Bounechada
et al. [
11] reported on the activity of a Pd-Rh (Pd/Rh = 39/1) TWC converter supported on stabilized Al
2O
3, promoted with Ce-Zr (Zr/Ce = 3.5) and wash coated on a ceramic honeycomb monolith, tested under fuel-lean (λ > 1), stoichiometric (λ = 1.00), and fuel-rich (λ < 1) conditions (gas composition: 0.15 vol.% CH
4, 0.6% CO, 0.1% H
2, 10% H
2O, 10.7% CO
2, 0.13% NO, 0–1.14% O
2; λ was varied by changing feed O
2 concentration; GHSV = 50,000 h
−1). At stationary conditions (constant λ; steady-state experiment), the CH
4 conversion was observed to continuously decrease under both stoichiometric (52 to 43% after 0.5 h reaction) and fuel-lean (from 62 to 59% after 0.5 h reaction) conditions, even though injecting a fuel-rich pulse during fuel-lean stationary operation increased the CH
4 conversion to its initial value at the onset of reaction. The authors attributed the deactivation under fuel-lean conditions to the inhibition effect of H
2O on the CH
4 oxidation reaction, whereas under stoichiometric conditions, partial reduction of PdO due to the lack of oxygen, may lead to a loss in PdO active sites for CH
4 oxidation. The authors also claimed that the presence of high oxygen capacity metals (Ce and Zr) in the catalyst made the reduction of PdO improbable under stoichiometric conditions. Under fuel-rich conditions, H
2O acts as an oxidant through water-gas shift and steam reforming reactions.
2.1. Water Concentration and Reaction Temperature Effects on CH4 Oxidation Activity of Pd Catalysts
With the growing interest in NGVs, recent studies have focused on effects of H
2O on Pd catalysts during CH
4 combustion [
16,
18,
30,
31,
32,
33,
34,
35,
36,
37,
38]. Deactivation or inhibition effects of H
2O are dependent upon several factors including catalyst formulation, reaction temperature, catalyst time-on-stream history, and H
2O concentration.
Table 1 summarizes selected data that show effects of H
2O added to the feed gas during CH
4 light-off experiments over Pd catalysts. The light-off temperature (here reported as the temperature corresponding to 30% CH
4 conversion during temperature programmed reaction,
T30) increases as the H
2O concentration increases, showing a clear inhibition effect that increases in magnitude with increasing H
2O concentration.
In several cases the effects of H
2O have been examined by measuring the CH
4 conversion at steady-state, with and without H
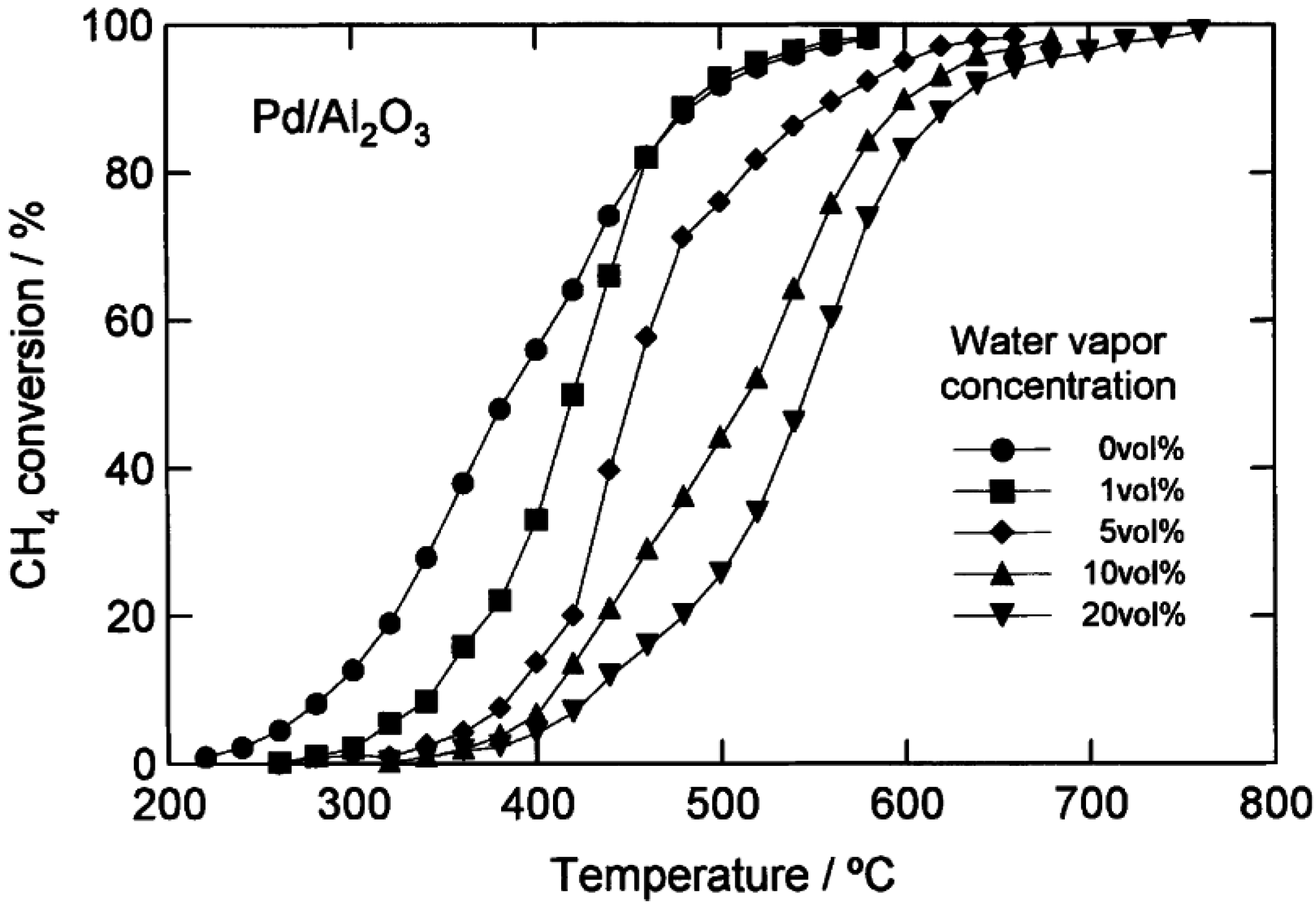
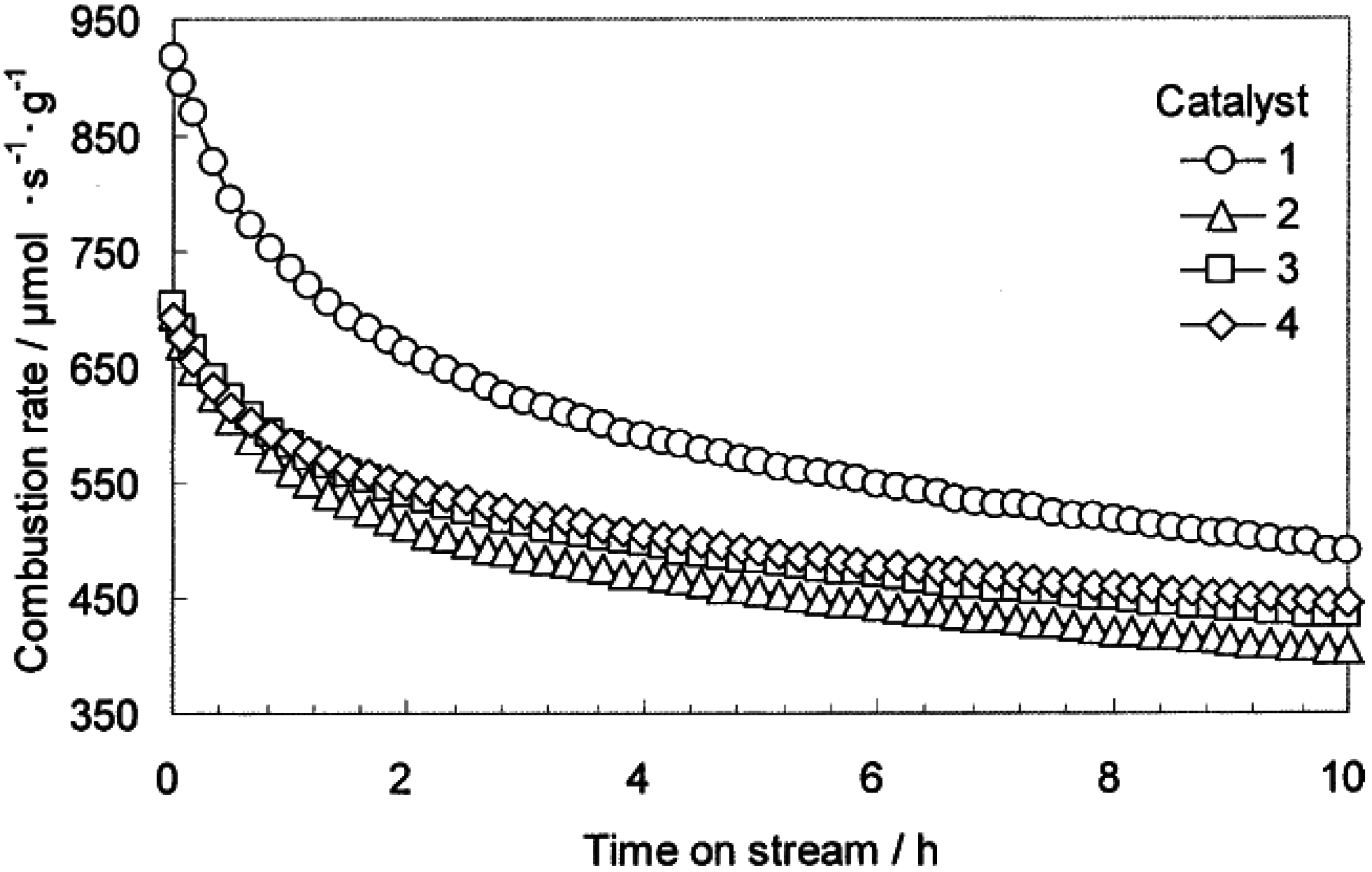
2O added to the feed gas. A typical set of data, reported by Persson
et al. [
35], is shown in
Figure 1 using several Pd/Al
2O
3 catalysts reacted at 500 °C. These data also show that added H
2O significantly suppresses CH
4 conversion, but the effect is at least partially reversible. Similar effects of H
2O addition have been reported in the literature, as summarized in
Table 2. These reports confirm that H
2O acts as an inhibitor of CH
4 oxidation over Pd catalysts and that upon removal of the H
2O from the CH
4/O
2 reactant, the inhibition is partially reversible [
31,
33].
Figure 1.
Effect of water vapor on the activity for CH
4 combustion over Pd/Al
2O
3 (■); 2:1 PdPt/Al
2O
3 (∆); and 1:1 PdPt/Al
2O
3 (o) at 500 °C; 5 vol.% of steam was added to the 1.5% CH
4/air feed gas, GHSV = 100,000 h
−1, for 5 h. [
35] Copyright© 2007 Elsevier.
Figure 1.
Effect of water vapor on the activity for CH
4 combustion over Pd/Al
2O
3 (■); 2:1 PdPt/Al
2O
3 (∆); and 1:1 PdPt/Al
2O
3 (o) at 500 °C; 5 vol.% of steam was added to the 1.5% CH
4/air feed gas, GHSV = 100,000 h
−1, for 5 h. [
35] Copyright© 2007 Elsevier.
Table 1.
Effect of H2O addition on T30 (temperature at 30% CH4 conversion) during temperature-programmed CH4 oxidation over Pd catalysts.
Table 1.
Effect of H2O addition on T30 (temperature at 30% CH4 conversion) during temperature-programmed CH4 oxidation over Pd catalysts.
| | Reference [16] | Reference [39] |
| 1.1% Pd/Al2O3 | 1.1% Pd/SnO2 | 1.1% Pd/Al2O3-36NiO | 0.9% Pd/ZrO2 a | 0.9% Pd/ZrO2 b |
| GHSV, h−1 | 48,000 | 50,000 |
| Dry feed gas composition, vol.% | 1% CH4/20% O2 in N2 | 0.4% CH4/0.05% CO/5% CO2/10% O2 in N2 |
| | T30, °C | T30, °C |
| Added H2O, vol.% | | | | | |
| 0 | 345 | 290 | 372 | 360 | 300 |
| 1 | 400 | 315 | 372 | - | - |
| 5 | 430 | 335 | 420 | - | - |
| 10 | 460 | 360 | 425 | 410 | 350 |
| 20 | 510 | 365 | 445 | - | - |
Table 2.
H2O inhibition over Pd catalysts during CH4 oxidation.
Table 2.
H2O inhibition over Pd catalysts during CH4 oxidation.
| Catalyst | Reaction conditions | Conc. vol % | Period a min | Water addition CH4 Conversion, % | Comments | Refs. |
| Temp °C | GHSV h−1 | Feed Gas mol % | Before H2O addition | During H2O addition | After H2O addition |
| 0.1 wt.% Pd/H-beta | 400 | 120,000 | 0.2% CH4/10%O2 in N2 | 10 | 100 | 75 | 15 | 58 | Conversion after 400 min TOS with periodic water addition | [38] |
| 1.3 wt.% Pd/HTNU-10c | 400 | 120,000 | 1% CH4/4%O2 in N2 | 5 | 900 | 43 | 8 | 40 | Conversion after 35 h TOS with periodic water addition | [33] |
| 5 wt.% Pd/Al2O3 | 500 | 100,000 | 1.5% CH4 in air | 5 | 300 | 95 | 13 | 30 | Initial activity | [35] |
| 2 wt.% Pd/Al2O3 | 550 | 160,000 | 0.4% CH4 in air | 10 | 60 | 95 | 79 | 92 | Conversion after 400 min TOS with periodic water addition | [32] |
| 1 wt.% Pd/Al2O3 | 600 | 160,000 | 0.4% CH4 in air | 8 | 60 | 95 | 90 | 93 | Conversion after 300 min TOS with periodic water addition | [36] |
Reaction temperature is another key variable affecting the role of H
2O addition. Although the data of
Table 2 cannot be compared directly because of the different operating conditions, they do show that at 600 °C, the decrease in CH
4 conversion with H
2O addition is much less significant than at lower temperatures (400 °C). Several authors have proposed that the deactivation is related to the reaction of H
2O with active PdO sites [
16,
18,
31,
40,
41], PdO + H
2O→Pd(OH)
2, resulting in the formation of inactive Pd(OH)
2, as first proposed by Cullis
et al. [
40]. Burch
et al. [
31] also reported a strong inhibitory effect of water on Pd catalysts up to 450 °C. However, at higher temperatures the negative influence of water on the activity was very small, suggesting that above 450 °C the reverse reaction (Pd(OH)
2→PdO + H
2O) occurs. Eriksson
et al. [
41] observed a significant decrease in CH
4 conversion over a much wider range of temperatures (200–800 °C) after adding 18% H
2O to a CH
4/O
2 feed over a Pd/ZrO
2 catalyst, which was likely due to the relatively high H
2O concentration used in this study. Different results were reported by Kikuchi
et al. [
16] when adding 1 vol.% H
2O during CH
4 oxidation over a Pd/Al
2O
3 catalyst,
i.e., a decrease in activity was observed up to about 450 °C and no H
2O inhibition was observed at higher temperatures. However, during addition of 20 vol.% H
2O, the inhibiting effect could be observed up to 600 °C, in qualitative agreement with Eriksson
et al. [
41].
