga, E.; Shibata, N. Direct nucleophilic difluoromethylation of aromatic isoxazoles activated by electron-withdrawing groups using (difluoromethyl)trimethylsilane. ScienceOpen Res. 2015. [Google Scholar] [CrossRef]
- Pietruszka, J. Synthesis and Properties of Oligocyclopropyl-Containing Natural Products and Model Compounds. Chem. Rev. 2003, 103, 1051–1070. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, W.A. Synthesis of cyclopropane containing natural products. Tetrahedron 2001, 57, 8589–8627. [Google Scholar] [CrossRef]
- Lebel, H.; Marcoux, J.F.; Molinaro, C.; Charet, A.B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Smith, H.W.; Jones, R.V.H.; Fieldhouse, R. Catalytic Asymmetric Cyclopropanation of Electron Deficient Alkenes Mediated By Chiral Sulfides. Chem. Commun. 1998, 1785, 1785–1786. [Google Scholar]
- Aggarwal, V.K.; Alonso, E.; Fang, G.; Ferrara, M.; Hynd, G.; Porcelloni, M. Application of Chiral Sulfides to Catalytic Asymmetric Aziridination and Cyclopropanation with In Situ Generation of the Diazo Compound. Angew. Chem. Int. Ed. 2001, 40, 1433–1436. [Google Scholar] [CrossRef]
- Zheng, J.C.; Liao, W.W.; Tang, Y.; Sun, X.L.; Dai, L.-X. The Michael Addition−Elimination of Ylides to α,β-Unsaturated Imines. Highly Stereoselective Synthesis of Vinylcyclopropanecarbal-dehydes and Vinylcyclopropylaziridines. J. Am. Chem. Soc. 2005, 127, 12222–12223. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cai, P.; Ye, S.; Sun, X.; Liao, W.; Li, K.; Tang, Y.; Wu, Y.D.; Dai, L.X. Enantioselective Synthesis of Vinylcyclopropanes and Vinylepoxides Mediated by Camphor-Derived Sulfur Ylides: Rationale of Enantioselectivity, Scope, and Limitation. J. Am. Chem. Soc. 2006, 128, 9730–9740. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Grange, E. Asymmetric Sulfonium Ylide Mediated Cyclopropanation: Stereocontrolled Synthesis of (+)-LY354740. Chem. Eur. J. 2006, 12, 568. [Google Scholar] [CrossRef]
- Special Issue on “Organocatalysis”; List, B. (Ed.) ACS: Washington, DC, USA, 2007; pp. 5413–5883.
- Houk, K.N.; List, B. Asymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 487. [Google Scholar]
- Bremeyer, N.; Smith, S.C.; Ley, S.V.; Gaunt, M.J. An Intramolecular Organocatalytic Cyclopropanation Reaction. Angew. Chem. Int. Ed. 2004, 43, 2681–2684. [Google Scholar] [CrossRef]
- Papageorgiou, C.D.; Cubillo de Dios, M.A.; Ley, S.V.; Gaunt, M.J. Enantioselective Organocatalytic Cyclopropanation via Ammonium Ylides. Angew. Chem. Int. Ed. 2004, 43, 4641–4644. [Google Scholar] [CrossRef]
- Johansson, C.C.C.; Bremeyer, N.; Ley, S.V.; Owen, D.R.; Smith, S.C.; Gaunt, M.J. Enantioselective Catalytic Intramolecular Cyclopropanation using Modified Cinchona Alkaloid Organocatalysts. Angew. Chem. Int. Ed. 2006, 45, 6024–6028. [Google Scholar] [CrossRef]
- Belyk, K.M.; Xiang, B.; Bulger, P.G.; Leonard, W.R.; Balsells, J.; Yin, J.; Chen, C. Enantioselective Synthesis of (1R,2S)-1-Amino-2-vinylcyclopropanecarboxylic Acid Ethyl Ester (Vinyl-ACCA-OEt) by Asymmetric Phase-Transfer Catalyzed Cyclopropanation of (E)-N-Phenylmethyleneglycine Ethyl Ester. Org. Proc. Res. Develop. 2010, 14, 692–700. [Google Scholar] [CrossRef]
- Herchl, R.; Waser, M. Asymmetric cyclopropanation of chalcones using chiral phase-transfer catalysts. Tetrahedron Lett. 2013, 54, 2472–2475. [Google Scholar] [CrossRef] [PubMed]
- Kunz, R.K.; MacMillan, D.W.C. Enantioselective Organocatalytic Cyclopropanations. The Identification of a New Class of Iminium Catalyst Based upon Directed Electrostatic Activation. J. Am. Chem. Soc. 2005, 127, 3240–3241. [Google Scholar] [CrossRef] [PubMed]
- McCooey, S.H.; McCabe, T.; Connon, S.J. Stereoselective Synthesis of Highly Functionalized Nitrocyclopropanes via Organocatalyic Conjugate Addition to Nitroalkenes. J. Org. Chem. 2006, 71, 7494–7497. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.M.; Longbottom, D.A.; Ley, S.V. A new asymmetric organocatalytic nitrocyclopropanation reaction. Chem. Commun. 2006, 4838–4840. [Google Scholar] [CrossRef]
