
Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology

 , , , and
, , , and
Abstract

1. Introduction
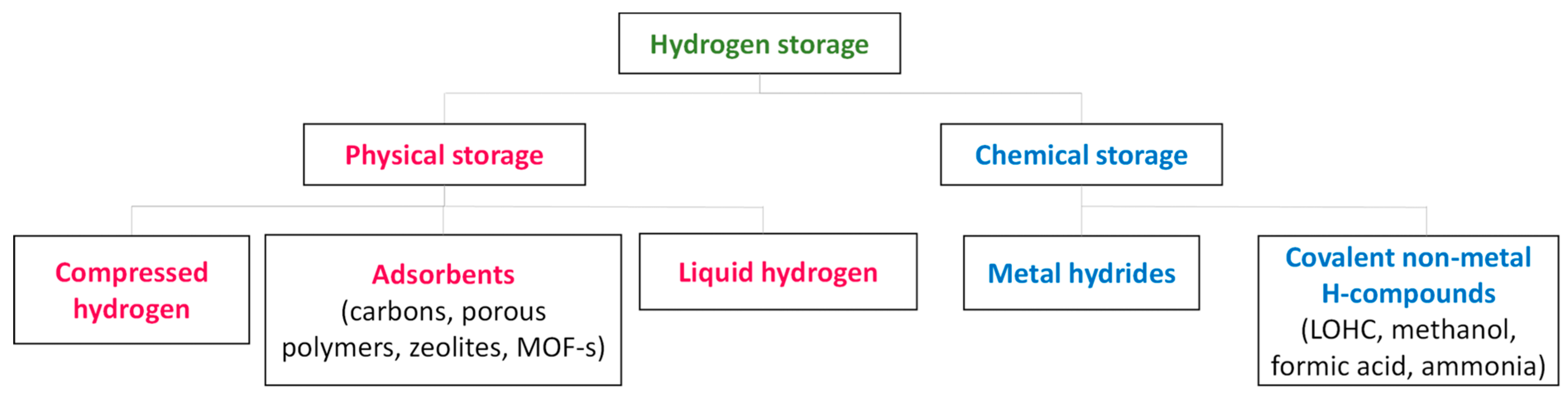
2. Options for the Storage and Transport of Hydrogen
3. Applications of Liquid Organic Hydrogen Carriers (LOHCs) for the Storage of Hydrogen
- Both the hydrogenated and dehydrogenated forms should have a melting point of below −30 °C to allow transport and storage in a liquid state.
- The boiling point of both forms is above 300 °C, as the low vapor pressure of the LOHC near room temperature facilitates the purification of the released hydrogen.
- The H2 storage capacity of the LOHC shall be greater than 56 kg/m3 or higher than 6 wt%.
- The desorption heat of hydrogen should be low (42–54 kJ/molH2) so that complete dehydrogenation can be achieved at low temperatures (<200 °C), even at 1 bar H2 pressure.
- Hydrogenation–dehydrogenation can be carried out with high selectivity over as many cycles as possible.
- They should fit into today′s fuel supply infrastructure.
