
Unlocking Synergistic Catalysis in NiP: Dual Role of Electronic Structure and Lewis Acidity for Enhanced Oxygen Evolution Reaction


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
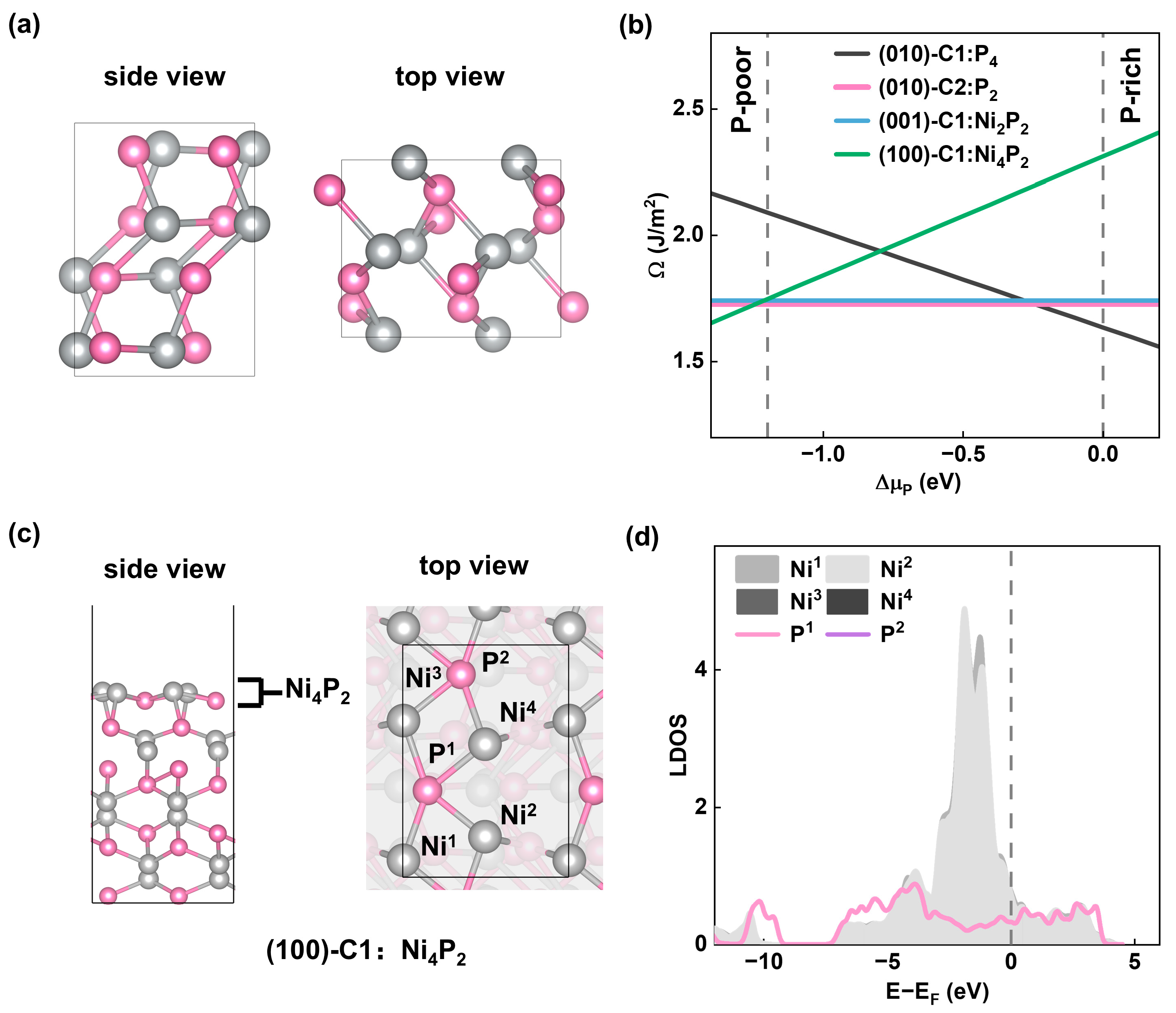
2.1. Stability of NiP Surfaces
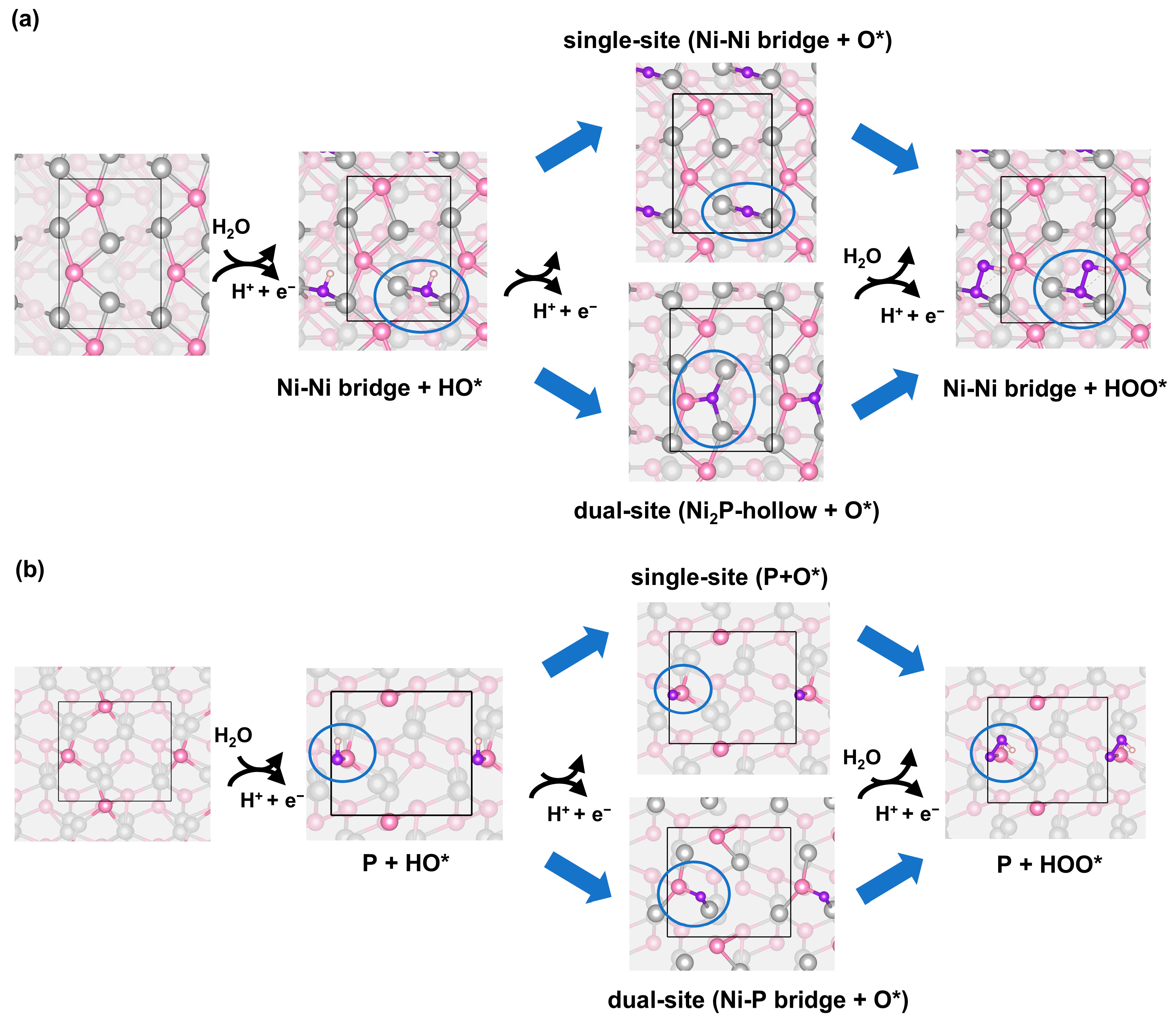
2.2. Stability of the Adsorption Structures
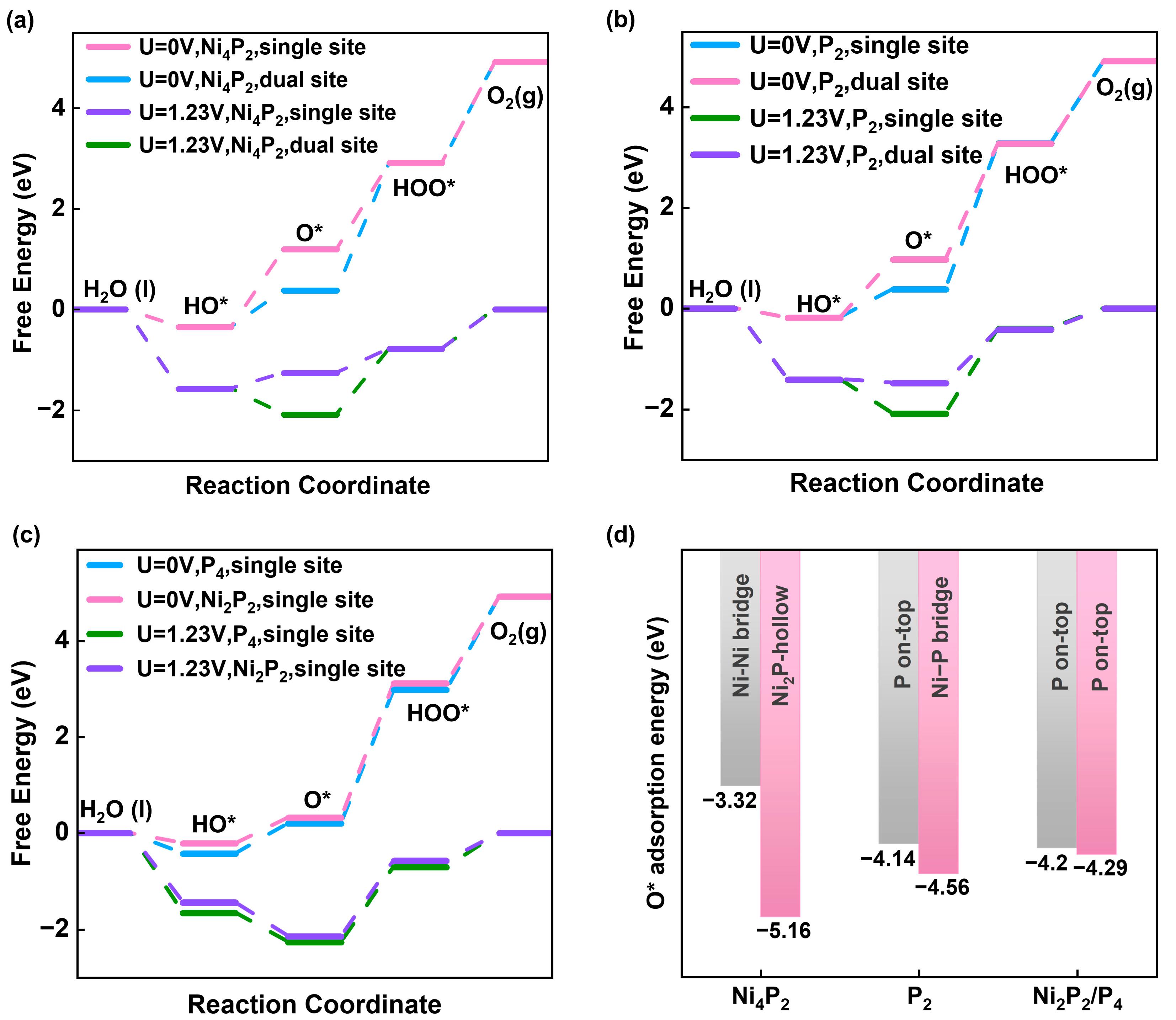
2.3. OER Pathways on NiP Surfaces
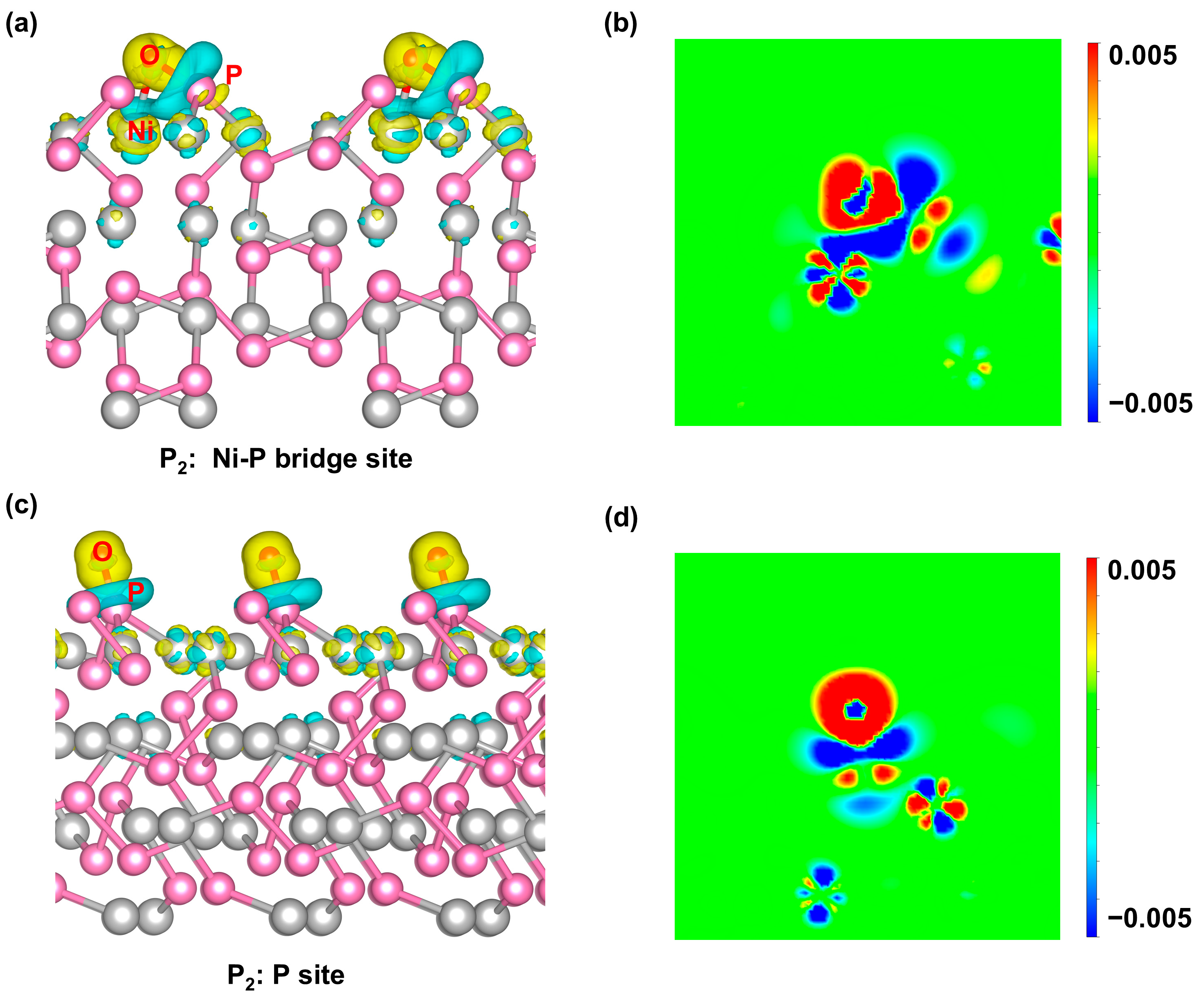
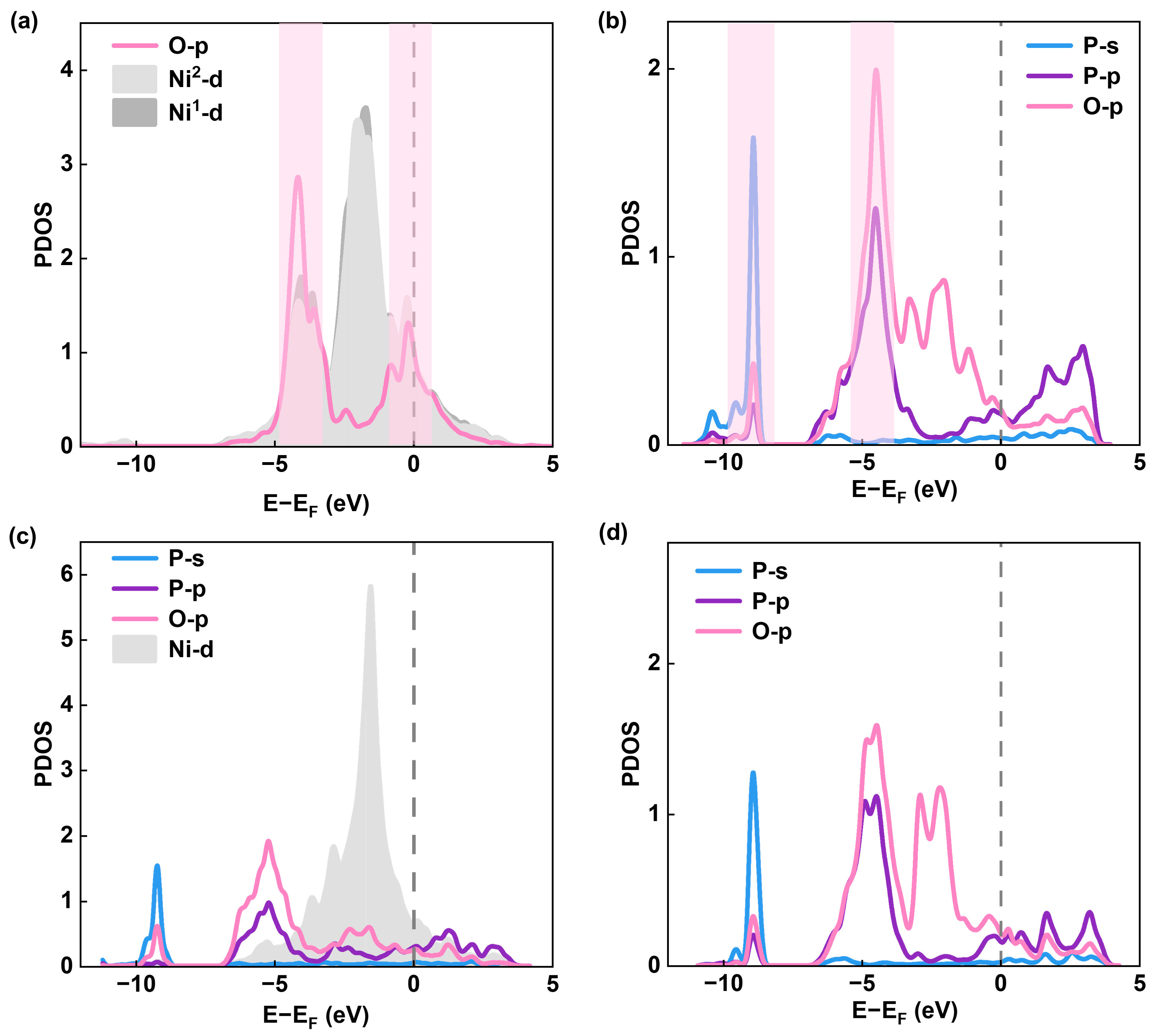
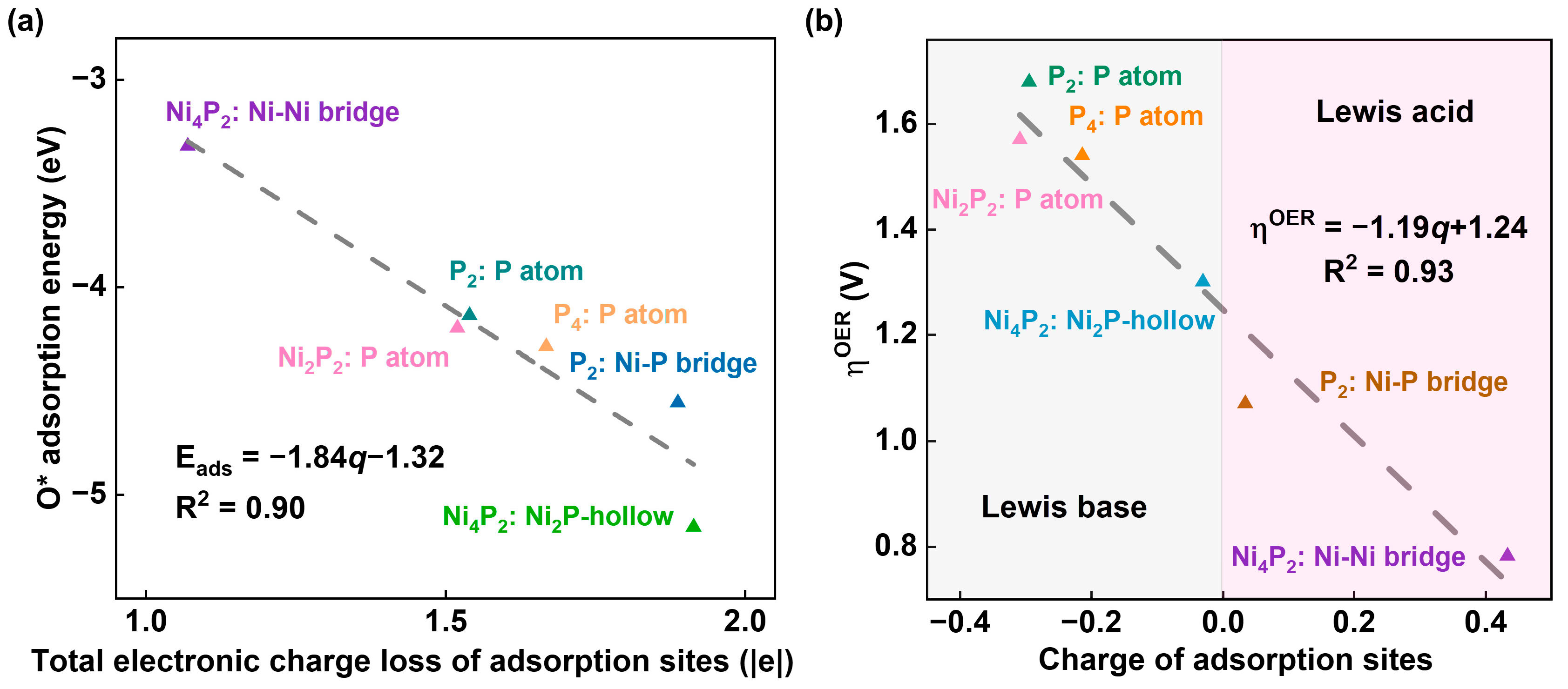
2.4. Investigation of Electrocatalytic OER Mechanism
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chow, J.; Kopp, R.J.; Portney, P.R. Energy resources and global development. Science 2003, 302, 1528–1531. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef]
- Shindell, D.; Smith, C.J. Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature 2019, 573, 408–411. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef]
- Rausch, B.; Symes, M.D.; Chisholm, G.; Cronin, L. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 2014, 345, 1326–1330. [Google Scholar] [CrossRef]
- Gao, M.-R.; Liang, J.-X.; Zheng, Y.-R.; Xu, Y.-F.; Jiang, J.; Gao, Q.; Li, J.; Yu, S.-H. An efficient molybdenum disulfide/cobalt diselenide hybrid catalyst for electrochemical hydrogen generation. Nat. Commun. 2015, 6, 5982. [Google Scholar] [CrossRef]
- Castelvecchi, D. How the hydrogen revolution can help save the planet—And how it can’t. Nature 2022, 611, 440–443. [Google Scholar] [CrossRef]
- Xue, Z.; Li, X.; Liu, Q.; Cai, M.; Liu, K.; Liu, M.; Ke, Z.; Liu, X.; Li, G. Interfacial Electronic Structure Modulation of NiTe Nanoarrays with NiS Nanodots Facilitates Electrocatalytic Oxygen Evolution. Adv. Mater. 2019, 31, e1900430. [Google Scholar] [CrossRef]
- Nong, H.N.; Falling, L.J.; Bergmann, A.; Klingenhof, M.; Tran, H.P.; Spöri, C.; Mom, R.; Timoshenko, J.; Zichittella, G.; Knop-Gericke, A.; et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 2020, 587, 408–413. [Google Scholar] [CrossRef]