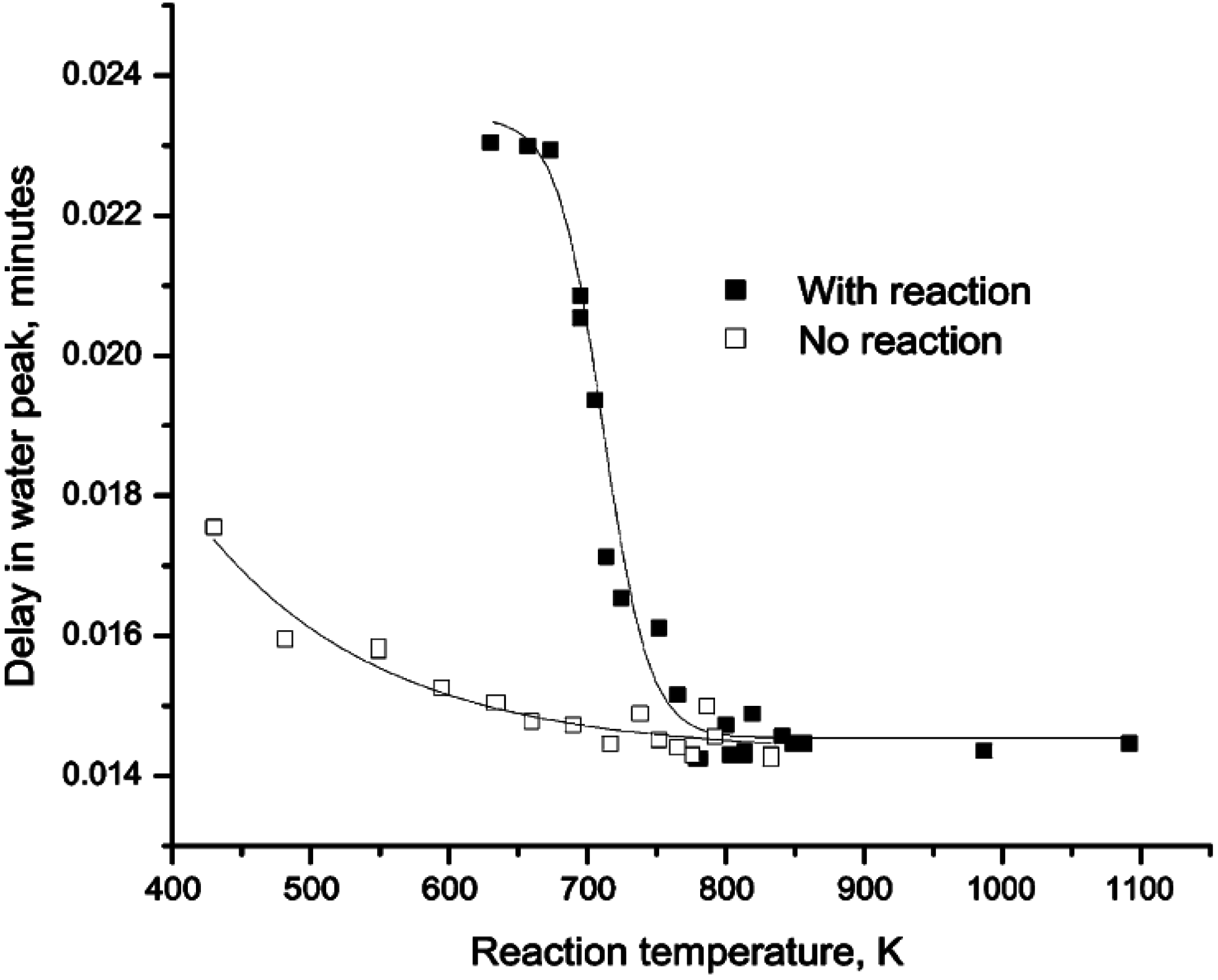
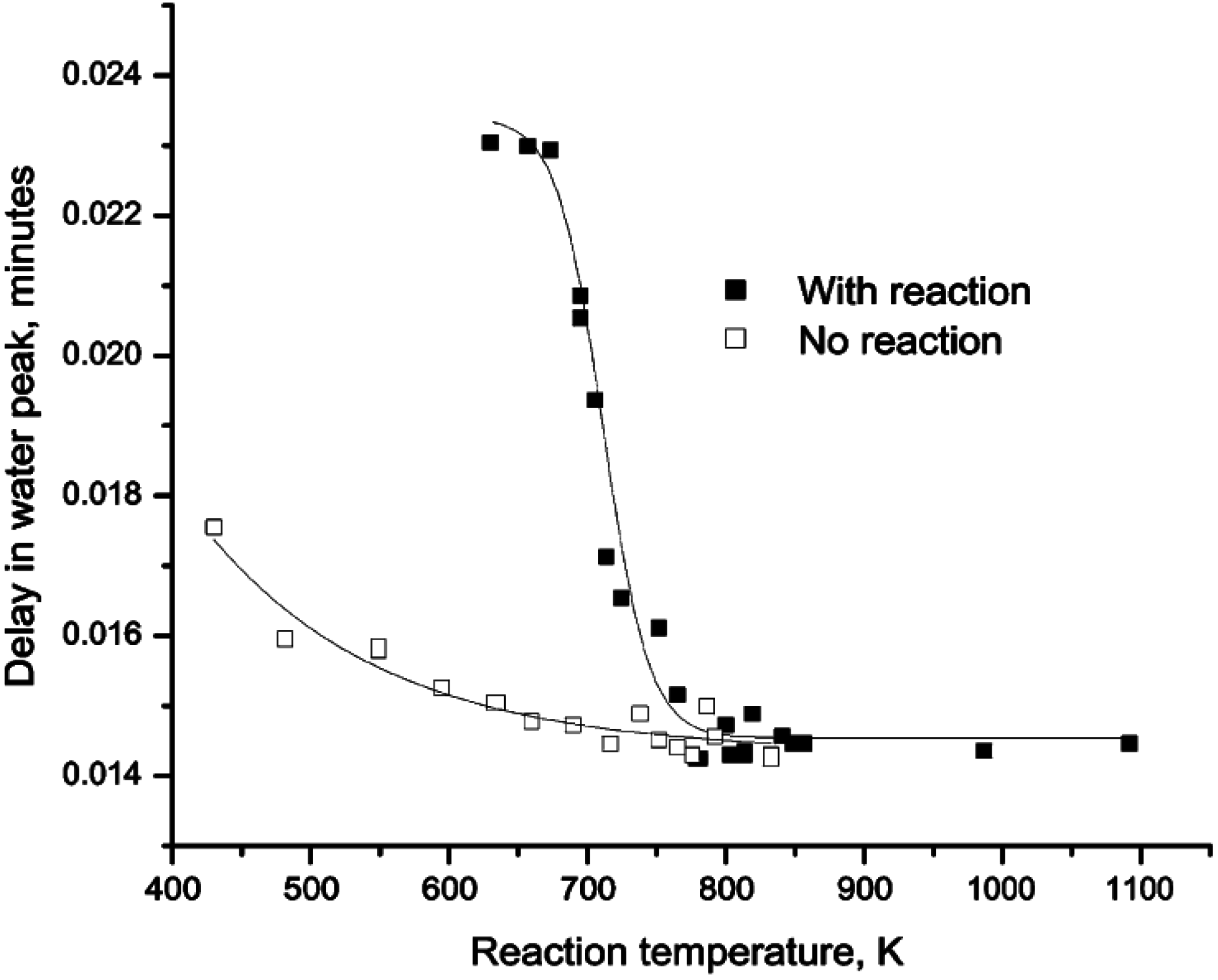
Figure 2.
Delay in the H
2O peak with respect to other products obtained by passing pulses of CH
4/O
2/He (closed square) and 1 vol.% O
2/3.45 vol.% H
2O/He (open square) over Pd/ZrO
2 at different temperatures. Reproduced with permission from [
42]. Copyright © 2001 Elsevier.
Figure 2.
Delay in the H
2O peak with respect to other products obtained by passing pulses of CH
4/O
2/He (closed square) and 1 vol.% O
2/3.45 vol.% H
2O/He (open square) over Pd/ZrO
2 at different temperatures. Reproduced with permission from [
42]. Copyright © 2001 Elsevier.
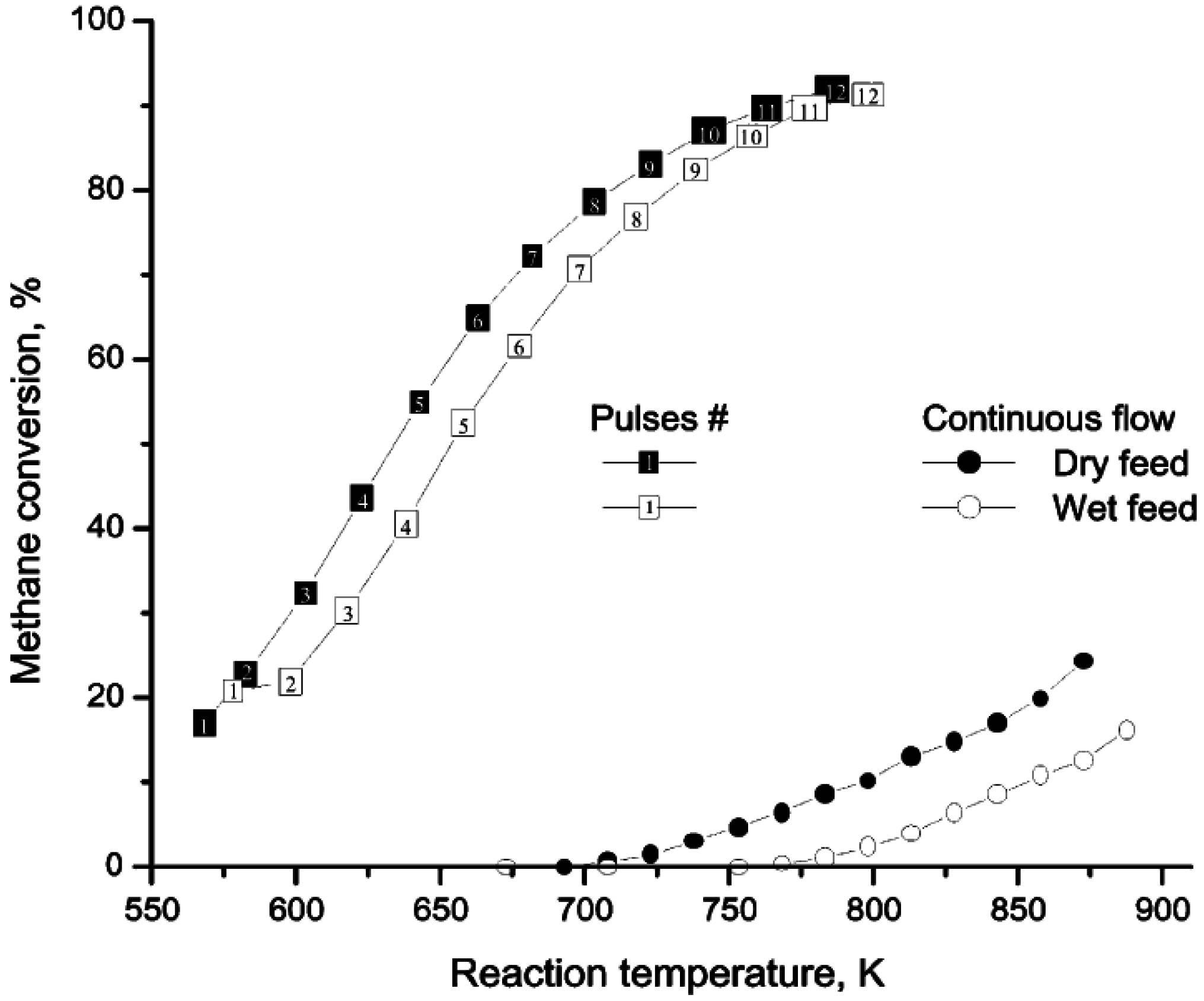
Further insight into the H
2O adsorption/desorption phenomena on Pd/ZrO
2 catalysts has been obtained using pulsed-flow experiments [
42,
43]. Accordingly, pulses of CH
4/O
2/He (1:4:95 vol %) were passed over a Pd/ZrO
2 catalyst at various temperatures and the products monitored by mass spectrometer. The time at which the peak maximum for H
2O appeared in each spectrum, compared to other products, was reported as the delay in the H
2O peak. The data (
Figure 2) show that the H
2O generated during CH
4 oxidation lags other products, suggesting a slow H
2O adsorption/desorption equilibrium which might include spillover to the support. As the temperature increases above 450 °C (723 K), the desorption rate of H
2O increases and the delay in the H
2O peak compared to the other products is insignificant. This behavior is in agreement with observations from other studies [
30,
31,
44] that the desorption rate of H
2O produced during CH
4 oxidation is slow and on the order of seconds below 450 °C, even though CO
2, the other product of reaction, desorbs very quickly. Increasing temperature above 450 °C removes the desorption time gap between CO
2 and H
2O, and thus, no inhibition by H
2O occurs. Ciuparu
et al. [
42] also pulsed gas containing 3.45 vol.% H
2O/O
2/He but no CH
4 (and hence no reaction) through the same catalyst bed (
Figure 2), showing that the H
2O generated from CH
4 oxidation lags the H
2O added to the feed. These data demonstrate that the adsorption/desorption of H
2O from the Pd catalyst surface is temperature dependent and reaches equilibrium at temperatures above ~450 °C (723 K), even for H
2O added in the gas phase.
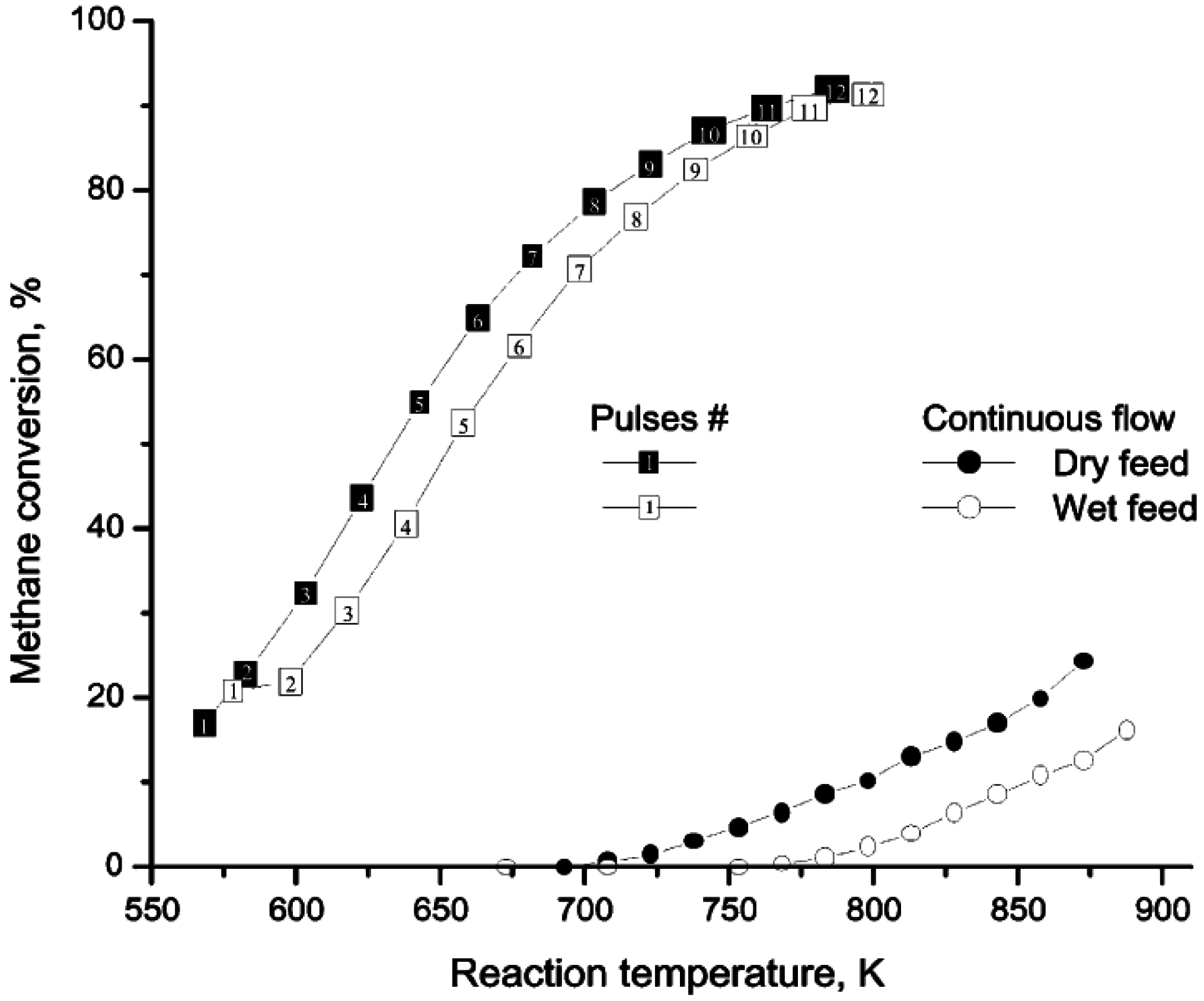
Figure 3 compares temperature-programmed-reaction (TPR) profiles for CH
4 oxidation obtained over a Pd/ZrO
2 catalyst, from both pulsed and continuous flow experiments with or without H
2O added [
42,
43]. The pulsed flow TPR profile was obtained by injecting pulses of the reaction mixture (1/4/95:CH
4/O
2/He for the “dry” feed and 1/4/95:CH
4/O
2/He saturated with ~2% H
2O for the “wet” feed) into a He stream every 3 min while ramping the temperature at 0.5 K min
−1. Between consecutive pulses the catalyst was purged in flowing He. The pulsed flow data of
Figure 3 show that at temperatures above 450 °C (723 K), there is no H
2O inhibition, since the conversions of “dry” and “wet” reaction mixtures are essentially the same. At <450 °C, inhibition is observed due to a low H
2O desorption rate. When H
2O is added to the gas phase, the H
2O adsorption rate is enhanced and the rate of desorption is further decreased. With continuous flow of reactants and a higher H
2O concentration, H
2O inhibition occurs at high temperatures due to re-adsorption. The addition of H
2O to the feed directs the equilibrium towards more H
2O adsorption on the surface and hence a greater decrease in catalyst activity during CH
4 oxidation.
Figure 3.
Temperature-programmed reactions during pulsed or continuous flow of reactants over Pd/ZrO
2 with or without H
2O in the feed. Reproduced with permission from [
42]. Copyright © 2001 Elsevier.
Figure 3.
Temperature-programmed reactions during pulsed or continuous flow of reactants over Pd/ZrO
2 with or without H
2O in the feed. Reproduced with permission from [
42]. Copyright © 2001 Elsevier.