- They should be cheap and easy to produce.
- They must meet the applicable toxicological and ecotoxicological requirements during transport and use, i.e., they must not be classified as hazardous substances.
4. The Main LOHC Materials
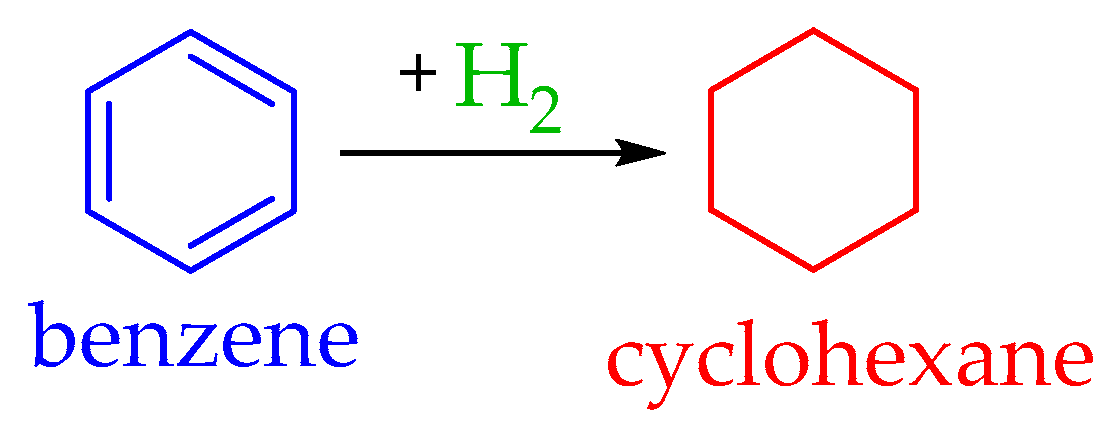
4.1. The Benzene–Cyclohexane System
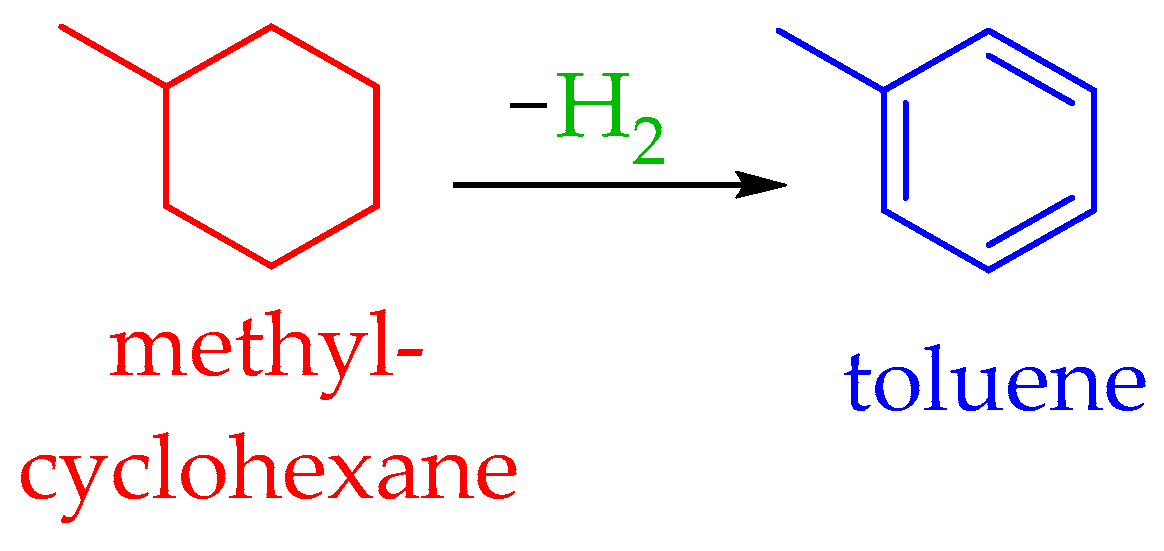
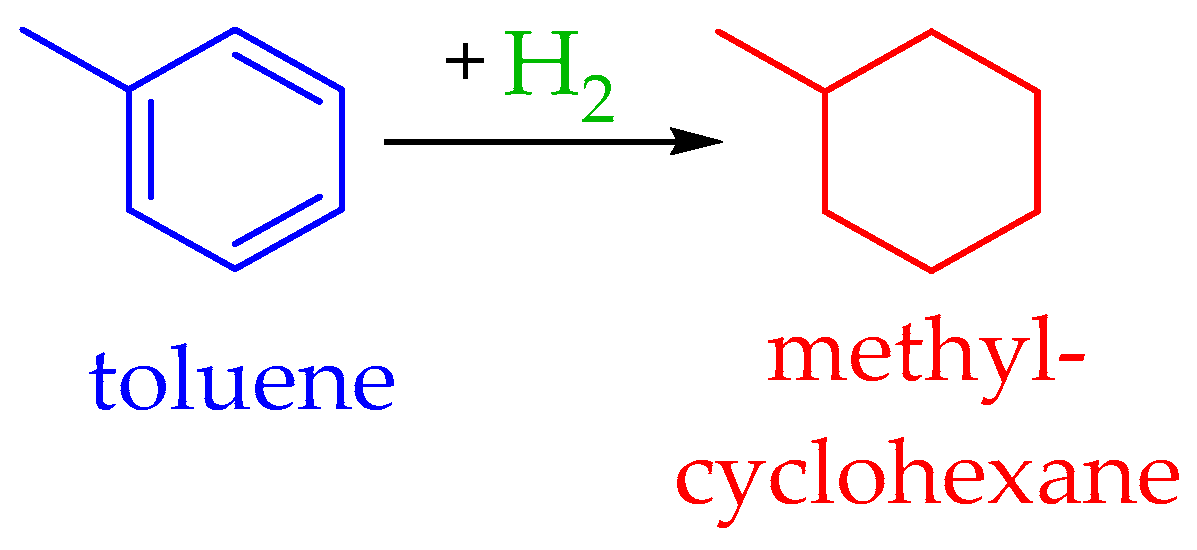
4.2. The Toluene–Methylcyclohexane System
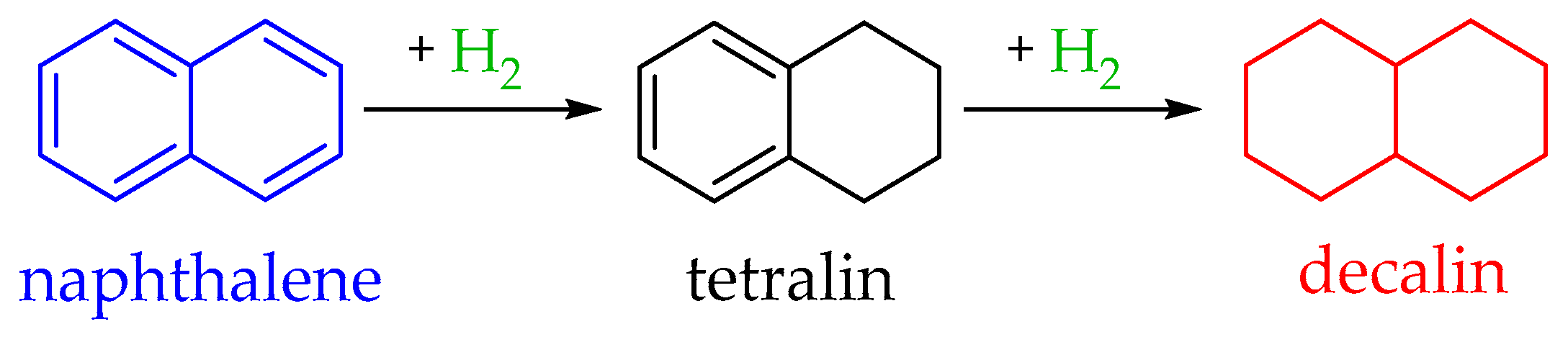
4.3. The Naphthalene–Decahydronaphthalene (Decalin) System
4.4. The Dibenzyltoluene–Perhydrodibenzyltoluene System
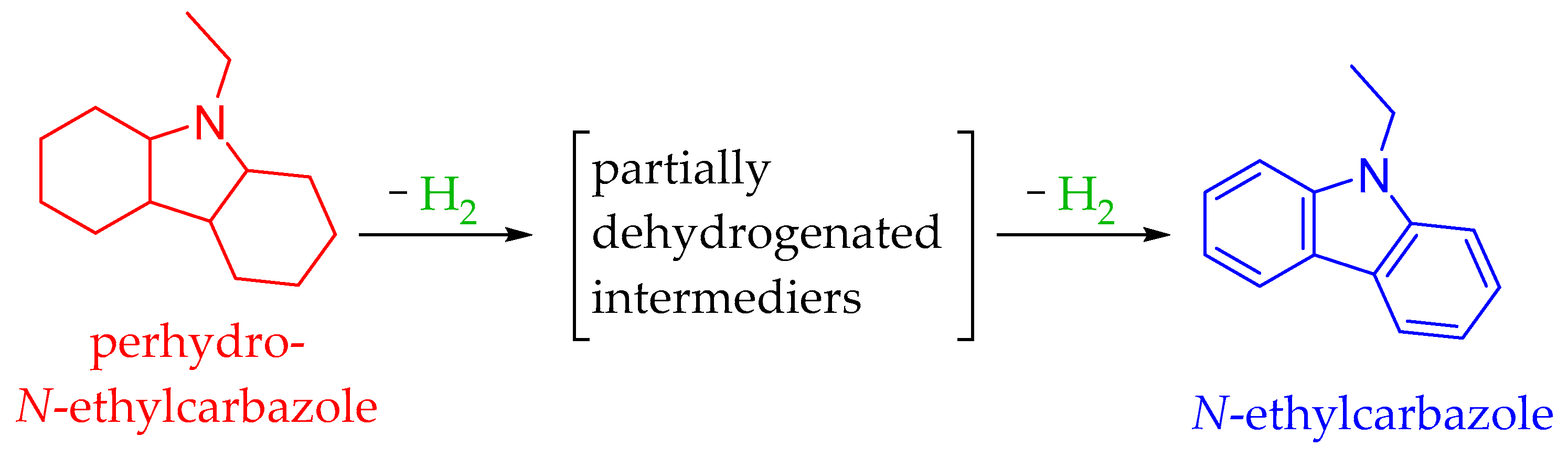
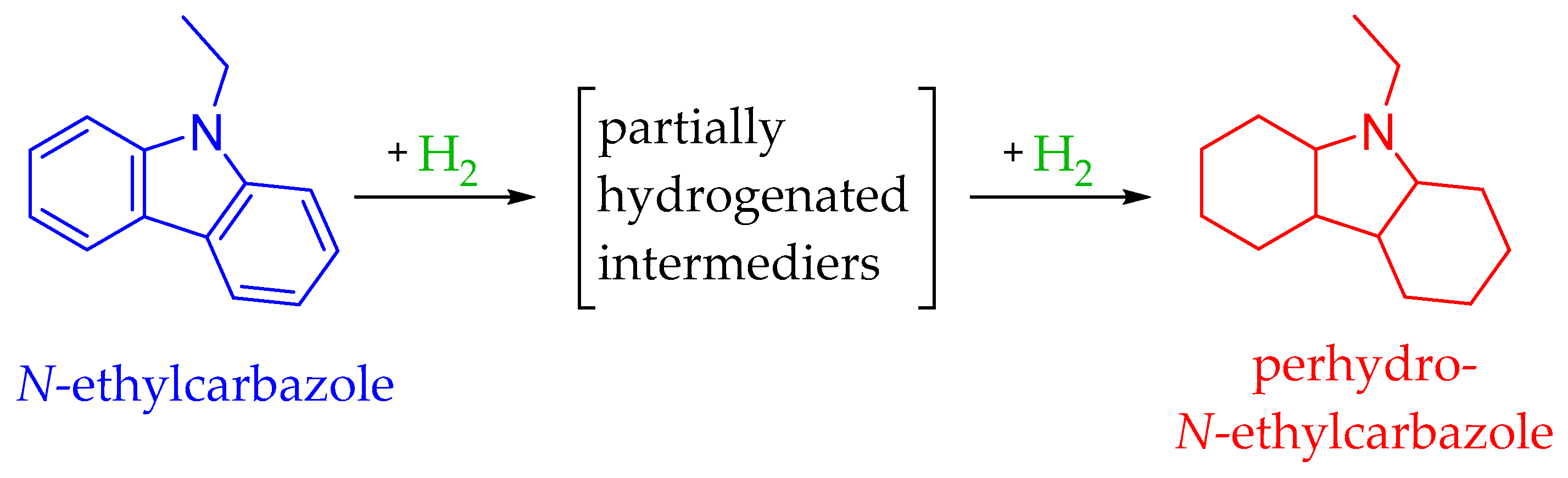
4.5. The N-Ethylcarbazole–Dodecahydro-N-Ethylcarbazole System
5. Catalysts for the LOHC Process
5.1. Background
5.2. Dehydrogenation of the Main LOHC Substances
5.2.1. Dehydrogenation of Cyclohexane to Benzene
5.2.2. Dehydrogenation of Methylcyclohexane to Toluene
5.2.3. Dehydrogenation of Decalin to Naphthalene
5.2.4. Dehydrogenation of Perhydrodibenzyltoluene to Dibenzyltoluene