- Xie, H.; Zu, L.; Li, H.; Wang, J.; Wang, W.J. Organocatalytic Enantioselective Cascade Michael-Alkylation Reactions: Synthesis of Chiral Cyclopropanes and Investigation of Unexpected Organocatalyzed Stereoselective Ring Opening of Cyclopropanes. J. Am. Chem. Soc. 2007, 129, 10886–10894. [Google Scholar] [CrossRef] [PubMed]
- Rios, R.; Sunden, H.; Vesely, J.; Zhao, G.L.; Dziedzic, P.; Cordova, A. A Simple Organocatalytic Enantioselective Cyclopropanation of α,β-Unsaturated Aldehydes. Adv. Synth. Catal. 2007, 349, 1028–1032. [Google Scholar] [CrossRef]
- Hartikka, A.; Slosarczyk, A.T.; Arvidsson, P.I. Application of novel sulfonamides in enantioselective organocatalyzed cyclopropanation. Tetrahedron 2007, 18, 1403–1409. [Google Scholar] [CrossRef]
- Wascholowski, V.; Hansen, H.M.; Longbottom, D.A.; Ley, S.V. A general organocatalytic enantioselective nitrocyclopropanation reaction. Synthesis 2008, 1269–1275. [Google Scholar]
- Ibrahem, I.; Zhao, G.L.; Rios, R.; Vesely, J.; Sunden, H.; Dziedzic, P.; Cordova, A. One-Pot Organocatalytic Domino Michael/α-Alkylation Reactions: Direct Catalytic Enantioselective Cyclopropanation and Cyclopentanation Reactions. Chem. Eur. J. 2008, 14, 7867–7879. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, J.; Lin, Z.; Wang, Y. Enantioselective Synthesis of Functionalized Nitrocyclopropanes by Organocatalytic Conjugate Addition of Bromonitroalkanes to α,β-Unsaturated Enones. Chem. Eur. J. 2009, 15, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Nie, S.; Dong, L.; Zhang, J.; Yan, M. Highly Enantioselective Synthesis of Nitrocyclopropanes via Organocatalytic Conjugate Addition of Bromomalonate to α,β-Unsaturated Nitroalkenes. Org. Lett. 2009, 11, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Sternativo, S.; Del Verme, F.; Testaferri, L.; Tiecco, M.A. New Stereoselective Synthesis of Cyclopropanes Containing Quaternary Stereocentres via Organocatalytic Michael Addition to Vinyl Selenones. Adv. Synth. Catal. 2009, 351, 1801–1806. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew.Chem Int. Ed. Engl. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Nakayama, K.; Ishida, T.; Shioiri, T. Asymmetric cyclopropanation reaction under phase-transfer catalyzed conditions. Tetrahedron: Lett. 1999, 40, 4215–4218. [Google Scholar] [CrossRef]
- Del Fiandra, C.; Piras, L.; Fini, F.; Disetti, P.; Moccia, M.; Adamo, M.F.A. Phase transfer catalyzed enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles. Chem Commun. 2012, 48, 3863–3865. [Google Scholar] [CrossRef]
- Gomez, B.E.; Linden, A.; López, R.; Mendiola, I.M.; Oiarbide, M.; Palomo, C. Asymmetric Aza-Henry Reaction Under Phase Transfer Catalysis: An Experimental and Theoretical Study. J. Am. Chem. Soc. 2008, 130, 7955–7966. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piras, L.; Moccia, M.; Cortigiani, M.; Adamo, M.F.A. Cyclopropanation of 5-(1-Bromo-2-phenyl-vinyl)-3-methyl-4-nitro-isoxazoles under Phase Transfer Catalysis (PTC) Conditions. Catalysts 2015, 5, 595-605. https://doi.org/10.3390/catal5020595
Piras L, Moccia M, Cortigiani M, Adamo MFA. Cyclopropanation of 5-(1-Bromo-2-phenyl-vinyl)-3-methyl-4-nitro-isoxazoles under Phase Transfer Catalysis (PTC) Conditions. Catalysts. 2015; 5(2):595-605. https://doi.org/10.3390/catal5020595
Chicago/Turabian StylePiras, Linda, Maria Moccia, Mauro Cortigiani, and Mauro F. A. Adamo. 2015. "Cyclopropanation of 5-(1-Bromo-2-phenyl-vinyl)-3-methyl-4-nitro-isoxazoles under Phase Transfer Catalysis (PTC) Conditions" Catalysts 5, no. 2: 595-605. https://doi.org/10.3390/catal5020595
APA StylePiras, L., Moccia, M., Cortigiani, M., & Adamo, M. F. A. (2015). Cyclopropanation of 5-(1-Bromo-2-phenyl-vinyl)-3-methyl-4-nitro-isoxazoles under Phase Transfer Catalysis (PTC) Conditions. Catalysts, 5(2), 595-605. https://doi.org/10.3390/catal5020595