The above observations are consistent with the following hypotheses: (1) product inhibition of CH
4 oxidation by H
2O on PdO catalysts occurs at temperatures below 450 °C; (2) product inhibition by H
2O is enhanced by its slow rate of desorption from the PdO catalyst relative to a higher rate of CH
4 oxidation; (3) PdO and H
2O may interact via the reversible reaction: PdO + H
2O↔Pd(OH)
2 yielding inactive Pd(OH)
2 and thus reversibly deactivating PdO as first proposed by Cullis
et al. [
40]; and (4) the extent of the CH
4 oxidation reaction increases with increasing temperature but is reduced with increasing H
2O concentration in the gas phase.
2.2. Catalyst Sintering by H2O
The possibility that addition of H
2O may degrade Pd catalysts through a sintering mechanism [
26] has also been investigated. According to Hansen
et al. [
45], the sintering rate of metal nanoparticles depends on their size. For nanoparticles <3 nm in diameter, Ostwald ripening is the most likely sintering mechanism. For larger particles (3–10 nm), both Ostwald ripening and particle migration and coalescence may occur, but the sintering rate is much slower than for the smaller particles [
45]. The particle sintering rate has also been shown to correlate with the vapor pressure of the surface species [
4]. Pd is unique among the PGMs in that the oxide (PdO) has a much lower vapor pressure than the metal (Pd), and consequently, one would expect a very low sintering rate of PdO by Ostwald ripening [
4]. The rate of sintering is also dependent on the support. Lamber
et al. [
46] suggested that on SiO
2 in the presence of H
2O, the formation of silanol (Si-OH) groups favors the migration and coalescence of Pd, whereas in the absence of H
2O, Ostwald ripening is favored. Sintering suppression has been demonstrated for Pt catalysts using supports that enhance metal-support interactions [
28]. Nagai
et al. [
47] demonstrated a correlation between the O electron density of the support, the strength of the Pt-O interaction and the resulting crystallite size. Thus, supports with a stronger metal-support interaction have a higher O electron density and yield smaller Pt crystallites in the order SiO
2 < Al
2O
3 < ZrO
2 < TiO
2 < CeO
2 [
28,
47].
Xu
et al. [
48] reported that the main deactivation mechanism of Pd/Al
2O
3 catalysts following exposure to 10 (
v/
v)% H
2O/N
2 at 900 °C for up to 200 h is Pd sintering. A substantial decrease in Pd dispersion from 3.7% to 0.9% over 7 wt.% Pd/Al
2O
3 and similar decreases at other Pd loadings after 96 h hydrothermal aging, were observed. As noted by Xu
et al. [
48], aging the catalyst at 900 °C ensures that PdO decomposition to Pd
0 is complete and consequently the more rapid sintering observed is relevant to the behavior of Pd
0 rather than PdO.
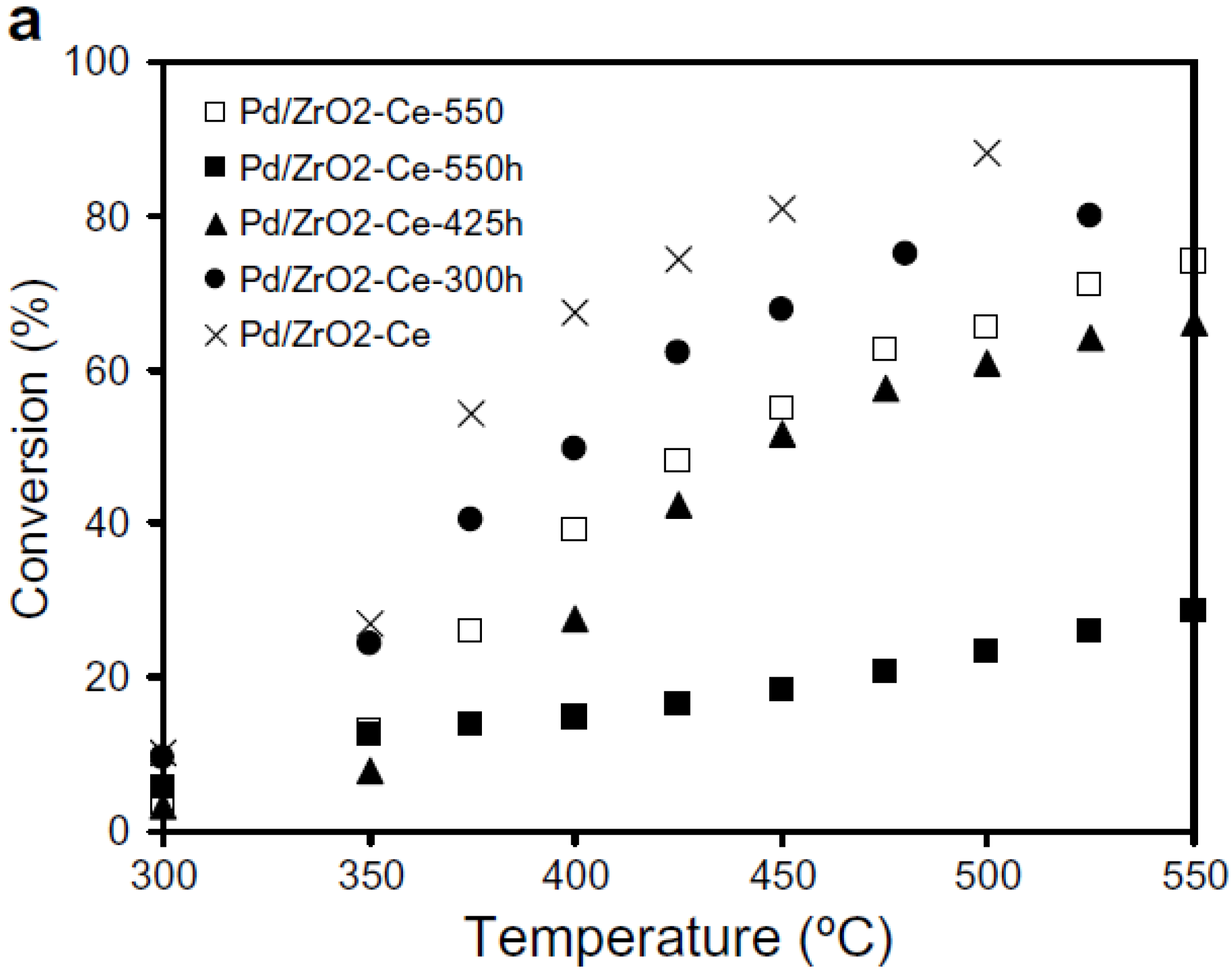
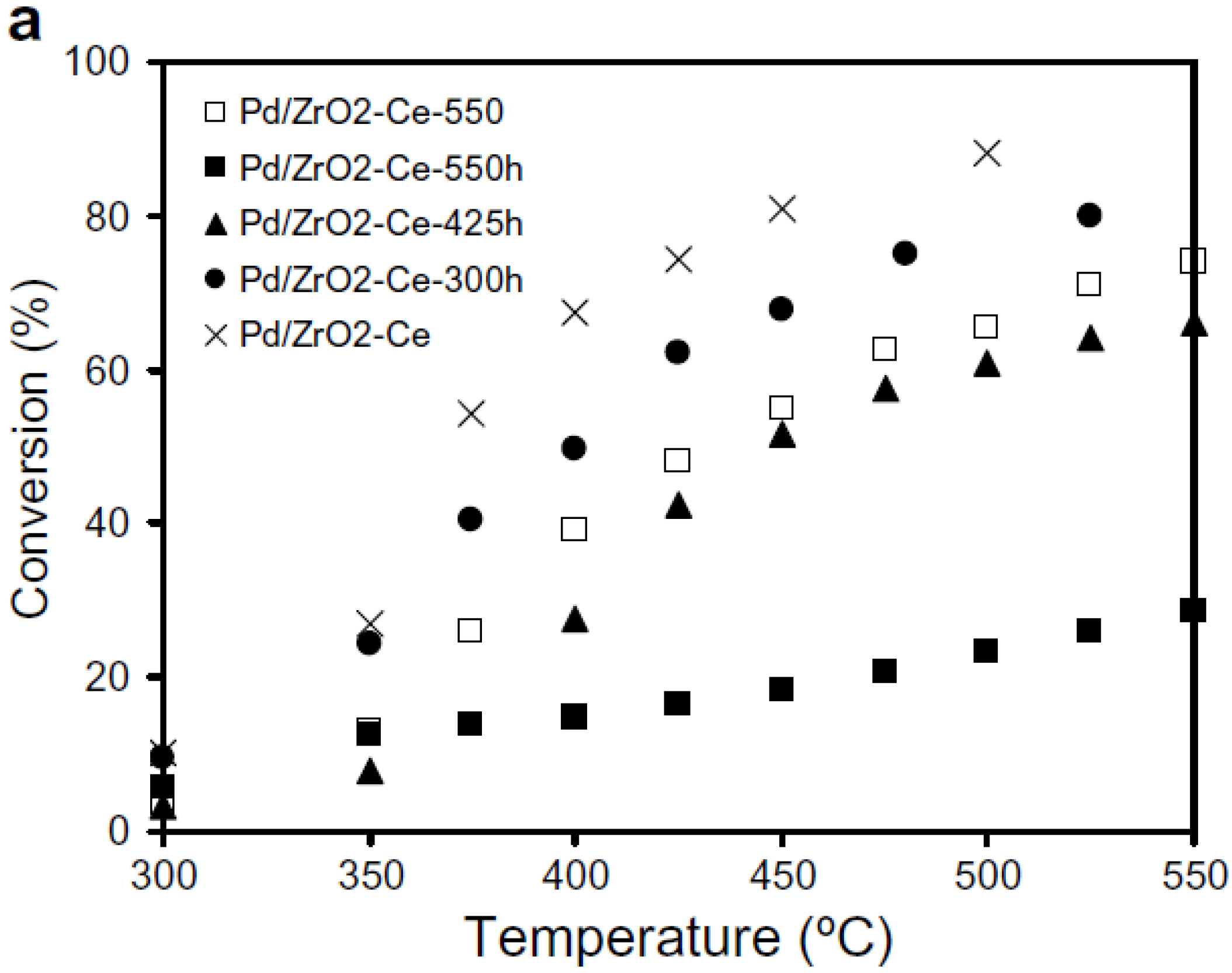
Escandon
et al. [
49] examined effects of hydrothermal aging at lower temperatures, where PdO is thermodynamically stable [
6]. A 1 wt.% Pd/ZrO
2-Ce catalyst was hydrothermally aged at 300, 425, and 550 °C in 2% H
2O/Air for 30 h, before being evaluated for CH
4 oxidation under lean-burn conditions (5000 ppmv CH
4 in dry air). The results, shown in
Figure 4, are compared with the same catalyst, thermally aged at 550 °C in dry air for 30 h (identified as Pd/ZrO
2-Ce-550 in
Figure 4) [
49]. A significant irreversible decrease in CH
4 conversion occurs and the extent of catalyst deactivation increases with aging temperature (
Figure 4). The
T50% increases from 375 °C for the fresh oxidized catalyst (identified as Pd/ZrO
2-Ce in
Figure 4), to 450 °C for the air-aged catalyst and to > 550 °C for the hydrothermally aged catalyst. Pd dispersion and BET surface area of the aged catalysts did not change [
49]. Comparing the activity results of the catalyst thermally aged in air (Pd/ZrO
2-Ce-550) with that aged in 2% H
2O/air at 550 °C (Pd/ZrO
2-Ce-550h), confirms that catalyst deactivation rate increases in the presence of H
2O. The stability of the hydrothermally aged catalysts during reaction was also evaluated, using both isothermal deactivation experiments at 500 °C and light-off measurements made after 50 h reaction with 5000 ppmv CH
4 in air. The catalysts aged in the presence of H
2O at 300 °C underwent a significant deactivation whereas the catalyst aged in the presence of H
2O at 425 °C was much more resistant to deactivation, and after 25 h time-on-stream was the most active of all the catalysts examined. XRD analysis of the catalysts showed that the more stable catalysts are associated with the most stable supports [
49].
Figure 4.
CH
4 conversion over fresh 1 wt.% Pd/ZrO
2-Ce catalyst compared to 1 wt.% Pd/ZrO
2-Ce thermally aged in air at 550 °C (Pd/ZrO
2-Ce-550) and hydrothermally aged at different temperatures in 2% H
2O/air (identified as Pd/ZrO
2-Ce-TTTh where TTT is the aging temperature in °C). Reproduced with permission from [
49]. Copyright© 2008 Elsevier.
Figure 4.
CH
4 conversion over fresh 1 wt.% Pd/ZrO
2-Ce catalyst compared to 1 wt.% Pd/ZrO
2-Ce thermally aged in air at 550 °C (Pd/ZrO
2-Ce-550) and hydrothermally aged at different temperatures in 2% H
2O/air (identified as Pd/ZrO
2-Ce-TTTh where TTT is the aging temperature in °C). Reproduced with permission from [
49]. Copyright© 2008 Elsevier.
In another study of CH
4 oxidation at low temperature (250–450 °C), a change in PdO dispersion was suggested as the main cause of deactivation of 0.5% Pd/Al
2O
3 and 0.5% Pd/SiO
2 catalysts [
50]. Dispersion decreased from 10% for the unused 0.5% Pd/SiO
2 catalyst to 5.6% for the catalyst reacted in 1% CH
4/air feed at 450 °C for 7 h, whereas for the 0.5% Pd/Al
2O
3 catalyst the corresponding changes in dispersion were 67% to 6.3%, respectively. These observations are in good agreement with that of Narui
et al. [
51], in which the PdO dispersion of a 0.5% Pd/Al
2O
3 catalyst decreased from 14% to 11% after 6 h reaction at 350 °C. Zhang
et al. [
52] investigated Pd catalysts supported on ZSM-5 and reported that catalyst stability is improved when CH
4 oxidation is carried out in the presence of H
2O at 430–480 °C, compared to the reaction in a dry feed. In both cases, the loss in catalyst activity could be related to reduced PdO dispersion, as determined by the Pd/Si ratio measured by XPS, but the loss in dispersion is smaller in the presence of H
2O [
52]. By contrast, Araya
et al. [
53] reported an insignificant drop in PdO dispersion (from 31.7% to 28.2%) of a Pd/SiO
2 catalyst after 96 h of reaction at 325 °C in 1.5% CH
4/6% O
2 in He, despite a significant decrease in CH
4 conversion from 32% to 22%. The extent of catalyst deactivation was found to further increase in the presence of 3% H
2O added to the feed.
Several studies have demonstrated that catalyst sintering can be reduced by encapsulating Pd/PdO nanoparticles in support materials. Sinter-resistant Pd catalysts have been prepared by atomic layer deposition of Al
2O
3 overlayers on Pd [
54], as well as by the synthesis of Pd/SiO
2 core-shell structures [
55,
56]. Cargnello
et al. [
22] reported a Pd/CeO
2 core-shell catalyst supported on Al
2O
3 for CH
4 oxidation that is about 200xs more active than an equivalent Pd-CeO
2/Al
2O
3 catalyst prepared by wet impregnation. The authors demonstrated that the Pd cores remain isolated even after heating the catalyst to 850 °C and that the CH
4 light-off curves (measured at GHSV of 200,000 h
−1 in a feed gas of 0.5% CH
4, 2% O
2 in Ar) are the same for the fresh catalyst and one that has been aged at 850 °C for 12 hours. The Pd nanoparticles encapsulated by CeO
2 enhance the metal-support interaction that leads to exceptionally high CH
4 oxidation activity and good thermal stability [
22].