5.2.5. Dehydrogenation of Dodecahydro-N-Ehylcarbazole to N-Ethylcarbazole
5.3. Hydrogenation of the Most Important LOHC Materials
5.3.1. Hydrogenation of Benzene to Cyclohexane
5.3.2. Hydrogenation of Toluene to Methylcyclohexane
5.3.3. Hydrogenation of Naphthalene to Decalin
5.3.4. Hydrogenation of Dibenzyltoluene to Perhydrodibenzyltoluene
5.3.5. Hydrogenation of N-ethylcarbazole to Dodecahydro-N-ethylcarbazole
6. Future Research Directions for LOHC Hydrogen Storage
6.1. Background
6.2. Directions for the Development of New LOHC Materials
6.3. Development of Hydrogenation and Dehydrogenation Catalysts
- Achieve the highest possible metal dispersion, which is most easily achieved on mesoporous supports with a high specific surface area. The fine tuning of the chemical properties of the support (e.g., acidity, basicity) is also an important issue. The method of metal deposition, the development of ideal pre-treatment conditions, and the doping of the support and/or the supported metal can also improve the efficiency of the catalyst.
- An important aspect is the acceleration of hydrogen spillover between the metal component and the LOHC material in the activated state on the support. This can be facilitated by increasing the number of acidic surface hydroxyl groups (Brønsted acidity) or by introducing a hydrogen transfer additive, which, as we have seen, facilitates the hydrogenation of the LOHC in the case of the LaNi5/LiH3 catalyst [98], since LiH3 is able to transfer atomic hydrogen to N-ethylcarbazole activated on Ni/Al2O3.
- By doping a support (e.g., with boron, nitrogen, phosphorus, or sulfur), the degree of interaction between the electron-rich benzene rings of LOHC materials and the Lewis acid sites of the catalyst can be increased, which can increase the rate of hydrogenation.
- By using two or more metals together, the synergistic effect between the metals can be exploited by creating electron-deficient sites on the catalyst surface, which then promote the adsorption of the electron-rich aromatic molecules.
7. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.; Lee, S.; Sung, S.; Gu, S.; Kim, J.; Lee, G.; Park, J.; Yip, A.C.K.; Choi, J. Zeolite Membrane-Based Low-Temperature Dehydrogenation of a Liquid Organic Hydrogen Carrier: A Key Step in the Development of a Hydrogen Economy. Adv. Sci. 2024, 11, e2403128. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Zhu, Y. Porous carbon-based materials for hydrogen storage: Advancement and challenges. J. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Langmi, H.W.; Ren, J.; North, B.; Mathe, M.; Bessarabov, D. Hydrogen Storage in Metal-Organic Frameworks: A Review. Electrochimica Acta 2014, 128, 368–392. [Google Scholar] [CrossRef]
- Germain, J.; Fréchet, J.M.J.; Svec, F. Nanoporous Polymers for Hydrogen Storage. Small 2009, 5, 1098–1111. [Google Scholar] [CrossRef]
- Weitkamp, J.; Fritz, M.; Ernst, S. Zeolites as media for hydrogen storage*1. Int. J. Hydrogen Energy 1995, 20, 967–970. [Google Scholar] [CrossRef]
- Adametz, P.; Müller, K.; Arlt, W. Efficiency of low-temperature adsorptive hydrogen storage systems. Int. J. Hydrogen Energy 2014, 39, 15604–15613. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, C.; Lai, Q.; Liu, W.; Wang, D.-W.; Aguey-Zinsou, K.-F. Tailoring magnesium based materials for hydrogen storage through synthesis: Current state of the art. Energy Storage Mater. 2018, 10, 168–198. [Google Scholar] [CrossRef]
- Graetz, J.; Reilly, J.; Yartys, V.; Maehlen, J.; Bulychev, B.; Antonov, V.; Tarasov, B.; Gabis, I. Aluminum hydride as a hydrogen and energy storage material: Past, present and future. J. Alloy. Compd. 2011, 509, S517–S528. [Google Scholar] [CrossRef]
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Baricco, M.; Bang, M.; Fichtner, M.; Hauback, B.; Linder, M.; Luetto, C.; Moretto, P.; Sgroi, M. SSH2S: Hydrogen storage in complex hydrides for an auxiliary power unit based on high temperature proton exchange membrane fuel cells. J. Power Sources 2017, 342, 853–860. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Jones, J.-P.; Prakash, G.K.S.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef]
- Pfromm, P.H. Towards sustainable agriculture: Fossil-free ammonia. J. Renew. Sustain. Energy 2017, 9, 034702. [Google Scholar] [CrossRef]
- Ajeebi, A.M.; Ali, A.; AlAmoudi, O.M.; Sanhoob, M.A.; Hossain, M.M.; Alghamdi, H.S.; Usman, M.; Zahir, H.; Shaikh, M.N. Alumina-Supported Bimetallic Catalysts with Ruthenium and CoNi for Enhanced Ammonia Decomposition. Sustain. Energy Fuels 2025. [Google Scholar] [CrossRef]
- Sørensen, R.Z.; Klerke, A.; Quaade, U.; Jensen, S.; Hansen, O.; Christensen, C.H. Promoted Ru on high-surface area graphite for efficient miniaturized production of hydrogen from ammonia. Catal. Lett. 2006, 112, 77–81. [Google Scholar] [CrossRef]
- Su, T.; Guan, B.; Zhou, J.; Zheng, C.; Guo, J.; Chen, J.; Zhang, Y.; Yuan, Y.; Xie, W.; Zhou, N.; et al. Review on Ru-Based and Ni-Based Catalysts for Ammonia Decomposition: Research Status, Reaction Mechanism, and Perspectives. Energy Fuels 2023, 37, 8099–8127. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—Recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Modisha, P.M.; Ouma, C.N.M.; Garidzirai, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Chilunda, M.D.; Talipov, S.A.; Farooq, H.M.U.; Biddinger, E.J. Electrochemical Cycling of Liquid Organic Hydrogen Carriers as a Sustainable Approach for Hydrogen Storage and Transportation. ACS Sustain. Chem. Eng. 2025, 13, 1174–1195. [Google Scholar] [CrossRef]
- Lebedeva, O.; Kultin, D.; Kalenchuk, A.; Kustov, L. Advances and prospects in electrocatalytic hydrogenation of aromatic hydrocarbons for synthesis of “loaded” liquid organic hydrogen carriers. Curr. Opin. Electrochem. 2023, 38, 101207. [Google Scholar] [CrossRef]