- Li, L.; Wang, P.; Shao, Q.; Huang, X. Recent Progress in Advanced Electrocatalyst Design for Acidic Oxygen Evolution Reaction. Adv. Mater. 2021, 33, e2004243. [Google Scholar] [CrossRef]
- Gao, J.; Tao, H.; Liu, B. Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media. Adv. Mater. 2021, 33, 2003786. [Google Scholar] [CrossRef] [PubMed]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Du, D.; Lin, Y. Robust noble metal-based electrocatalysts for oxygen evolution reaction. Chem. Soc. Rev. 2019, 48, 3181–3192. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, M.-C.; Lee, K.B.; Choi, H.; Zhang, L.; Sohn, J.I. Nonprecious High-Entropy Chalcogenide Glasses-Based Electrocatalysts for Efficient and Stable Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis. Adv. Energy Mater. 2023, 13, 2301420. [Google Scholar] [CrossRef]
- Kong, S.; Li, A.; Long, J.; Adachi, K.; Hashizume, D.; Jiang, Q.; Fushimi, K.; Ooka, H.; Xiao, J.; Nakamura, R. Acid-stable manganese oxides for proton exchange membrane water electrolysis. Nat. Catal. 2024, 7, 252–261. [Google Scholar] [CrossRef]
- Pu, Z.; Liu, T.; Amiinu, I.S.; Cheng, R.; Wang, P.; Zhang, C.; Ji, P.; Hu, W.; Liu, J.; Mu, S. Transition-Metal Phosphides: Activity Origin, Energy-Related Electrocatalysis Applications, and Synthetic Strategies. Adv. Funct. Mater. 2020, 30, 2004009. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, L.; Wang, M.; Qi, Y.; Guo, D.; Li, H.; Chen, X.; Wang, S. Transition Metal-Based Electrocatalysts for Seawater Oxidation. Adv. Mater. Interfaces 2022, 9, 2201486. [Google Scholar] [CrossRef]
- Guo, T.; Li, L.; Wang, Z. Recent Development and Future Perspectives of Amorphous Transition Metal-Based Electrocatalysts for Oxygen Evolution Reaction. Adv. Energy Mater. 2022, 12, 2200827. [Google Scholar] [CrossRef]