2.3. Effects of Support
The data of
Table 1 show that the inhibition of CH
4 oxidation by H
2O on Pd catalysts is dependent upon the support. Pd/Al
2O
3 shows significantly more inhibition with 10% H
2O added to the feed than either the Pd/SnO
2 or Pd/ZrO
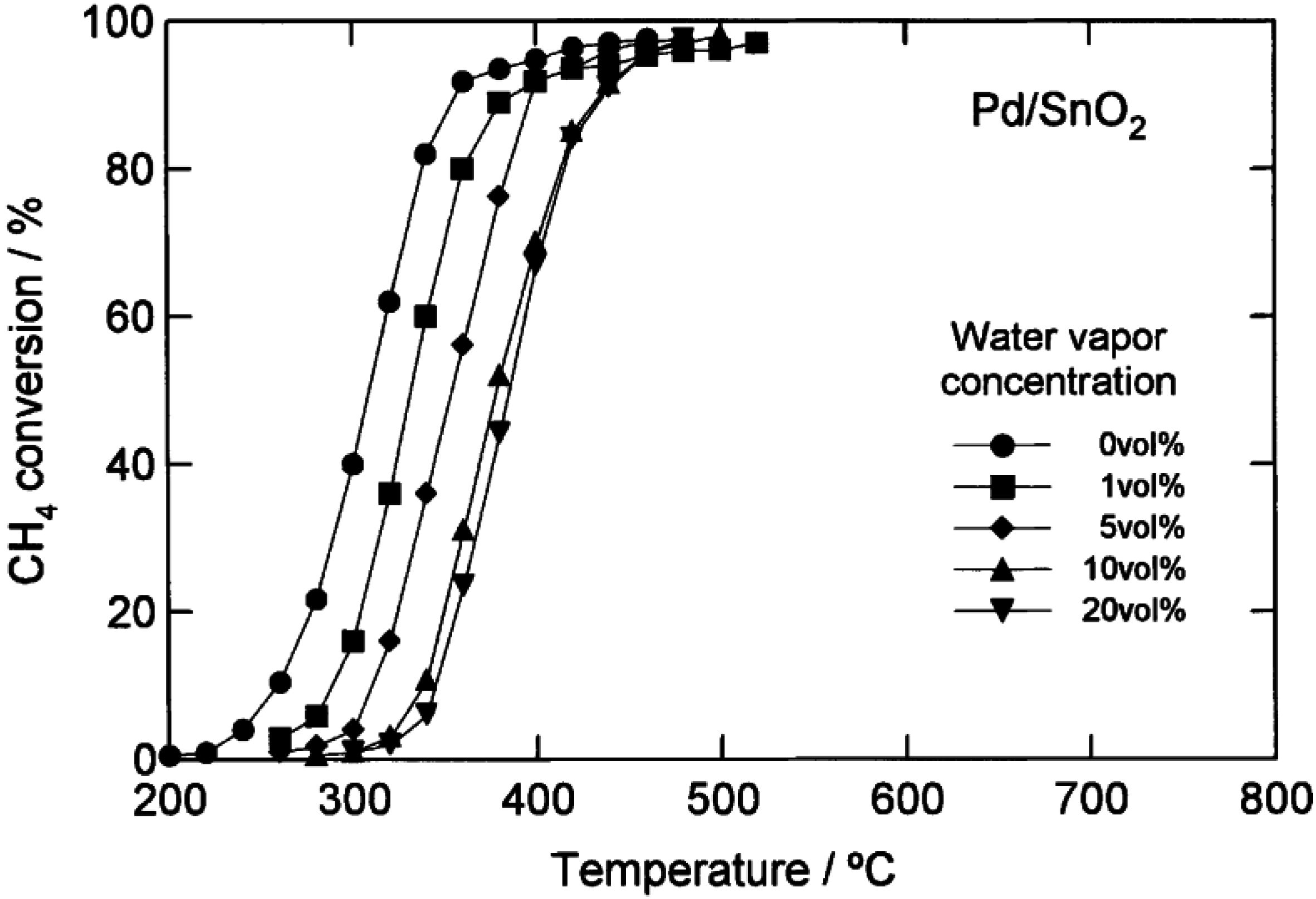
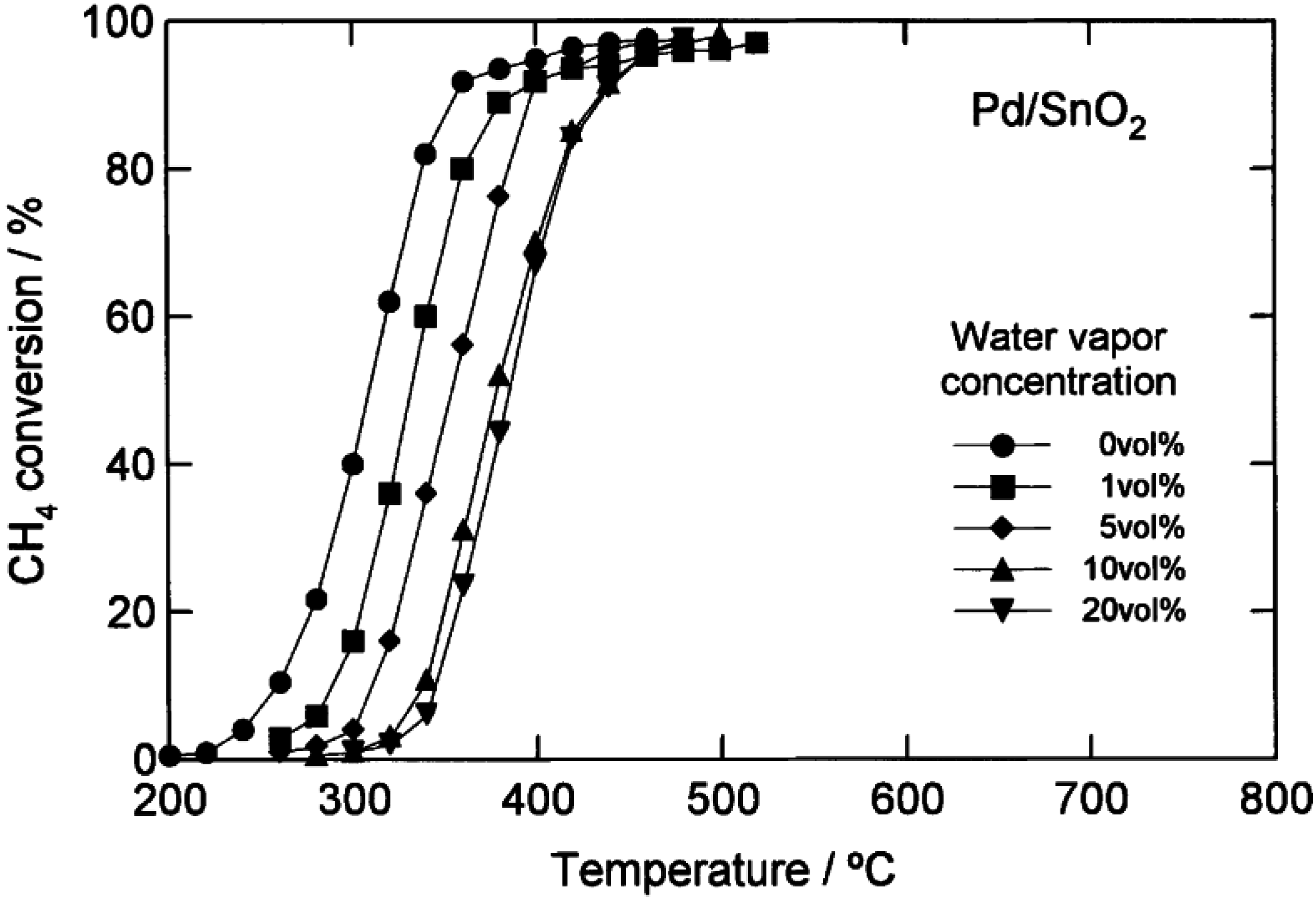
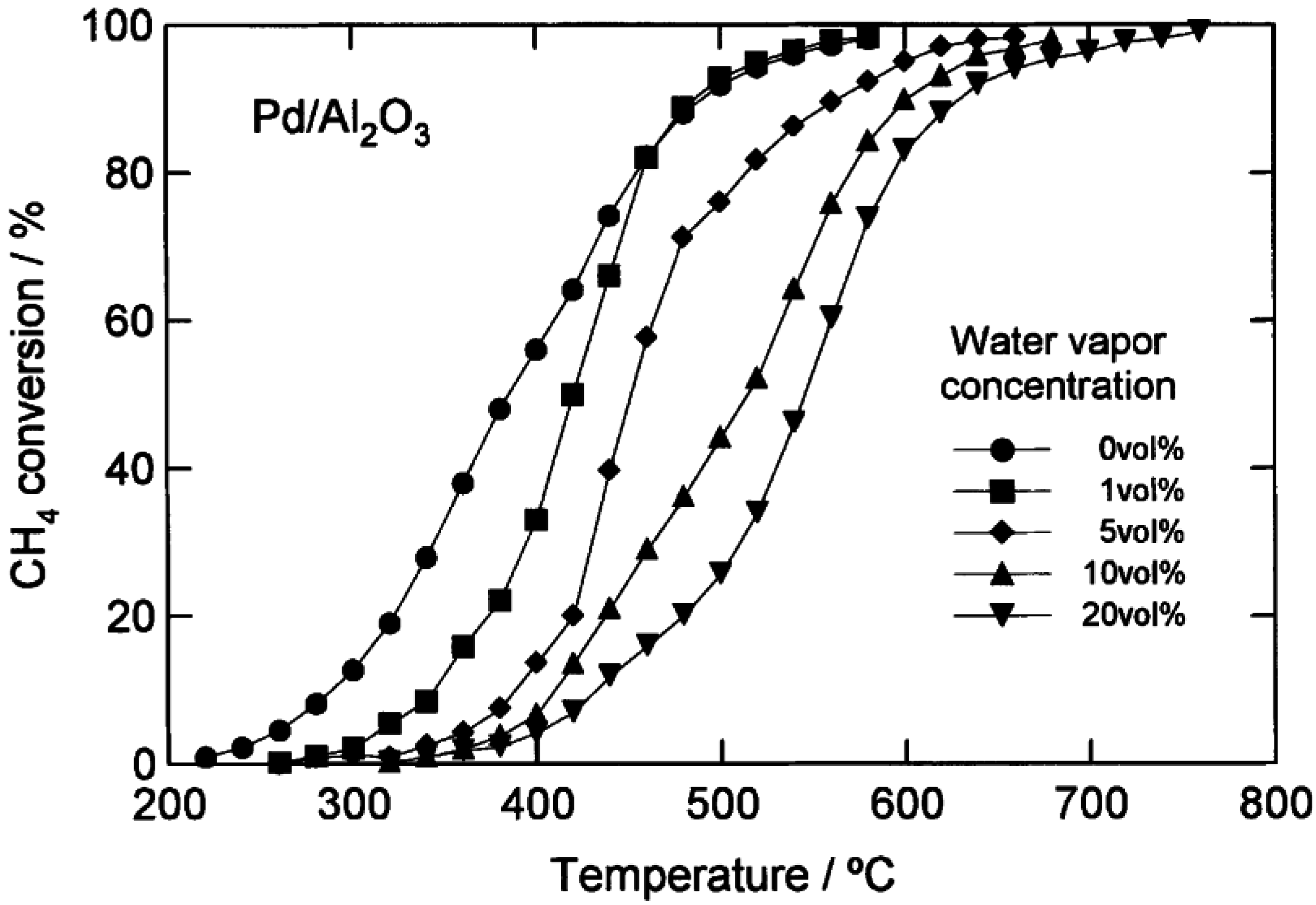
2 catalysts. More detailed data from Kikuchi
et al. [
16] comparing CH
4 light-off curves for a 1.1 wt.% Pd/Al
2O
3 catalyst and a 1.1 wt.% Pd/SnO
2 catalyst with H
2O added to the feed over a range of concentrations (1–20 vol.%), are shown in
Figure 5 and
Figure 6. By increasing the H
2O concentration, the CH
4 light-off curves for both catalysts shift to higher temperatures. However, the temperature shift is larger over the Pd/Al
2O
3 catalyst than the Pd/SnO
2. The authors completed a simplified kinetic analysis of the CH
4 oxidation rate data to show that the enthalpy of adsorption of H
2O is strongest on the Pd/Al
2O
3 catalyst (∆H
ad ~ −49 kJ/mol), from which they concluded that the significant loss in activity of the Pd/Al
2O
3 in the presence of H
2O is due to a high coverage of the active sites by H
2O [
16]. These results could also be interpreted according to the more recent proposals by Schwartz
et al. [
44,
57], that hydroxyl accumulation on the support hinders oxygen migration and exchange, and hence CH
4 oxidation. The strong adsorption of H
2O determined by kinetic analysis on the Pd/Al
2O
3 catalyst [
16] is consistent with a large hydroxyl accumulation on the catalyst surface that could inhibit the O exchange.
Figure 5.
Catalytic combustion of CH
4 over 1.1 wt.% Pd/SnO
2 with different amounts of water added (vol.%). Reaction conditions: CH
4, 1 vol.%; O
2, 20 vol.%; H
2O, 0–20 vol.%; N
2, balance; GHSV 48,000 h
−1. Reprinted with permission from [
16]. Copyright© 2002 Elsevier.
Figure 5.
Catalytic combustion of CH
4 over 1.1 wt.% Pd/SnO
2 with different amounts of water added (vol.%). Reaction conditions: CH
4, 1 vol.%; O
2, 20 vol.%; H
2O, 0–20 vol.%; N
2, balance; GHSV 48,000 h
−1. Reprinted with permission from [
16]. Copyright© 2002 Elsevier.
Figure 6.
Catalytic combustion of CH
4 over 1.1 wt.% Pd/Al
2O
3 with different amounts of water added (vol.%). Reaction conditions: CH
4, 1 vol.%; O
2, 20 vol.%; H
2O, 0–20 vol.%; N
2, balance; GHSV 48,000 h
−1. Reprinted with permission from [
16]. Copyright © 2002 Elsevier.
Figure 6.
Catalytic combustion of CH
4 over 1.1 wt.% Pd/Al
2O
3 with different amounts of water added (vol.%). Reaction conditions: CH
4, 1 vol.%; O
2, 20 vol.%; H
2O, 0–20 vol.%; N
2, balance; GHSV 48,000 h
−1. Reprinted with permission from [
16]. Copyright © 2002 Elsevier.
The rate of deactivation during CH
4 oxidation in the presence of H
2O has been shown to be reduced by using a support with high oxygen surface mobility. At temperatures below 450 °C, Ciuparu
et al. [
30] reported the inhibition effect of H
2O to be dependent upon the oxygen mobility of the support. Comparing PdO supported on oxides with increasing surface oxygen mobility: Al
2O
3 < ZrO
2 < Ce
0.1Zr
0.9O
2, they show that the resistance to H
2O inhibition during CH
4 oxidation increases in the same order. The deactivation rate of PdO was also compared over Al
2O
3, MgO, and TiO
2 supports by Schwartz
et al. [
44,
57] at temperatures <450 °C. Deactivation is shown to be a consequence of reduced oxygen mobility due to hydroxyl adsorption. They also reported that PdO/MgO catalyst has a slower deactivation rate compared with Al
2O
3 and TiO
2 supports because of the higher oxygen surface mobility on the MgO [
44,
57]. However, Pd catalysts dispersed on other supports such as MCM-41, which have high surface area (1113 m
2/g) and lower oxygen mobility than MgO and Al
2O
3, did not deactivate either, suggesting that other factors also play a role, depending on the catalyst and the support.