- Craig, V.S. Gas Solubility of Electrolytes. In Encyclopedia of Applied Electrochemistry; Springer New York: New York, NY, USA, 2014; pp. 927–931. [Google Scholar] [CrossRef]
- Lebedeva, O.K.; Kultin, D.Y.; Kustov, L.M. Electrochemical Behavior of Benzene, Diphenyl, and p-Terphenyl in Room-Temperature Ionic Liquid N-Butylpyridinium Chloride-AlCl3. Russ. J. Phys. Chem. A 2021, 95, 217–220. [Google Scholar] [CrossRef]
- Lee, C.J.; Kim, T.; Song, J.; Yoon, S.J.; Oh, K.-H.; Yu, D.M.; Lee, S.-Y.; So, S. An efficient toluene barrier membrane for high-performance direct toluene hydrogenation via an electrochemical process. J. Mater. Chem. A 2024, 13, 4090–4099. [Google Scholar] [CrossRef]
- Sievi, G.; Geburtig, D.; Skeledzic, T.; Bösmann, A.; Preuster, P.; Brummel, O.; Waidhas, F.; Montero, M.A.; Khanipour, P.; Katsounaros, I.; et al. Towards an efficient liquid organic hydrogen carrier fuel cell concept. Energy Environ. Sci. 2019, 12, 2305–2314. [Google Scholar] [CrossRef]
- Cho, J.; Kim, B.; Venkateshalu, S.; Chung, D.Y.; Lee, K.; Choi, S.-I. Electrochemically Activatable Liquid Organic Hydrogen Carriers and Their Applications. J. Am. Chem. Soc. 2023, 145, 16951–16965. [Google Scholar] [CrossRef]
- Tan, R.; Ji, Q.; Ling, Y.; Li, L. Advances in liquid organic hydrogen carriers: Developing efficient dehydrogenation strategies. Chem. Commun. 2024, 60, 8186–8203. [Google Scholar] [CrossRef]
- D’ambra, F.; Gébel, G. Literature review: State-of-the-art hydrogen storage technologies and Liquid Organic Hydrogen Carrier (LOHC) development. Sci. Technol. Energy Transit. 2023, 78, 32. [Google Scholar] [CrossRef]
- Dürr, S.; Müller, M.; Jorschick, H.; Helmin, M.; Bösmann, A.; Palkovits, R.; Wasserscheid, P. Carbon Dioxide-Free Hydrogen Production with Integrated Hydrogen Separation and Storage. ChemSusChem 2016, 10, 42–47. [Google Scholar] [CrossRef]
- Jorschick, H.; Bösmann, A.; Preuster, P.; Wasserscheid, P. Charging a Liquid Organic Hydrogen Carrier System with H2/CO2 Gas Mixtures. ChemCatChem 2018, 10, 4329–4337. [Google Scholar] [CrossRef]
- Hydrogenious Technologies GmbH, Erlangen, Germany. Available online: https://hydrogenious.net/ (accessed on 8 October 2024).
- Verevkin, S.P.; Emel’yanenko, V.N.; Heintz, A.; Stark, K.; Arlt, W. Liquid Organic Hydrogen Carriers: An Upcoming Alternative to Conventional Technologies. Thermochemical Studies. Ind. Eng. Chem. Res. 2012, 51, 12150–12153. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Smith, K.J. An overview of the kinetics and catalysis of hydrogen storage on organic liquids. Can. J. Chem. Eng. 2013, 91, 1477–1490. [Google Scholar] [CrossRef]
- Okada, Y.; Sasaki, E.; Watanabe, E.; Hyodo, S.; Nishijima, H. Development of dehydrogenation catalyst for hydrogen generation in organic chemical hydride method. Int. J. Hydrogen Energy 2006, 31, 1348–1356. [Google Scholar] [CrossRef]
- Kumar, A.; Daw, P.; Milstein, D. Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics. Chem. Rev. 2021, 122, 385–441. [Google Scholar] [CrossRef]
- Zhou, M.; Miao, Y.; Gu, Y.; Xie, Y. Recent Advances in Reversible Liquid Organic Hydrogen Carrier Systems: From Hydrogen Carriers to Catalysts. Adv. Mater. 2024, 36, e2311355. [Google Scholar] [CrossRef]
- Kariya, N.; Fukuoka, A.; Utagawa, T.; Sakuramoto, M.; Goto, Y.; Ichikawa, M. Efficient hydrogen production using cyclohexane and decalin by pulse-spray mode reactor with Pt catalysts. Appl. Catal. A Gen. 2003, 247, 247–259. [Google Scholar] [CrossRef]
- Kariya, N.; Fukuoka, A.; Ichikawa, M. Efficient evolution of hydrogen from liquid cycloalkanes over Pt-containing catalysts supported on active carbons under “wet–dry multiphase conditions”. Appl. Catal. A Gen. 2002, 233, 91–102. [Google Scholar] [CrossRef]