- Han, L.; Dong, S.; Wang, E. Transition-Metal (Co, Ni, and Fe)-Based Electrocatalysts for the Water Oxidation Reaction. Adv. Mater. 2016, 28, 9266–9291. [Google Scholar] [CrossRef]
- Ledendecker, M.; Krick Calderon, S.; Papp, C.; Steinruck, H.P.; Antonietti, M.; Shalom, M. The synthesis of nanostructured Ni5 P4 films and their use as a non-noble bifunctional electrocatalyst for full water splitting. Angew. Chem. Int. Ed. Engl. 2015, 54, 12361–12365. [Google Scholar] [CrossRef]
- Sun, H.; Yan, Z.; Liu, F.; Xu, W.; Cheng, F.; Chen, J. Self-Supported Transition-Metal-Based Electrocatalysts for Hydrogen and Oxygen Evolution. Adv. Mater. 2020, 32, e1806326. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, M.J.; Kim, J.J. Modulating the active sites of nickel phosphorous by pulse-reverse electrodeposition for improving electrochemical water splitting. Appl. Catal. B Environ. 2022, 308, 121226. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Q.; Qu, X.; Wang, X.; Chi, J.; Wang, L. Manipulating Electron Redistribution in Ni2P for Enhanced Alkaline Seawater Electrolysis. Adv. Mater. 2024, 36, e2307395. [Google Scholar] [CrossRef]
- Jiang, M.; Zhai, H.; Chen, L.; Mei, L.; Tan, P.; Yang, K.; Pan, J. Unraveling the Synergistic Mechanism of Bi-Functional Nickel–Iron Phosphides Catalysts for Overall Water Splitting. Adv. Funct. Mater. 2023, 33, 2302621. [Google Scholar] [CrossRef]
- Xu, T.; Jiao, D.; Zhang, L.; Zhang, H.; Zheng, L.; Singh, D.J.; Zhao, J.; Zheng, W.; Cui, X. Br-induced P-poor defective nickel phosphide for highly efficient overall water splitting. Appl. Catal. B Environ. 2022, 316, 121686. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Hao, H.; Yuan, M.; Lv, Z.; Xu, L.; Wei, B. In situ unraveling surface reconstruction of Ni5P4@FeP nanosheet array for superior alkaline oxygen evolution reaction. Appl. Catal. B Environ. 2022, 305, 121033. [Google Scholar] [CrossRef]