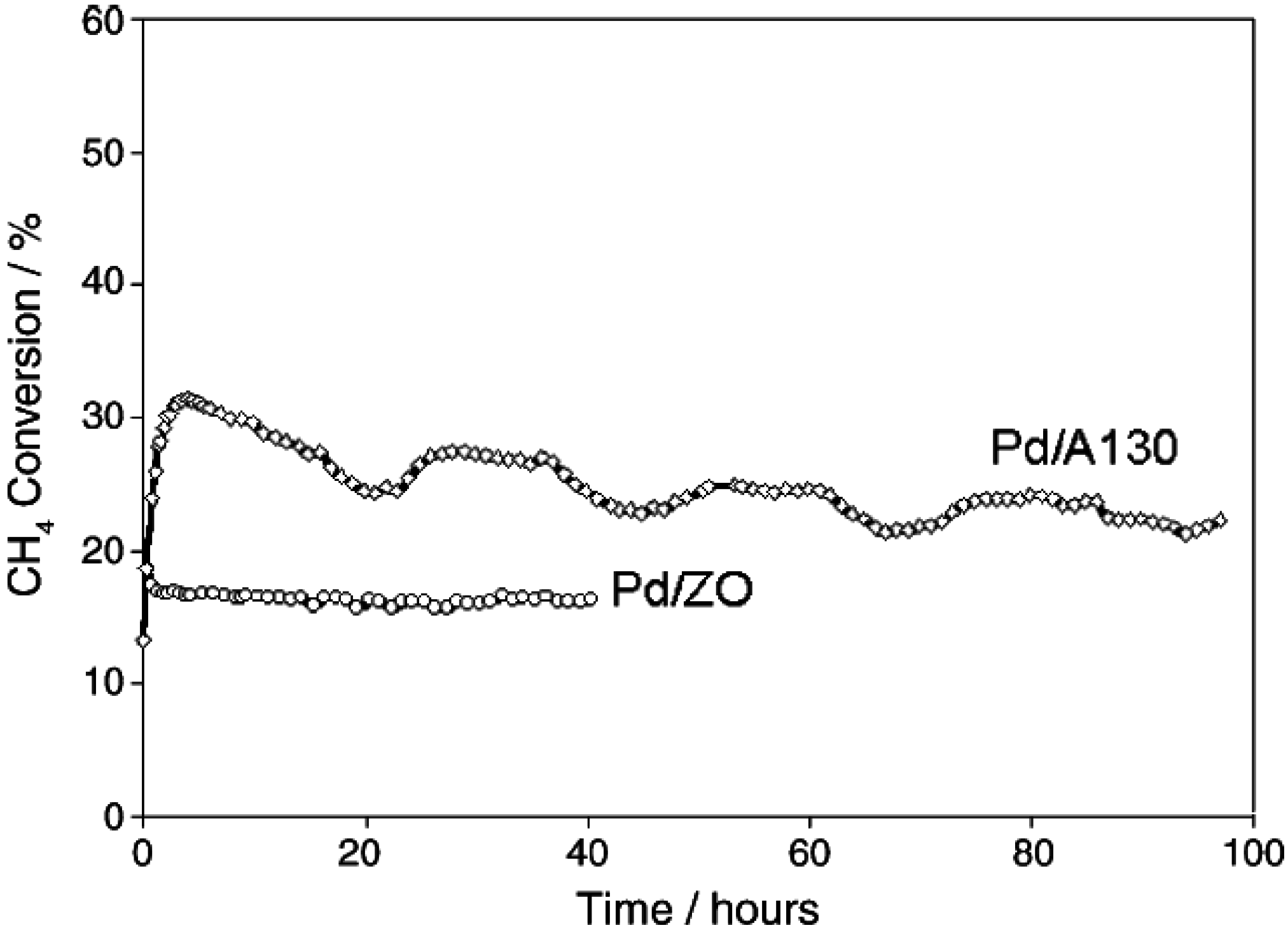
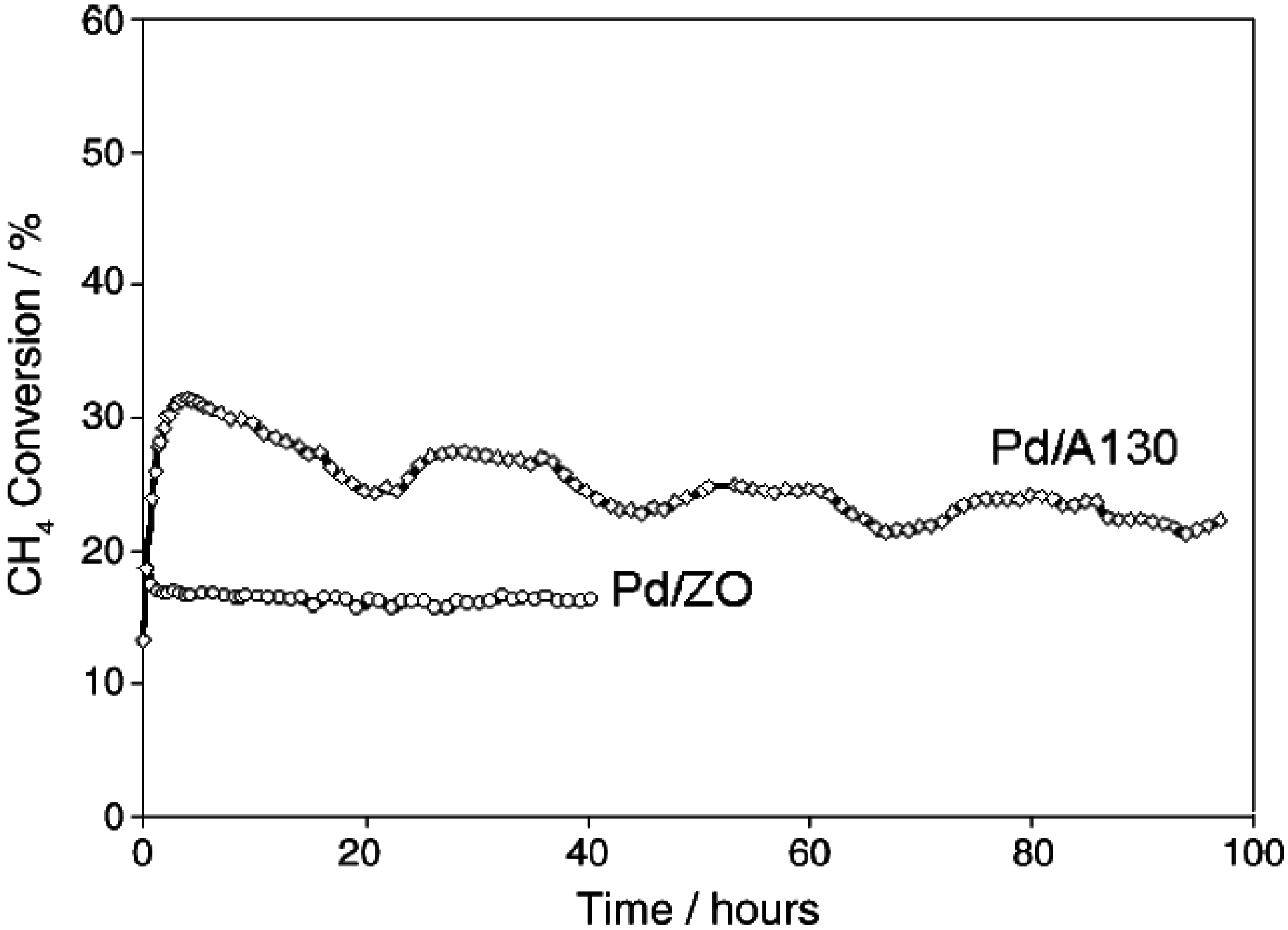
Another study compared the stability of Pd/SiO
2 and Pd/ZrO
2 during CH
4 oxidation using a dry feed gas [
53]. The data (
Figure 7) show that the Pd/ZrO
2 is stable after 40 h time-on-stream, while the CH
4 conversion over the Pd/SiO
2 catalyst increases from 13% to 32% in the first 3 h, and then decreases to 22% after 96 h (see
Figure 7). Although the Pd/ZrO
2 catalyst is more stable than the Pd/SiO
2 catalyst, its conversion is lower than for the Pd/SiO
2 catalyst. The lower deactivation rate observed on the Pd/ZrO
2 is consistent with the higher oxygen mobility of this catalyst compared to Pd/SiO
2, as noted above.
Metal-support interactions, support stability and the tendency of the support to encapsulate Pd, may also play a role in the deactivation of Pd catalysts during CH
4 oxidation. Gannouni
et al. [
58] compared Pd catalysts supported on silica and mesoporous aluminosilicas and showed that, according to the light-off curves measured with 1% CH
4, 4% O
2 in He, CH
4 oxidation activity is enhanced on the pure silica support, whereas on the aluminosilica, the beneficial effect of Al
3+ on metal dispersion and catalytic activity is counterbalanced by partial metal encapsulation. Above 500 °C in the presence of H
2O, the structural collapse of the support, metal sintering, and metal encapsulation by the support all occur [
58]. Similar effects were reported with SiO
2 supports by Zhu
et al. [
59]. SiO
2 desorbs chemisorbed H
2O (silanol groups –Si-OH) at ~397 °C [
46] and the formation of hydroxides according to the reaction:
is feasible at temperatures above 700 °C [
60,
61]. Hydroxyl mobility can change the extent of metal-support interactions [
45,
46]. Zhu
et al. [
59] reported the encapsulation of PdO by SiO
2 during CH
4 oxidation at only 325 °C. The authors suggested that silica migration by (i) formation of a palladium silicide during H
2 reduction at 650 °C that is subsequently oxidized during CH
4 oxidation and (ii) migration of SiO
2 during CH
4 oxidation caused by the water formed during reaction, are important related factors facilitating the encapsulation of PdO by the SiO
2. Migration of SiO
2 onto the metal crystallites in other catalyst systems containing H
2O has also been reported in the literature [
46,
62].
Figure 7.
Methane conversion over time of Pd/ZrO
2 and Pd/Aerosil130 catalysts. Reaction conditions: 1.5% CH
4; 6% O
2; total flow = 90 cm
3min
−1, balanced in He; temperature = 325 °C; catalyst mass = 0.2 g. Reprinted with permission from [
53]. Copyright © 2005 Elsevier.
Figure 7.
Methane conversion over time of Pd/ZrO
2 and Pd/Aerosil130 catalysts. Reaction conditions: 1.5% CH
4; 6% O
2; total flow = 90 cm
3min
−1, balanced in He; temperature = 325 °C; catalyst mass = 0.2 g. Reprinted with permission from [
53]. Copyright © 2005 Elsevier.
Yoshida
et al. [
63] also examined the effects of various metal oxide supports of Pd on the low temperature oxidation of CH
4 as summarized in
Table 3. The catalytic activity varies with the support, but the support oxides with moderate acid strength (Al
2O
3 and SiO
2) give maximum CH
4 conversion. For these catalysts higher activity corresponds to a higher oxidation state of Pd (bulk PdO). The lower activity of Pd on basic supports is attributed to the formation of binary oxides from PdO and the support (such as Pd/MgO
x), in spite of a high Pd oxidation state.
The effect of metal oxides added to Pd/Al
2O
3 to improve the hydrothermal stability has been reported by Liu
et al. [
36] who showed in particular, that the addition of NiO or MgO improved the hydrothermal stability of Pd/Al
2O
3 through the formation of NiAl
2O
4 and MgAl
3O
4 spinel structures. According to the authors, the spinel results in weakened support acidity that suppresses the formation of Pd(OH)
2 during hydrothermal aging.
Table 3.
Effect of support on properties of 5 wt.% Pd catalysts and their CH
4 oxidation conversion. Data adapted from [
63].
Table 3.
Effect of support on properties of 5 wt.% Pd catalysts and their CH4 oxidation conversion. Data adapted from [63].
| Support | Support Acid Strength | Pd Dispersion | CH4 conversion a, % |
|---|
| | (Ho) | Fresh | Used | |
| MgO | 22.3 | 0.21 | 0.20 | 12 |
| ZrO2 | 9.3 | 0.41 | 0.12 | 3 |
| Al2O3 | 3.3 | 0.35 | 0.20 | 59 |
| SiO2 | -5.6 | 0.09 | 0.11 | 58 |
| SiO2-ZrO2 | -8.2 | 0.16 | 0.13 | 20 |
| SiO2-Al2O3 | -11.9 | 0.12 | 0.06 | 10 |
| SO42−-ZrO2 | -13.6 | - | 0.02 | 11 |
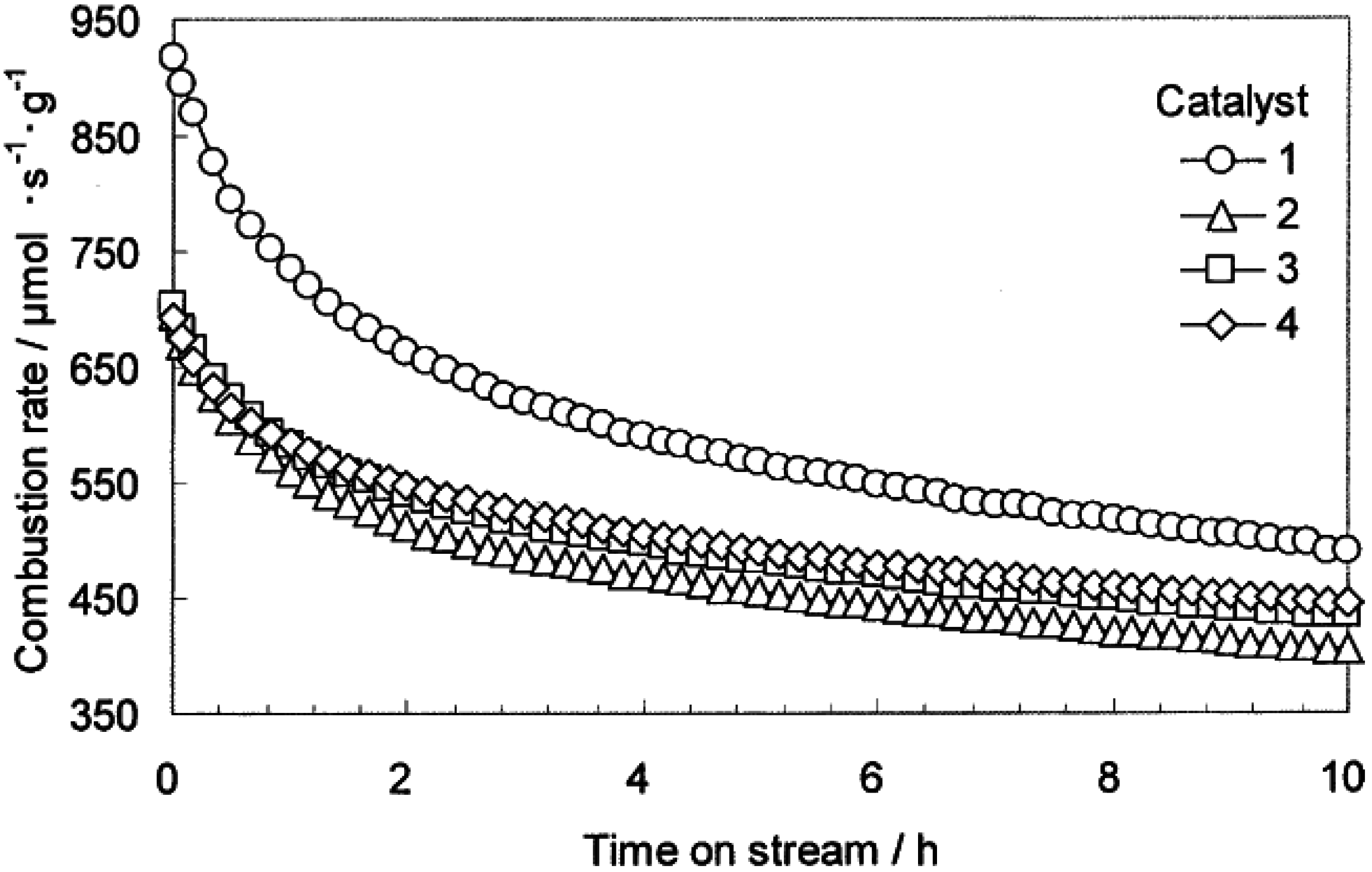
A comparison of initial CH
4 oxidation activity as a function of temperature for Pd-Pt catalysts on Al
2O
3, ZrO
2, LaMnAl
11O
19, Ce-ZrO
2, and Y-ZrO
2 was reported by Persson
et al. [
64]. Monolith catalysts were tested in a tubular quartz flow reactor at atmospheric pressure in 1.5 vol.% CH
4 in dry air and at a space velocity of 250,000 h
−1. In steady-state experiments, reaction temperature was set initially at 470 °C and then increased to 720 °C stepwise in 50 °C increments, with 1-h holds at each temperature. The Pd-Pt/Al
2O
3 catalyst had the highest activity at lower temperatures (470–570 °C), while the Pd-Pt/Ce-ZrO
2 catalyst had the highest activity between 620 °C and 800 °C [
64]. The authors suggested that the higher surface area of the Al
2O
3 compared to the other supports (e.g., 90 m
2/g for Al
2O
3 versus 10 m
2/g for Ce-ZrO
2) accounts for the higher activity of Pd-Pt/Al
2O
3 at lower temperatures, due to higher dispersion of Pd-Pt oxides, while at higher reaction temperatures the Pd-Pt catalyst probably undergoes reduction to the metal. A combination of lower activity for Pd metal and its propensity for rapid sintering probably explain the lower activity. The authors also suggested that the Ce-ZrO
2 likely enhances the stability of the PdO, similar to the enhanced stability observed on CeO
2 [
30]. In addition, ZrO
2 has high oxygen mobility [
30] and the ability to re-oxidize metallic Pd into PdO should be higher. Indeed, Pd/alumina is re-oxidized very slowly, whereas Pd supported on ceria-stabilized ZrO
2, is re-oxidized more rapidly.