- Xia, Z.; Lu, H.; Liu, H.; Zhang, Z.; Chen, Y. Cyclohexane dehydrogenation over Ni-Cu/SiO2 catalyst: Effect of copper addition. Catal. Commun. 2017, 90, 39–42. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Kariya, N.; Ichikawa, M. Dehydrogenation of Cyclohexane Over Ni Based Catalysts Supported on Activated Carbon using Spray-pulsed Reactor and Enhancement in Activity by Addition of a Small Amount of Pt. Catal. Lett. 2005, 105, 83–87. [Google Scholar] [CrossRef]
- Wu, H.; Wang, H.; Lv, Y.; Wu, Y.; Wang, Y.; Luo, Q.; Hui, Y.; Liu, L.; Zhang, M.; Hou, K.; et al. Ultra-small Metallic Nickel Nanoparticles on Dealuminated Zeolite for Active and Durable Catalytic Dehydrogenation. Angew. Chem. Int. Ed. Engl. 2024, 64, e202420306. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, X.; Liu, S. Hydrogen production by catalytic dehydrogenation of methylcyclohexane over Pt catalysts supported on pyrolytic waste tire char. Int. J. Hydrogen Energy 2011, 36, 8902–8907. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Ma, Y.; Ren, W.; Dai, X.; Chang, H.; Zhu, X. Morphological regulation of Pt/CeO2 and its catalytic dehydrogenation of methylcyclohexane in fixed bed reactor. Int. J. Hydrogen Energy 2024, 83, 1338–1348. [Google Scholar] [CrossRef]
- Jang, M.; Choi, S.; Kim, Y.; Cha, J.; Kim, A.-R.; Jeong, H.; Kim, Y.; Choi, S.H.; Nam, S.W.; Lim, J.; et al. Effect of CeO2 redox properties on the catalytic activity of Pt-CeOx over irreducible SiO2 support for methylcyclohexane (MCH) dehydrogenation. Appl. Surf. Sci. 2023, 627, 157134. [Google Scholar] [CrossRef]
- Koskin, A.P.; Stepanenko, S.A.; (Bykova), M.V.A.; Bulavchenko, O.A.; Gerasimov, E.Y.; Lysikov, A.I.; Yeletsky, P.M.; Kaichev, V.V.; Yakovlev, V.A. The origin of extraordinary selectivity in dehydrogenation of methylcyclohexane over Ni-Sn-based catalysts. Chem. Eng. J. 2023, 476, 146629. [Google Scholar] [CrossRef]
- Gao, J.; Li, N.; Zhang, D.; Ma, H.; Zhan, X.; Zhao, S.; Zhao, Y. Nix/TiO2 catalysts for enhancing the selectivity of methylcyclohexane dehydrogenation. Mol. Catal. 2024, 560, 114148. [Google Scholar] [CrossRef]
- Song, M.; Zhang, R.; Zhang, B.; Zhai, Z.; Liu, G. Dynamic stable Pt13 clusters anchored on isolated ZnOx nanorafts for efficient cycloparaffin dehydrogenation. Appl. Catal. B Environ. 2024, 363, 124787. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Z.; Chen, W.; Zhang, Q.; Chang, S.; Pan, M.; Lv, X.; Chang, H. Three-dimensional porous Pt/N-MXene catalyst for dehydrogenation of a liquid organic hydrogen carrier. Chem. Eng. J. 2025, 505, 159769. [Google Scholar] [CrossRef]
- He, Z.; Li, K.; Chen, T.; Feng, Y.; Villalobos-Portillo, E.; Marini, C.; Lo, T.W.B.; Yang, F.; Zhang, L.; Liu, L. High-purity hydrogen production from dehydrogenation of methylcyclohexane catalyzed by zeolite-encapsulated subnanometer platinum-iron clusters. Nat. Commun. 2025, 16, 1–14. [Google Scholar] [CrossRef]
- Shi, W.; Oda, A.; Yamamoto, Y.; Harada, S.; Ohtsu, T.; Sawabe, K.; Satsuma, A. Encapsulated Platinum–Tin Nanoparticles in Silicalite-1 Zeolite for Methylcyclohexane Dehydrogenation. ACS Sustain. Chem. Eng. 2025, 13, 3608–3621. [Google Scholar] [CrossRef]
- Kim, K.; Oh, J.; Kim, T.W.; Park, J.H.; Han, J.W.; Suh, Y.-W. Different catalytic behaviors of Pd and Pt metals in decalin dehydrogenation to naphthalene. Catal. Sci. Technol. 2017, 7, 3728–3735. [Google Scholar] [CrossRef]
- Patil, S.P.; Pande, J.V.; Biniwale, R.B. Non-noble Ni–Cu/ACC bimetallic catalyst for dehydrogenation of liquid organic hydrides for hydrogen storage. Int. J. Hydrogen Energy 2013, 38, 15233–15241. [Google Scholar] [CrossRef]