- Liu, K.; Wang, F.; He, P.; Shifa, T.A.; Wang, Z.; Cheng, Z.; Zhan, X.; He, J. The Role of Active Oxide Species for Electrochemical Water Oxidation on the Surface of 3d-Metal Phosphides. Adv. Energy Mater. 2018, 8, 1703290. [Google Scholar] [CrossRef]
- Menezes, P.W.; Indra, A.; Das, C.; Walter, C.; Göbel, C.; Gutkin, V.; Schmeiβer, D.; Driess, M. Uncovering the Nature of Active Species of Nickel Phosphide Catalysts in High-Performance Electrochemical Overall Water Splitting. ACS Catal. 2017, 7, 103–109. [Google Scholar] [CrossRef]
- Stern, L.-A.; Feng, L.; Song, F.; Hu, X. Ni2P as a Janus catalyst for water splitting: The oxygen evolution activity of Ni2P nanoparticles. Energy Environ. Sci. 2015, 8, 2347–2351. [Google Scholar] [CrossRef]
- Kim, B.K.; Kim, S.-K.; Cho, S.K.; Kim, J.J. Enhanced catalytic activity of electrodeposited Ni-Cu-P toward oxygen evolution reaction. Appl. Catal. B Environ. 2018, 237, 409–415. [Google Scholar] [CrossRef]
- Gao, L.; Cui, X.; Sewell, C.D.; Li, J.; Lin, Z. Recent advances in activating surface reconstruction for the high-efficiency oxygen evolution reaction. Chem. Soc. Rev. 2021, 50, 8428–8469. [Google Scholar] [CrossRef]
- Jiang, H.; He, Q.; Li, X.; Su, X.; Zhang, Y.; Chen, S.; Zhang, S.; Zhang, G.; Jiang, J.; Luo, Y.; et al. Tracking Structural Self-Reconstruction and Identifying True Active Sites toward Cobalt Oxychloride Precatalyst of Oxygen Evolution Reaction. Adv. Mater. 2019, 31, e1805127. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cheng, Y.; Wen, Y.; Wang, Y.; Yan, X.; Wei, J.; He, S.; Zhou, J. CoP/Fe-Co9S8 for Highly Efficient Overall Water Splitting with Surface Reconstruction and Self-Termination. Adv. Sci. 2022, 9, e2204742. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.B.; Wexler, R.B.; Whitaker, M.J.; Izett, E.J.; Calvinho, K.U.D.; Hwang, S.; Rucker, R.; Wang, H.; Li, J.; Garfunkel, E.; et al. Climbing the Volcano of Electrocatalytic Activity while Avoiding Catalyst Corrosion: Ni3P, a Hydrogen Evolution Electrocatalyst Stable in Both Acid and Alkali. ACS Catal. 2018, 8, 4408–4419. [Google Scholar] [CrossRef]