Since H
2O adsorption on the Pd and/or the support is an important step in inhibiting CH
4 oxidation over Pd, support hydrophobicity may be expected to impact the inhibition effect of H
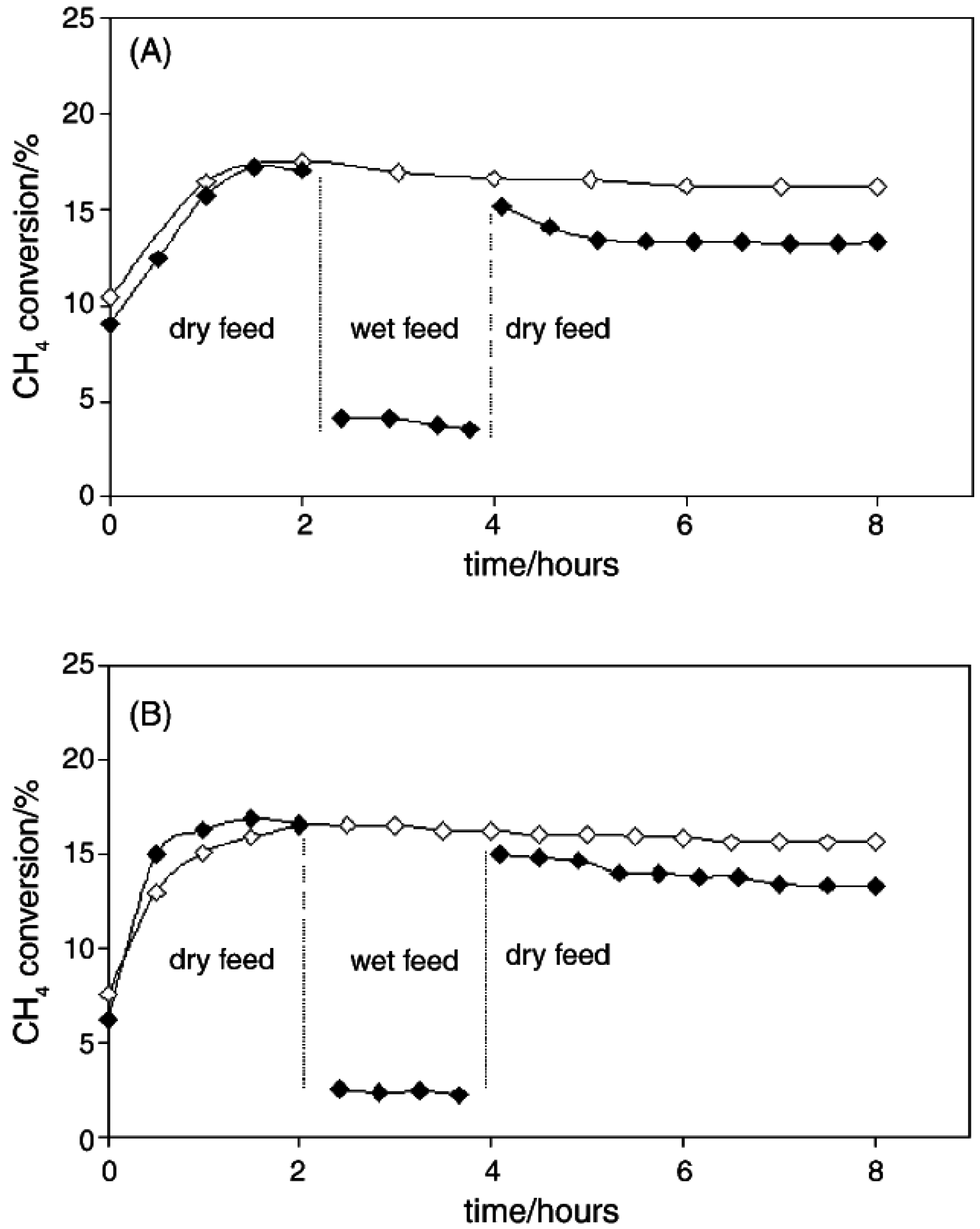
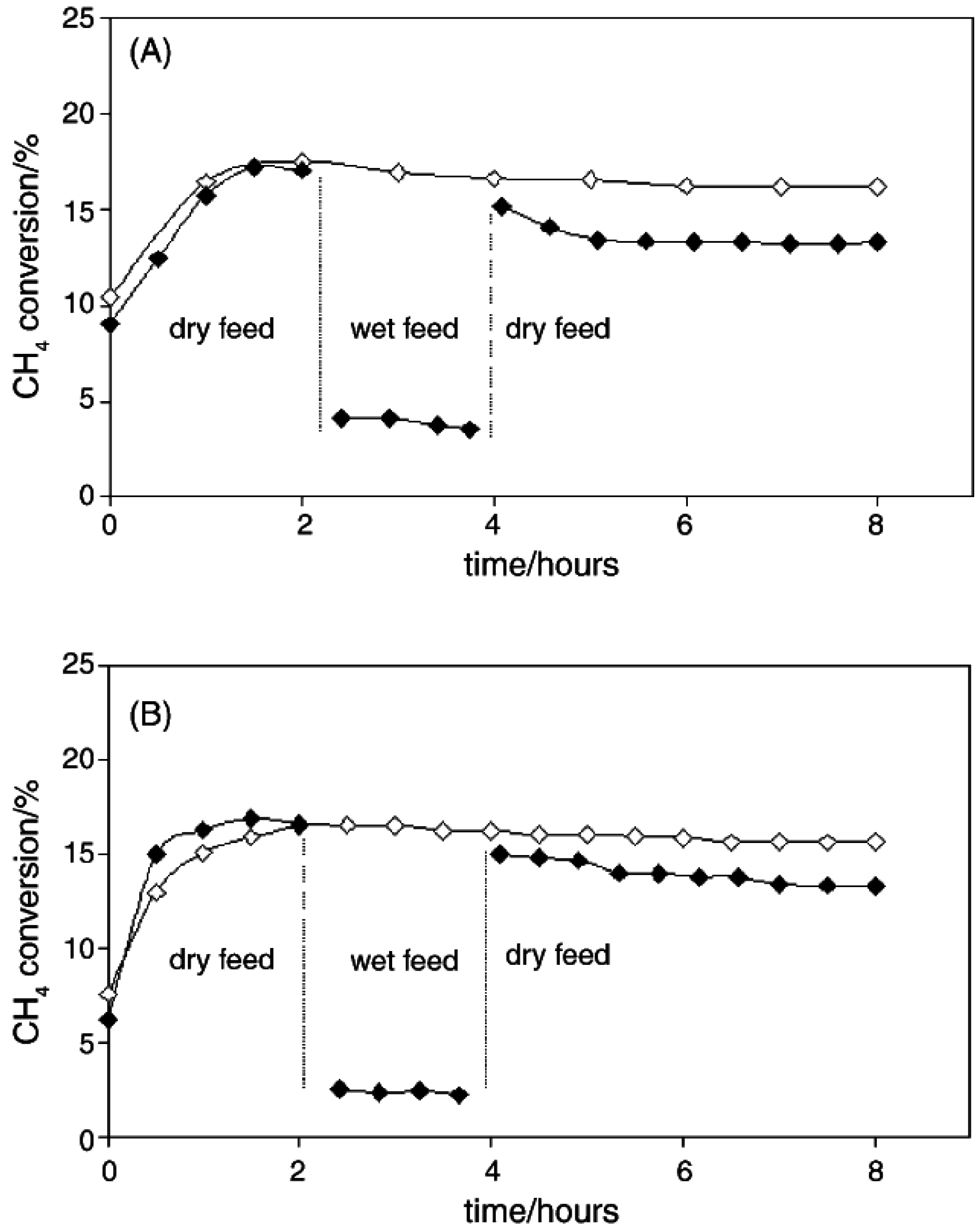
2O. Araya
et al. [
53] studied this effect on the deactivation of Pd-based catalysts by preparing 1 wt.% Pd on two different commercial silicas, Aerosil130 and Aerosil R972. The Aerosil R972 is hydrophobic since the OH groups have been replaced by methyl groups. Both 1% Pd/A130 and 1% Pd/R972 were tested at 325 °C in a gaseous mixture of 1.5% CH
4 and 6% O
2 in He at a total flow rate of 90 cm
3 min
−1 with addition of 3% H
2O after 2 h As shown in
Figure 8, the effect of H
2O addition to the feed gas is approximately the same for the hydrophobic silica, Pd/R972, and the hydrophilic Pd/A130. In both cases, a large decrease in CH
4 conversion is observed with the introduction of H
2O to the reactor. The authors reported a reaction order with respect to H
2O of −0.25 for both Pd/A130 and Pd/R972, emphasizing that the hydrophobicity of the support does not affect the extent of H
2O inhibition observed on either catalyst.
Figure 8.
(A) Pd/Aerosil130 catalyst, (
B) Pd/R972 catalyst. Reaction conditions: total flow = 90 cm
3 (STP) min
−1, temperature = 325 °C; catalyst mass = 0.2 g. Open symbols: dry feed 1.5% CH
4; 6% O
2;balance He; closed symbol: wet feed 1.5% CH
4; 6% O
2 with 3% H
2O, balance He. Reproduced with permission from [
53]. Copyright © 2005 Elsevier.
Figure 8.
(A) Pd/Aerosil130 catalyst, (
B) Pd/R972 catalyst. Reaction conditions: total flow = 90 cm
3 (STP) min
−1, temperature = 325 °C; catalyst mass = 0.2 g. Open symbols: dry feed 1.5% CH
4; 6% O
2;balance He; closed symbol: wet feed 1.5% CH
4; 6% O
2 with 3% H
2O, balance He. Reproduced with permission from [
53]. Copyright © 2005 Elsevier.
2.4. H2O Inhibition and Hydroxyl Formation
Although Pd(OH)
2 has been postulated as a cause for deactivation of PdO catalysts in the presence of H
2O [
18,
31,
32,
40], and while this mechanism is consistent with many of the observations discussed above, recent evidence obtained from FTIR and isotopic labeling experiments that monitor the formation and conversion of hydroxyls on the catalyst surface during reaction, suggest an alternative mechanism of deactivation.
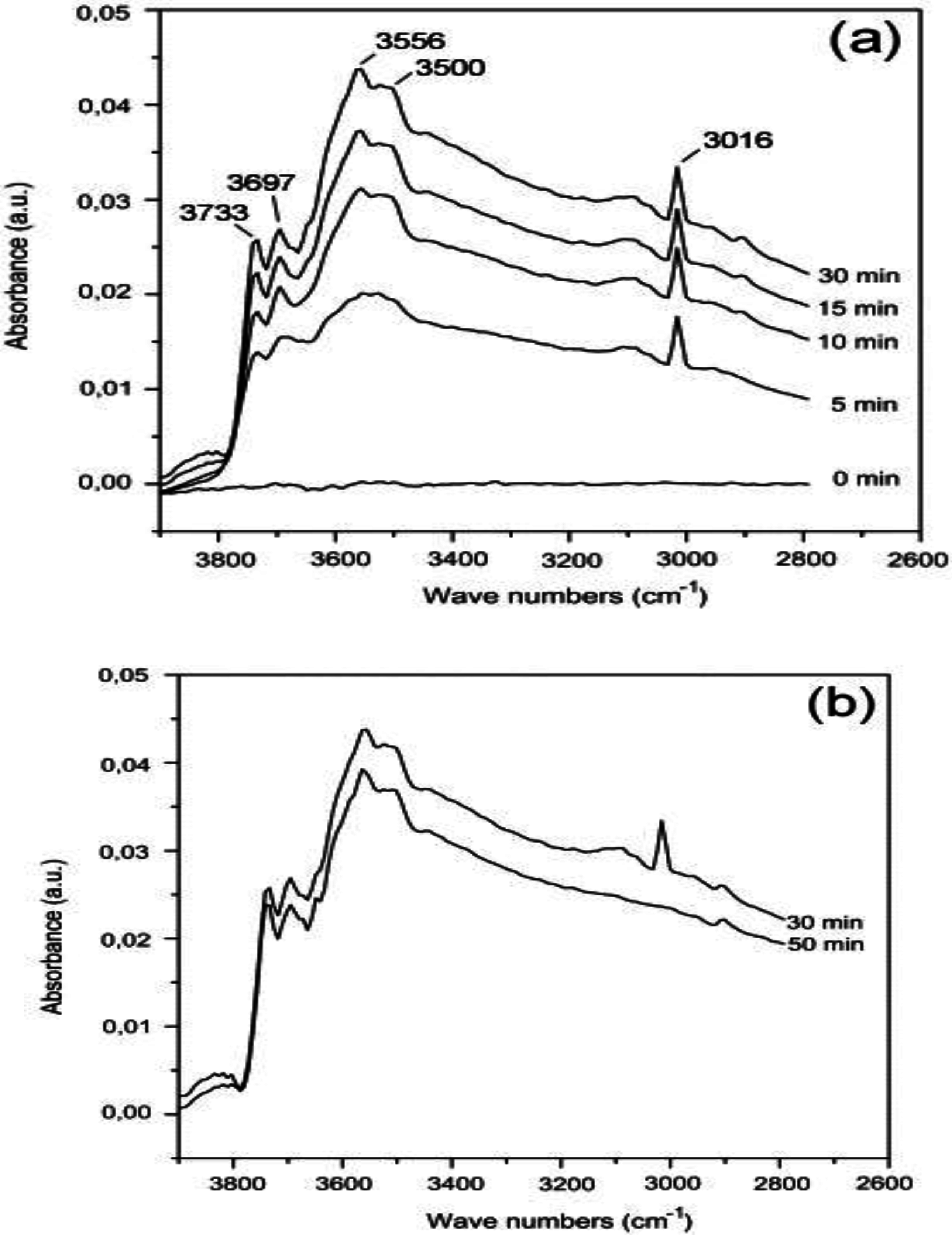
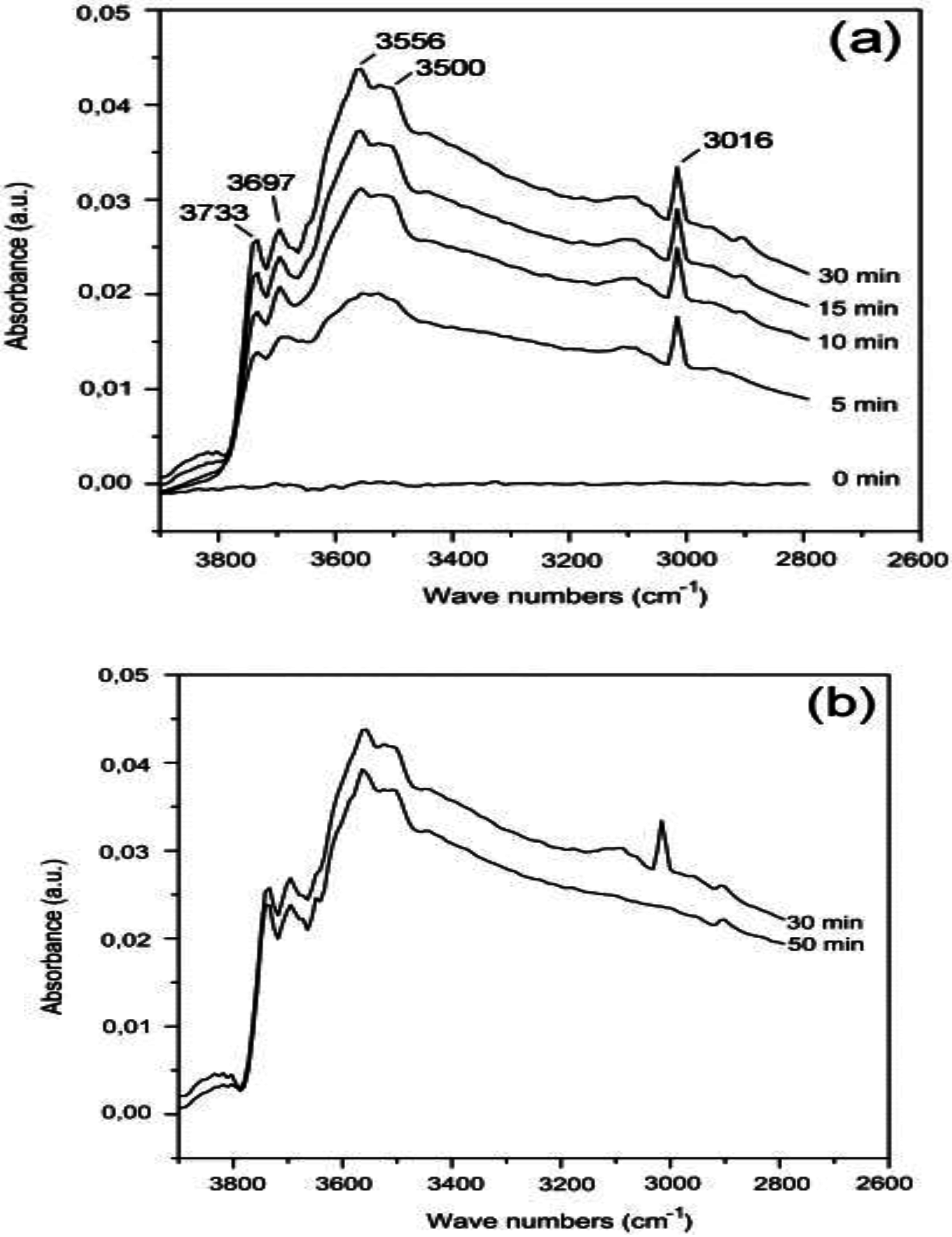
Using DRIFTS, Persson
et al. [
35] reported an increase in signal intensity from surface hydroxyls weakly H-bonded to the support (3200–3800 cm
−1) [
65] after introducing 1.5% CH
4 in air to a PdO/Al
2O
3 catalyst at low temperature (200 °C;
Figure 9). The peak at 3016 cm
−1 in
Figure 9a, assigned to gas phase CH
4, increases with time-on-stream because of catalyst deactivation. The hydroxyls have characteristic adsorptions at 3733, 3697, 3556 and 3500 cm
−1, with the hydroxyls at 3697 and 3733 cm
−1 assigned to bridged and terminal isolated hydroxyl species, respectively. Upon CH
4 removal from the feed (
Figure 9b), the peaks associated with OH species remain, highly consistent with a slow desorption of OH species produced during CH
4 oxidation. Hence, Persson
et al. [
35] suggested that catalyst deactivation on PdO/Al
2O
3 might be due to the formation and accumulation of hydroxyls on the catalyst surface, bound either to the PdO, Al
2O
3 or the interface between the two [
30]. Gao
et al. [
32] reported similar hydroxyl bands at 3733, 3697, 3556 and 3500 cm
−1 during lean-burn CH
4 oxidation (0.4% CH
4 in air) at 250 °C. The FTIR spectra from reaction with 2 vol.% H
2O added to the CH
4-O
2 feed also yield a broad band at 3445 cm
−1 that is associated with OH species on Al
2O
3 [
32].
Figure 9.
FTIR spectra of 5 wt.% Pd/Al
2O
3 at 200 °C (
a) during the CH
4-O
2 reaction; (
b) desorption when CH
4 was removed. Reproduced with permission from [
35]. Copyright© 2007 Elsevier.
Figure 9.
FTIR spectra of 5 wt.% Pd/Al
2O
3 at 200 °C (
a) during the CH
4-O
2 reaction; (
b) desorption when CH
4 was removed. Reproduced with permission from [
35]. Copyright© 2007 Elsevier.