- Banerjee, S.; Kakekhani, A.; Wexler, R.B.; Rappe, A.M. Relationship between the Surface Reconstruction of Nickel Phosphides and Their Activity toward the Hydrogen Evolution Reaction. ACS Catal. 2023, 13, 4611–4621. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, F. Main Descriptors To Correlate Structures with the Performances of Electrocatalysts. Angew. Chem. Int. Ed. Engl. 2022, 61, e202111026. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, L.; Li, Z.; Nie, G.; Cao, X.; Fang, Z.; Wang, X.; Ramakrishna, S.; Long, Y.; Jiao, L. Regulating Hydrogen/Oxygen Species Adsorption via Built-in Electric Field -Driven Electron Transfer Behavior at the Heterointerface for Efficient Water Splitting. Angew. Chem. Int. Ed. Engl. 2024, 63, e202400888. [Google Scholar] [CrossRef]
- Wexler, R.B.; Martirez, J.M.P.; Rappe, A.M. Active Role of Phosphorus in the Hydrogen Evolving Activity of Nickel Phosphide (0001) Surfaces. ACS Catal. 2017, 7, 7718–7725. [Google Scholar] [CrossRef]
- Wexler, R.B.; Martirez, J.M.P.; Rappe, A.M. Stable Phosphorus-Enriched (0001) Surfaces of Nickel Phosphides. Chem. Mater. 2016, 28, 5365–5372. [Google Scholar] [CrossRef]
- Liyanage, I.A.; Flores, A.V.; Gillan, E.G. Tunable Synthesis of Metal-Rich and Phosphorus-Rich Nickel Phosphides and Their Comparative Evaluation as Hydrogen Evolution Electrocatalysts. Inorg. Chem. 2023, 62, 4947–4959. [Google Scholar] [CrossRef]
- He, J.; Morales-García, Á.; Bludský, O.; Nachtigall, P. The surface stability and equilibrium crystal morphology of Ni2P nanoparticles and nanowires from an ab initio atomistic thermodynamic approach. CrystEngComm 2016, 18, 3808–3818. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Feng, Y.; Allen, C.S.; Wan, C.; Volosskiy, B.; Li, M.; Zhao, Z.; Wang, Y.; Sun, H.; et al. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 2018, 1, 63–72. [Google Scholar] [CrossRef]