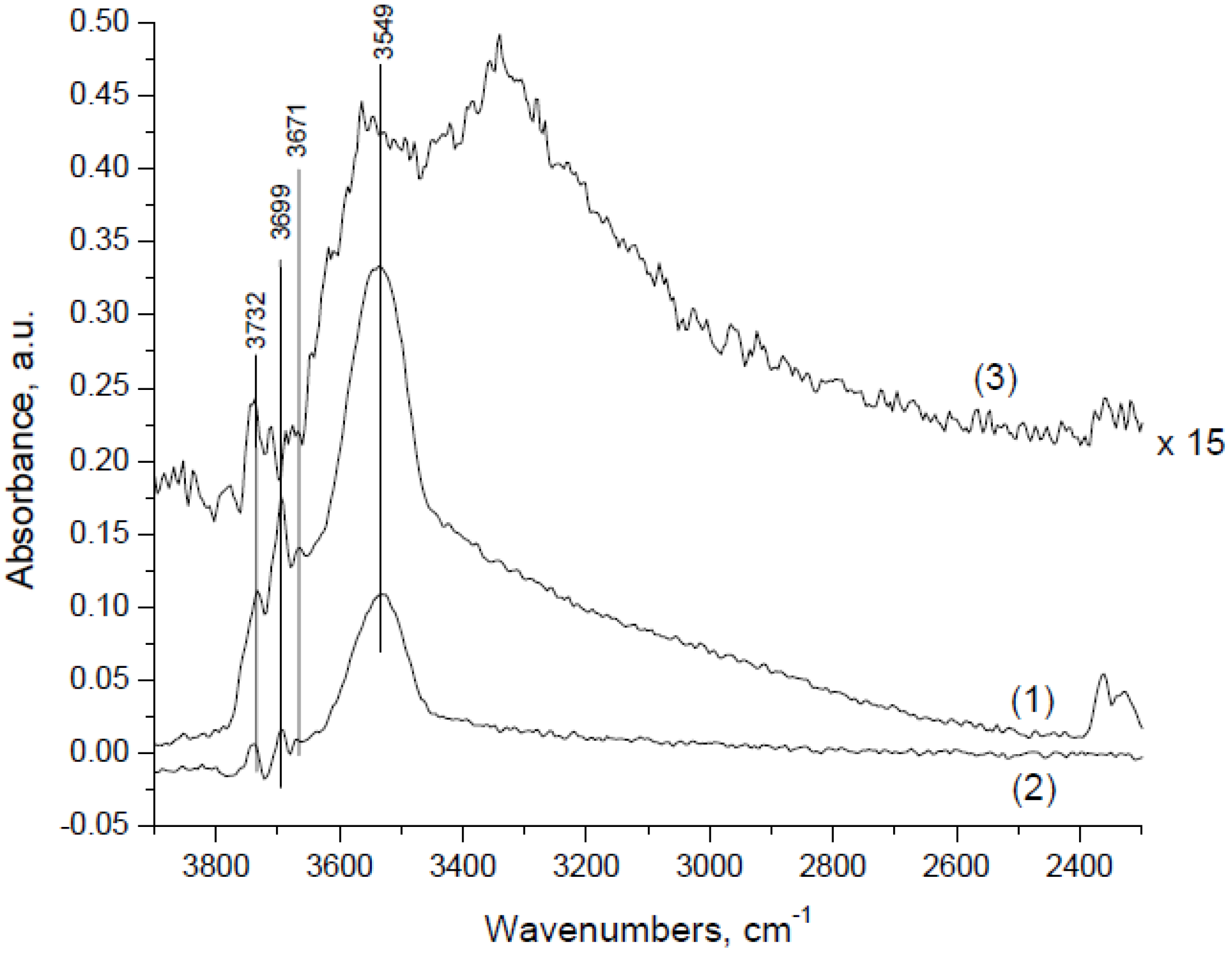
Figure 10.
FTIR spectra at highest surface coverage and 350 °C on (1) PdO/Al
2O
3 during CH
4-O
2 reaction, (2) PdO/Al
2O
3 and (3) Al
2O
3 when injecting H
2O pulses. Reproduced with permission from [
30]. Copyright © 2004 Elsevier.
Figure 10.
FTIR spectra at highest surface coverage and 350 °C on (1) PdO/Al
2O
3 during CH
4-O
2 reaction, (2) PdO/Al
2O
3 and (3) Al
2O
3 when injecting H
2O pulses. Reproduced with permission from [
30]. Copyright © 2004 Elsevier.
Ciuparu
et al. [
30] also identified three well-defined peaks at 3732 (OH
I), 3699 (OH
II), and 3549 (OH
III) cm
−1 associated with surface hydroxyls generated during CH
4 oxidation on a PdO/Al
2O
3 catalyst (3.5 wt % Pd) at 350 °C using a feed gas of 0.128% CH
4 and 17.3% O
2 in He/N
2 (
Figure 10). The spectrum was compared to that measured at the same temperature when injecting pulses of ~3% H
2O into an air flow over the PdO/Al
2O
3 catalyst and the Al
2O
3 support (see
Figure 10). Since Al
2O
3 has been shown to have a significantly lower hydroxyl coverage compared to PdO/Al
2O
3 when injecting H
2O pulses at 350 °C (the spectrum of Al
2O
3 is magnified by a factor of 15 in
Figure 10), they concluded that the three peaks are associated with the presence of OH adsorbed on the PdO catalyst surface. The higher hydroxyl coverage during CH
4 oxidation compared to pulse injection of H
2O onto the PdO/Al
2O
3 catalyst, indicates that (1) adsorbed H
2O is dissociated on the surface of PdO/Al
2O
3 and (2) hydroxyls formed from H
2O pulses are less strongly bound to the surface than hydroxyls produced by the CH
4 oxidation reaction.
Since the frequencies of the OH
I and OH
II species are shifted to higher wave numbers for OH species more weakly bound to Pd, Ciuparu
et al. [
30] suggested that the high frequency peaks (OH
I, OH
II) can be assigned to terminal and bridged hydroxyl species, respectively, and the low frequency peak at ~3549 cm
−1 with broad maximum values can be associated with OH species bound to different sites (multi-bound OHs; OH
III) (
Figure 10). Transient temperature experiments show that the hydroxyl binding energy increases in the order OH
I < OH
II < OH
III [
30].
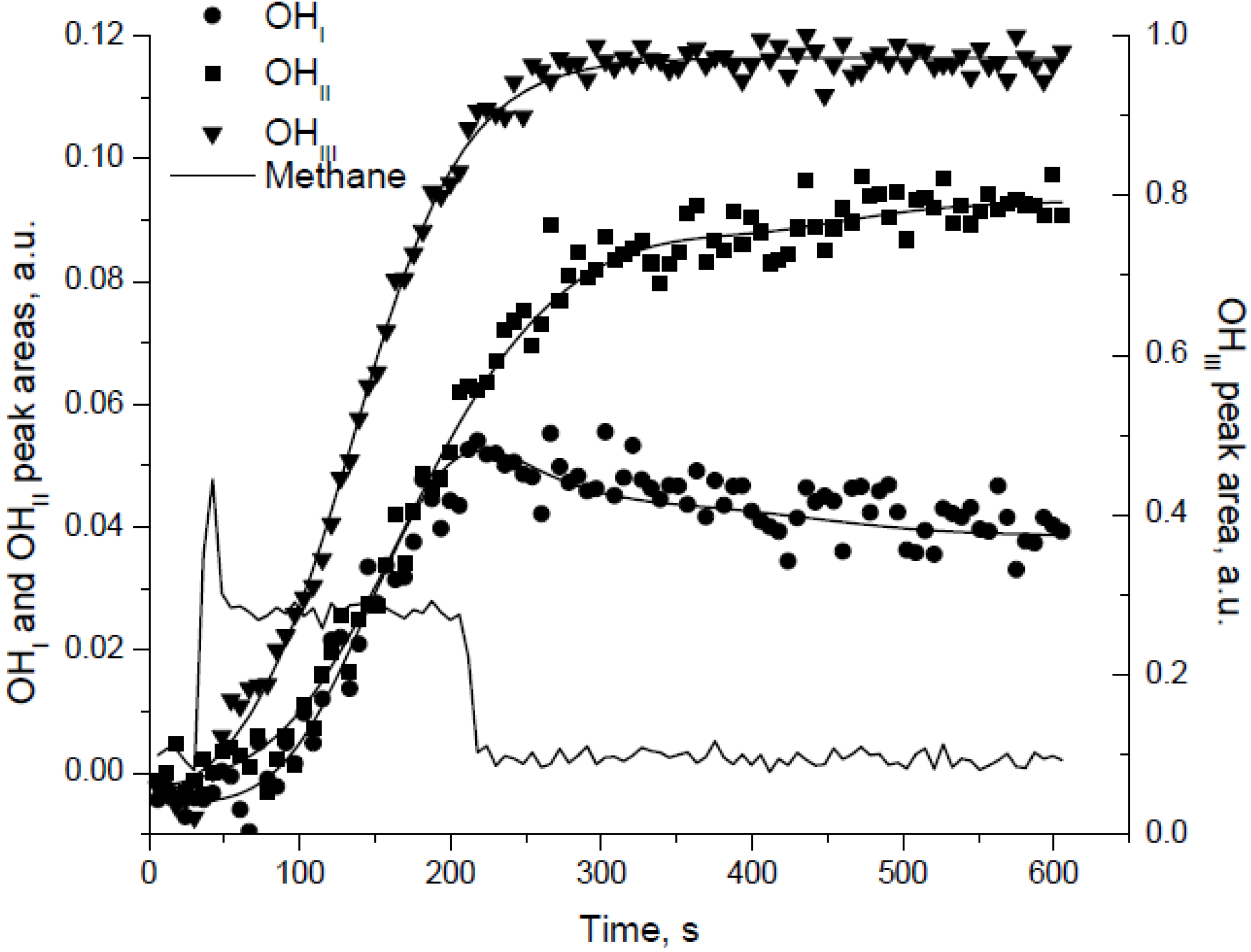
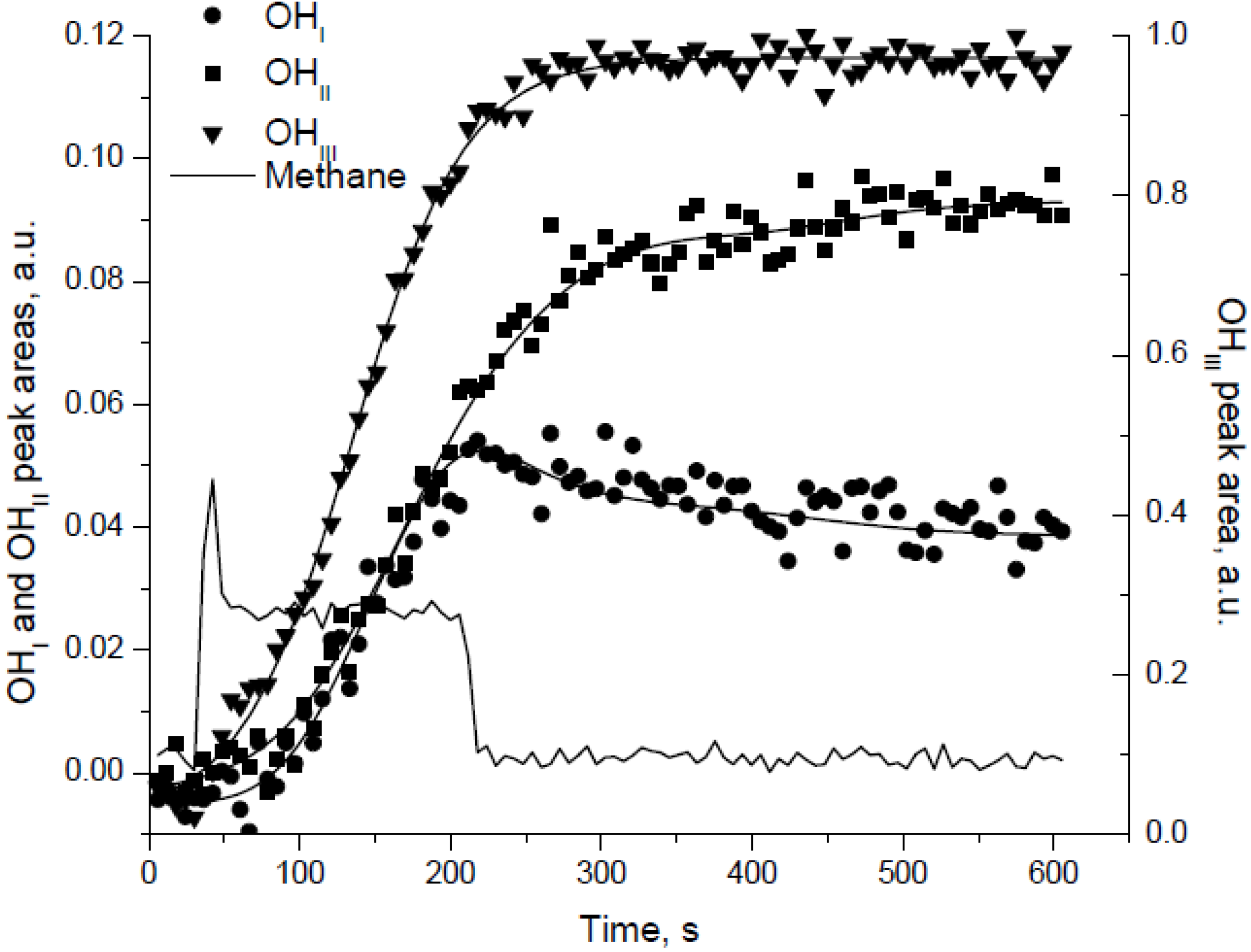
The peak areas of the terminal, bridged, and multi-bound hydroxyls were monitored with time-on-stream at different temperatures during reaction, as illustrated by
Figure 11 for reaction at 175 °C [
30]. Upon removal of CH
4 from the feed, the peak areas for the bridged and multi-bound OH species continue to increase, whereas the area of the terminal OH species decreases (
Figure 11). This decrease is attributed to the conversion of terminal OH species to bridged or multi-bound OH species. Based on the intensities of the various hydroxyl species at different temperatures, the authors proposed the inter-conversion among the OH species as:
where only terminal OH species recombine and desorb as H
2O and the transformation of bridged OH species to terminal OH species is the rate determining step (RDS) for hydroxyl desorption and hence low temperature CH
4 oxidation [
30]. Importantly the authors show that the surface coverage by the hydroxyls (
Figure 11) correlates with the activity loss at low temperature, meaning that the activity loss and surface coverage have similar timescales, from which they conclude that the former is likely an effect of the latter [
30].
Figure 11.
The normalized peak areas of different surface OH species generated during lean-CH
4-O
2 reaction at 175 °C. Reproduced with permission from [
30]. Copyright © 2004 Elsevier.
Figure 11.
The normalized peak areas of different surface OH species generated during lean-CH
4-O
2 reaction at 175 °C. Reproduced with permission from [
30]. Copyright © 2004 Elsevier.
FTIR spectra measured during CH
4 oxidation at 325 °C with 0.1% CH
4/4%O
2 in He over a series of 3 wt.% PdO catalysts supported on Al
2O
3, MgO, TiO
2 and MCM-41 [
44] show that the hydroxyl coverage is dependent on the support. On Al
2O
3, well defined peaks similar to those identified by Ciuparu
et al. [
30] are observed, but no common peak among all catalysts that would provide evidence for Pd-OH bond formation, are present. Furthermore the large contribution from OH bonding on the supports makes it impossible to directly identify the presence of Pd(OH)
2 on these supports [
32,
44]. However, by using
18O isotopic labeling and FTIR, the authors demonstrate that peaks associated with the accumulation of hydroxyls on PdO are not present at 325 °C. Hence, the more recent evidence suggests that deactivation by Pd(OH)
2 formation is unlikely, in agreement with the experimental observation that Pd(OH)
2/C decomposes in N
2 at about 250 °C [
66]. In addition, evidence from temperature-programmed desorption studies of H
2O adsorbed on PdO(101) thin films, suggests the formation of an OH-H
2O complex at low temperature (<127 °C) and low coverage (< ½ monolayer), whereas H
2O preferentially chemisorbs in molecular form at higher coverages [
67].
Schwartz
et al. [
44] showed, however, that catalyst deactivation during CH
4 oxidation correlates with hydroxyl accumulation on the oxide support. The redox mechanism for CH
4 combustion on Pd/PdO generally assumes dissociation of a CH
4 molecule to yield a methyl fragment and a hydroxyl group (CH
4 + Pd-O + Pd-*→Pd-OH + Pd-CH
3, where Pd-* represents a vacancy) [
68,
69]. H atoms are abstracted sequentially from the methyl group by neighboring Pd-O to form surface hydroxyl groups (Pd-OH). Recombination of surface hydroxyls yields water and a surface vacancy (2Pd-OH→H
2O + Pd-O +Pd-*), that is regenerated by oxygen (2Pd-* + O
2→2Pd-O) [
68,
69]. Based on their experimental studies, Schwartz
et al. [
44,
57], proposed that during lean-burn CH
4 oxidation, O
2 molecules dissociate on Pd-* sites and exchange with oxygen on the support so that Pd active sites are re-oxidized with oxygen atoms from the support during the catalytic reaction as follows:
and overall:
where S represents the support, S-* is an O vacancy on the support and O
s represents an O atom associated with the solid oxide. This proposed mechanism suggests the possibility that a primary cause for catalyst deactivation is hydroxyl accumulation on the support, which hinders oxygen migration and exchange processes.