- Kuo, D.-Y.; Paik, H.; Kloppenburg, J.; Faeth, B.; Shen, K.M.; Schlom, D.G.; Hautier, G.; Suntivich, J. Measurements of Oxygen Electroadsorption Energies and Oxygen Evolution Reaction on RuO2(110): A Discussion of the Sabatier Principle and Its Role in Electrocatalysis. J. Am. Chem. Soc. 2018, 140, 17597–17605. [Google Scholar] [CrossRef]
- He, F.; Zheng, Q.; Yang, X.; Wang, L.; Zhao, Z.; Xu, Y.; Hu, L.; Kuang, Y.; Yang, B.; Li, Z.; et al. Spin-State Modulation on Metal-Organic Frameworks for Electrocatalytic Oxygen Evolution. Adv. Mater. 2023, 35, e2304022. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, X.; Sun, Y.; Huang, M.; Fan, J.; Xu, S.; Li, H. Low Ruthenium Content Confined on Boron Carbon Nitride as an Efficient and Stable Electrocatalyst for Acidic Oxygen Evolution Reaction. Angew. Chem. Int. Ed. Engl. 2023, 62, e202308704. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, H.; Mebs, S.; Dau, H.; Driess, M.; Wang, Z.; Kang, Z.; Menezes, P.W. Reviving Oxygen Evolution Electrocatalysis of Bulk La-Ni Intermetallics via Gaseous Hydrogen Engineering. Adv. Mater. 2023, 35, e2208337. [Google Scholar] [CrossRef]
- Ren, X.; Zhai, Y.; Wang, P.; Xu, Z.; Gao, S.; Chen, X.; Gu, Q.; Wang, B.; Li, J.; Liu, S.F. Surface Restructuring of Zeolite-Encapsulated Halide Perovskite to Activate Lattice Oxygen Oxidation for Water Electrolysis. Adv. Mater. 2023, 35, e2301166. [Google Scholar] [CrossRef]
- Li, C.F.; Zhao, J.W.; Xie, L.J.; Wu, J.Q.; Ren, Q.; Wang, Y.; Li, G.R. Surface-Adsorbed Carboxylate Ligands on Layered Double Hydroxides/Metal-Organic Frameworks Promote the Electrocatalytic Oxygen Evolution Reaction. Angew. Chem. Int. Ed. Engl. 2021, 60, 18129–18137. [Google Scholar] [CrossRef]
- Shah, K.; Dai, R.; Mateen, M.; Hassan, Z.; Zhuang, Z.; Liu, C.; Israr, M.; Cheong, W.C.; Hu, B.; Tu, R.; et al. Cobalt Single Atom Incorporated in Ruthenium Oxide Sphere: A Robust Bifunctional Electrocatalyst for HER and OER. Angew. Chem. Int. Ed. Engl. 2022, 61, e202114951. [Google Scholar] [CrossRef]
- Hajiyani, H.; Pentcheva, R. Surface Termination and Composition Control of Activity of the CoxNi1–xFe2O4(001) Surface for Water Oxidation: Insights from DFT+U Calculations. ACS Catal. 2018, 8, 11773–11782. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Wang, H.; Huang, B.; Dai, Y. Multifunctional electrocatalyst PtM with low Pt loading and high activity towards hydrogen and oxygen electrode reactions: A computational study. Appl. Catal. B Environ. 2019, 255, 117743. [Google Scholar] [CrossRef]
- Xu, Q.; Jiang, H.; Zhang, H.; Hu, Y.; Li, C. Heterogeneous interface engineered atomic configuration on ultrathin Ni(OH)2/Ni3S2 nanoforests for efficient water splitting. Appl. Catal. B Environ. 2019, 242, 60–66. [Google Scholar] [CrossRef]
- Zhang, S.L.; Guan, B.Y.; Lu, X.F.; Xi, S.; Du, Y.; Lou, X.W.D. Metal Atom-Doped Co3O4 Hierarchical Nanoplates for Electrocatalytic Oxygen Evolution. Adv. Mater. 2020, 32, e2002235. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Hao, Y.; Yin, L.; Kuo, C.H.; Chen, H.Y.; Li, L.; Peng, S. Lewis Acid Driving Asymmetric Interfacial Electron Distribution to Stabilize Active Species for Efficient Neutral Water Oxidation. Adv. Mater. 2024, 36, e2308925. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227–1230. [Google Scholar] [CrossRef]
- Ramer, N.J.; Rappe, A.M. Designed nonlocal pseudopotentials for enhanced transferability. Phys. Rev. B 1999, 59, 12471–12478. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Larsson, E.J.A.K. An X-ray investigation of the Ni-P system and the crystal structures of NiP and NiP2. Ark. Kemi 1965, 23, 335–365. [Google Scholar]