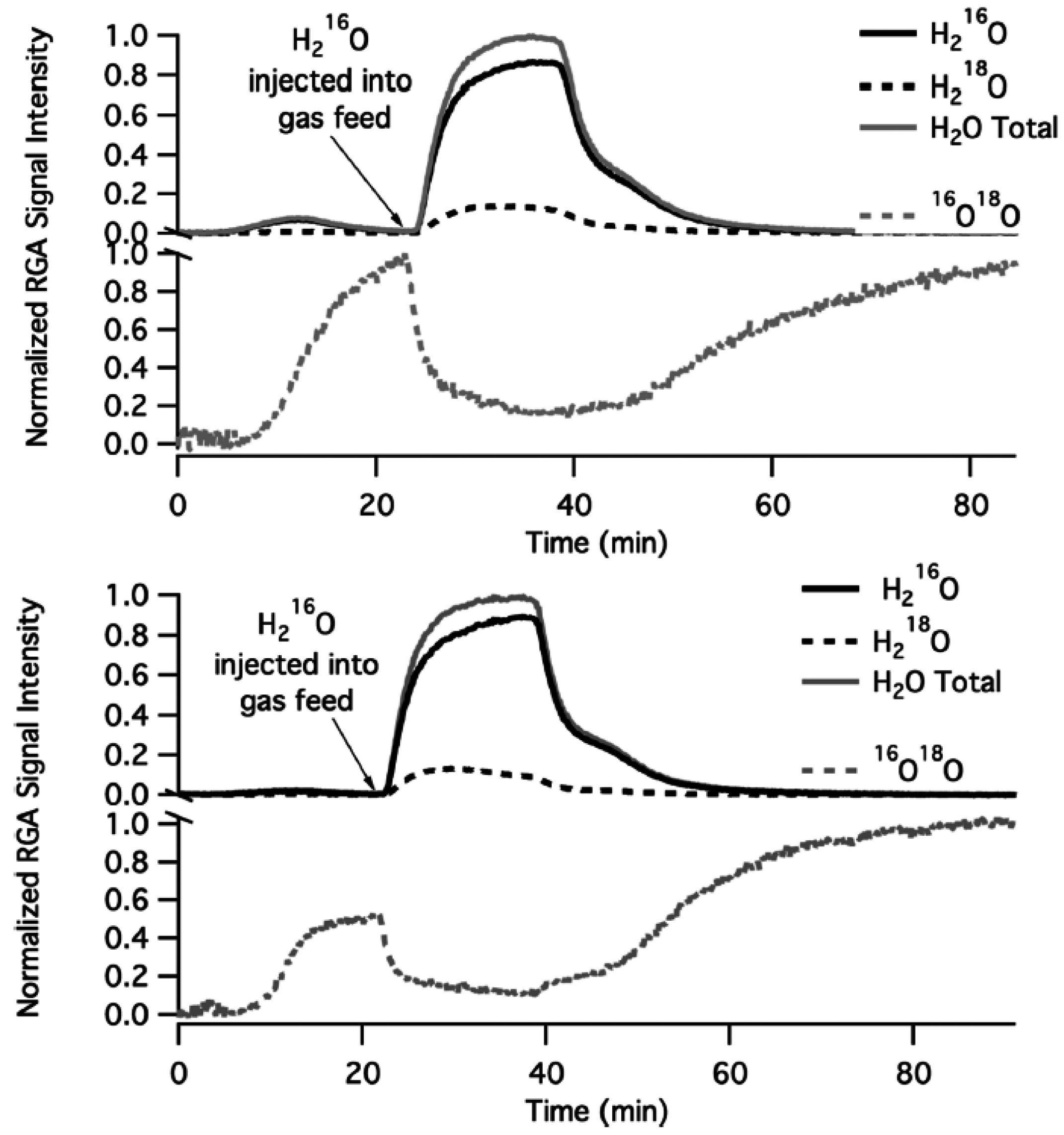
Evidence for O exchange with the support is provided by the isotopic labeling experiments summarized in
Figure 12, during which Pd
18O/Al
216O
3 and Pd
18O/Mg
16O were exposed to
18O
2/He flow at 400 °C [
57]. An increase in
16O
18O signal intensity with time is proposed to arise from oxygen exchange with the catalyst support [
44]. The
16O
18O signal (see lower, separate dashed line in
Figure 12) is reduced when H
216O is injected to the feed and is recovered when H
216O is removed. Apparently, hydroxyl groups tend to migrate to the oxide support rather than desorb. By increasing the concentration of hydroxyl groups, through addition and dissociation of H
2O, oxygen exchange of Pd-* active sites with the oxide support (S-O
s) is interrupted. Thus, the number of PdO sites participating in the CH
4 oxidation reaction decreases with time, as H
2O dissociates and OH coverage of the support increases, with a consequent decrease in CH
4 conversion [
44]. This proposed mechanism of catalyst deactivation is believed to occur at temperatures below 450 °C. Finally, the authors note that the rate of deactivation on Pd/Al
2O
3 catalysts, with higher concentrations of hydroxyl during reaction, is higher than on catalysts containing a support with higher oxygen mobility (Pd/MgO) [
44,
57].
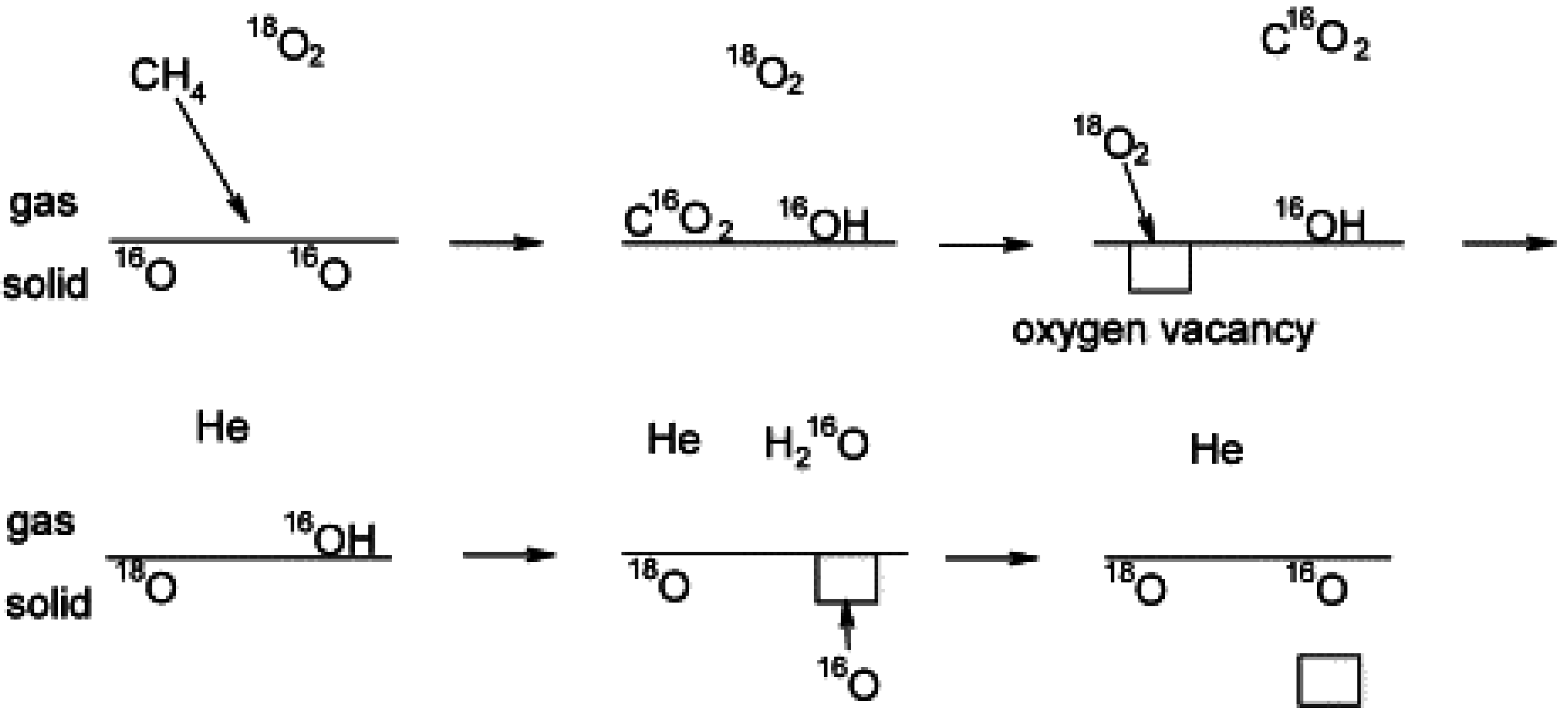
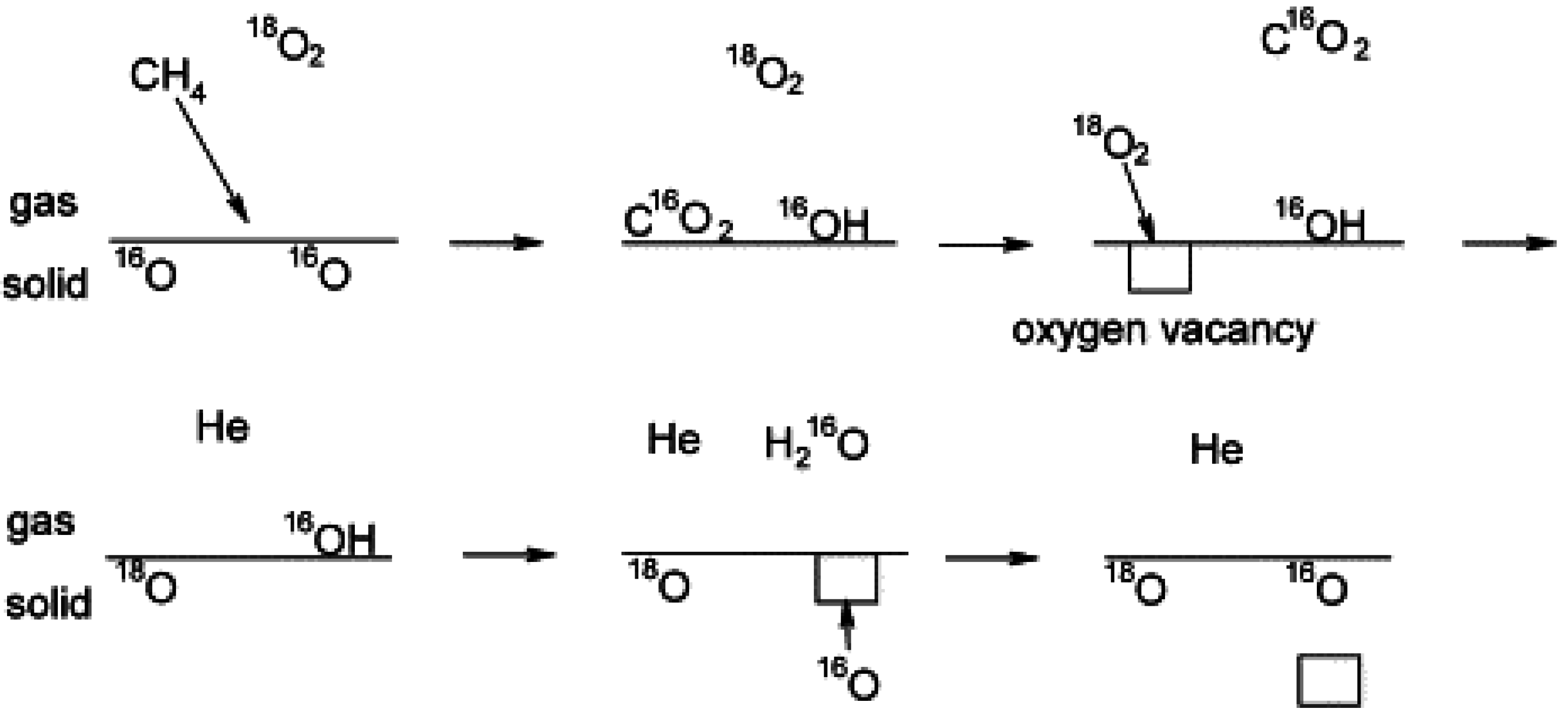
Ciuparu
et al. [
70] also reported on pulsed experiments with
18O
2 over pure Pd and Pd/ZrO
2 catalysts, oxidized before reaction, to clarify the effect of hydroxyls on the surface oxygen exchange. They determined that due to the slow recombination of hydroxyls and hence H
2O desorption from the Pd catalyst surface during CH
4 oxidation (2Pd-OH→H
2O + Pd-O +Pd-*), the isotopic exchange of oxygen with the Pd sites (see
Figure 13) occurs before H
2O desorption from the surface. The oxygen vacancies on the PdO surface resulting from H
2O desorption are thus rapidly filled by oxygen from the PdO bulk or oxide support (Pd-* + S-O
s↔Pd-O
s + S-*). In fact, in this unsteady-state experiment, the labeled oxygen pulsed through the catalyst bed, is purged from the reactor before H
2O is desorbed [
70]. These observations are in agreement with the studies of Schwartz
et al. [
44,
57] already discussed and confirm that the accumulation of hydroxyls on the Pd catalyst surface impedes the oxygen exchange and limits Pd catalyst activity.
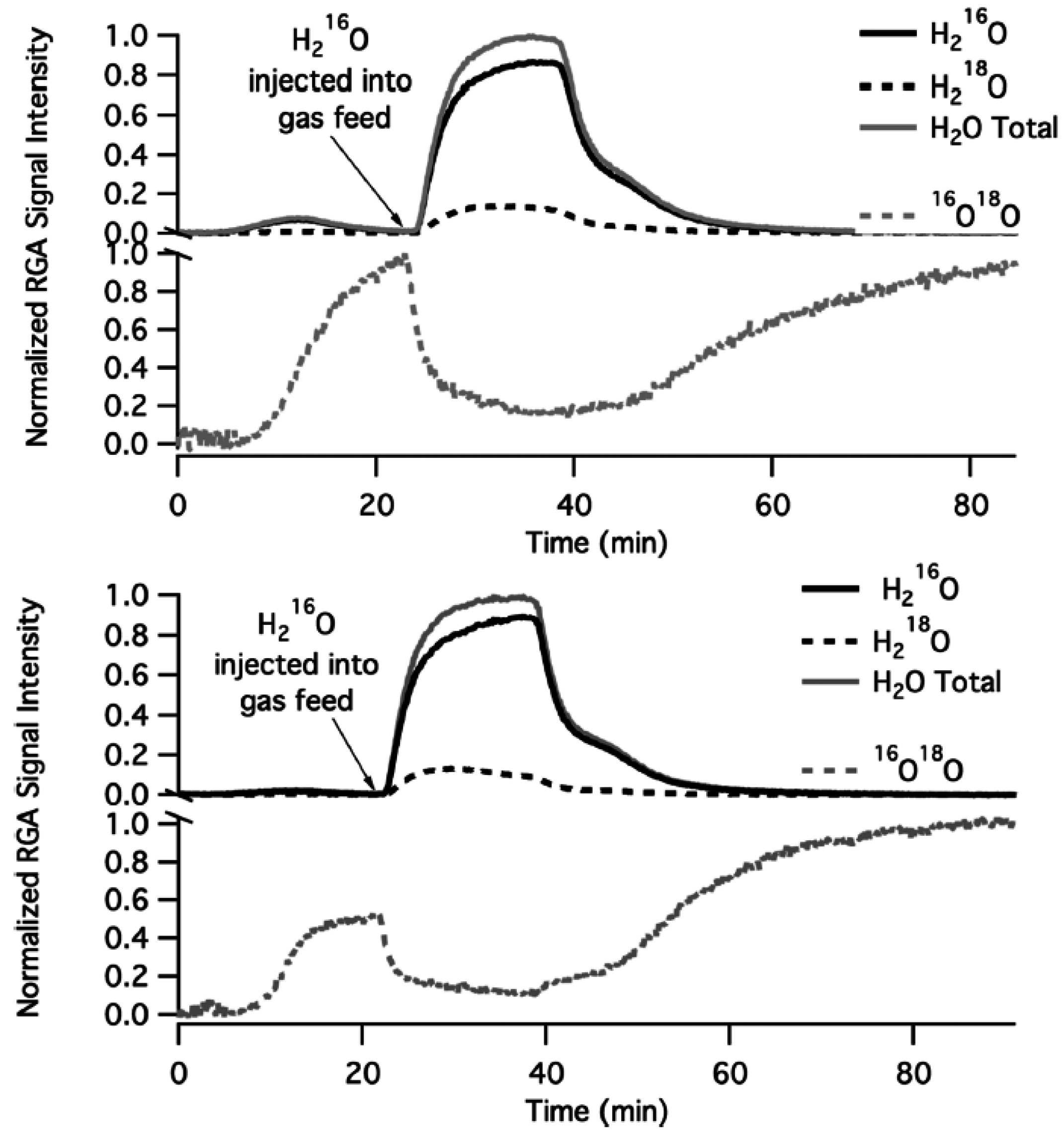
Figure 12.
Oxygen exchange of (a) 3 wt.% Pd
18O/Al
216O
3 (top) and (b) 3 wt.% Pd
18O/Mg
16O (bottom) with catalyst supports in a flow of
18O
2/He at 400 °C. H
216O was injected at some time to probe its effect on oxygen exchange. Reproduced with permission from [
44]. Copyright ©2012 American Chemical Society.
Figure 12.
Oxygen exchange of (a) 3 wt.% Pd
18O/Al
216O
3 (top) and (b) 3 wt.% Pd
18O/Mg
16O (bottom) with catalyst supports in a flow of
18O
2/He at 400 °C. H
216O was injected at some time to probe its effect on oxygen exchange. Reproduced with permission from [
44]. Copyright ©2012 American Chemical Society.
Figure 13.
Schematic of oxygen exchange during CH
4 oxidation using labeled pulsed experiments. Reproduced with permission from [
70]. Copyright © 2002 Elsevier.
Figure 13.
Schematic of oxygen exchange during CH
4 oxidation using labeled pulsed experiments. Reproduced with permission from [
70]. Copyright © 2002 Elsevier.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}