- Bengtsson, L. Dipole correction for surface supercell calculations. Phys. Rev. B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Wang, L.; Hao, Y.; Deng, L.; Hu, F.; Zhao, S.; Li, L.; Peng, S. Rapid complete reconfiguration induced actual active species for industrial hydrogen evolution reaction. Nat. Commun. 2022, 13, 5785. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Wen, Y.; Ni, F.; Zhang, B.; Peng, H. Boosting Neutral Water Oxidation through Surface Oxygen Modulation. Adv. Mater. 2020, 32, e2002297. [Google Scholar] [CrossRef]
- Han, M.; Wang, N.; Zhang, B.; Xia, Y.J.; Li, J.; Han, J.R.; Yao, K.L.; Gao, C.C.; He, C.N.; Liu, Y.C.; et al. High-Valent Nickel Promoted by Atomically Embedded Copper for Efficient Water Oxidation. ACS Catal. 2020, 10, 9725–9734. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Norskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. Chemcatchem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2(110)as a function of oxygen pressure. Phys. Rev. B 2001, 65, 035406. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Li, J.; Yan, J.; Rappe, A.M.; Yang, J. Unlocking Synergistic Catalysis in NiP: Dual Role of Electronic Structure and Lewis Acidity for Enhanced Oxygen Evolution Reaction. Catalysts 2025, 15, 457. https://doi.org/10.3390/catal15050457
Liang J, Li J, Yan J, Rappe AM, Yang J. Unlocking Synergistic Catalysis in NiP: Dual Role of Electronic Structure and Lewis Acidity for Enhanced Oxygen Evolution Reaction. Catalysts. 2025; 15(5):457. https://doi.org/10.3390/catal15050457
Chicago/Turabian StyleLiang, Jiazhou, Jiawei Li, Jiani Yan, Andrew M. Rappe, and Jing Yang. 2025. "Unlocking Synergistic Catalysis in NiP: Dual Role of Electronic Structure and Lewis Acidity for Enhanced Oxygen Evolution Reaction" Catalysts 15, no. 5: 457. https://doi.org/10.3390/catal15050457
APA StyleLiang, J., Li, J., Yan, J., Rappe, A. M., & Yang, J. (2025). Unlocking Synergistic Catalysis in NiP: Dual Role of Electronic Structure and Lewis Acidity for Enhanced Oxygen Evolution Reaction. Catalysts, 15(5), 457. https://doi.org/10.3390/catal15050457