- Auer, F.; Hupfer, A.; Bösmann, A.; Szesni, N.; Wasserscheidpeter, P. Influence of the nanoparticle size on hydrogen release and side product formation in liquid organic hydrogen carrier systems with supported platinum catalysts. Catal. Sci. Technol. 2020, 10, 6669–6678. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Kim, T.; Han, G.; Lee, J.; Lee, K.; Bae, J. Pt/CeO2 catalyst synthesized by combustion method for dehydrogenation of perhydro-dibenzyltoluene as liquid organic hydrogen carrier: Effect of pore size and metal dispersion. Int. J. Hydrogen Energy 2021, 46, 5520–5529. [Google Scholar] [CrossRef]
- Park, T.I.; Lee, S.H.; Lee, K.-Y. Characteristics of La-doped Pt/Al2O3 catalyst prepared by solvent-deficient method and effect on enhancement of dehydrogenation of perhydrodibenzyltoluene. Korean J. Chem. Eng. 2023, 40, 97–103. [Google Scholar] [CrossRef]
- Musavuli, K.C.; Modisha, P.; Everson, R.C.; Malakhov, A.; Bessarabov, D. Metal Foam as Surface-Extended Catalyst Support Structure for Process Intensification in the Dehydrogenation of Perhydro-Dibenzyltoluene on a Pt/Al2O3 Catalyst. Catalysts 2025, 15, 44. [Google Scholar] [CrossRef]
- Wang, B.; Yan, T.; Chang, T.; Wei, J.; Zhou, Q.; Yang, S.; Fang, T. Palladium supported on reduced graphene oxide as a high-performance catalyst for the dehydrogenation of dodecahydro-N-ethylcarbazole. Carbon 2017, 122, 9–18. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Bai, X. Catalytic dehydrogenation of liquid organic hydrogen carrier dodecahydro-N-ethylcarbazole over palladium catalysts supported on different supports. Environ. Sci. Pollut. Res. 2020, 27, 36172–36185. [Google Scholar] [CrossRef]
- Chen, X.; Li, G.; Gao, M.; Dong, Y.; Yang, M.; Cheng, H. Wet-impregnated bimetallic Pd-Ni catalysts with enhanced activity for dehydrogenation of perhydro-N-propylcarbazole. Int. J. Hydrogen Energy 2020, 45, 32168–32178. [Google Scholar] [CrossRef]
- Fan, Y.; Xu, Y.; Wang, P.; Liu, W.; Zhang, J. Continuous dehydrogenation of dodecahydro-N-ethylcarbazole for hydrogen production in a micro-packed bed reactor. Int. J. Hydrogen Energy 2025, 103, 787–796. [Google Scholar] [CrossRef]
- Lee, G.; Jeong, Y.; Kim, B.-G.; Han, J.S.; Jeong, H.; Bin Na, H.; Jung, J.C. Hydrogen production by catalytic decalin dehydrogenation over carbon-supported platinum catalyst: Effect of catalyst preparation method. Catal. Commun. 2015, 67, 40–44. [Google Scholar] [CrossRef]
- Díaz, E.; Rapado-Gallego, P.; Ordóñez, S. Effect of light alkanes and aromatics on decalin dehydrogenation over noble metal catalysts: A new strategy for the development of naphthalene-based LOHCs. Fuel 2023, 353, 129168. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, B.; Hou, G.; Liu, G.; Zhang, X. Thermally Stable Pt Clusters Anchored on Mg–Al–Ti Composite Metal Oxide for Efficient Cycloalkane Dehydrogenation. Energy Fuels 2024, 38, 18955–18964. [Google Scholar] [CrossRef]
- Wang, F.; Luo, M.; Liu, Q.; Shao, C.; Yang, Z.; Liu, X.; Guo, J. Preparation of Pt/MgAl2O4 Decalin Dehydrogenation Catalyst for Chemical Hydrogen Storage Application. Catal. Lett. 2023, 154, 191–205. [Google Scholar] [CrossRef]
- Lei, N.; Qiu, S.; Zhang, L.; Zhao, G.; Wang, S.; Liu, K. Synergistic Effect between Pt and CrOx on Reversible Hydrogenation and Dehydrogenation of Dibenzyltoluene. Ind. Eng. Chem. Res. 2025, 64, 4761–4770. [Google Scholar] [CrossRef]
- Auer, F.; Solymosi, T.; Erhardt, C.; Collados, C.C.; Thommes, M.; Wasserscheid, P. Enhancing the power density of hydrogen release from LOHC systems by high Pt loadings on hierarchical alumina support structures. Int. J. Hydrogen Energy 2024, 100, 1282–1290. [Google Scholar] [CrossRef]
- Li, L.; Yang, Z.; Xiong, H.; Ma, M.; Zhang, R.; Jiang, Z. High-Concentration N Vacancy of S-Doped C3N4 Regulates the Electronic Structure of Pd to Promote the Dehydrogenation of Dodecahydro-N-ethylcarbazole. ACS Appl. Mater. Interfaces 2025, 17, 15287–15300. [Google Scholar] [CrossRef]
- Park, B.G.; Lee, J.; Lee, Y.; Lee, H.; Kim, J.; Nam, E.; Bae, J.-S.; Han, J.W.; An, K. Pd-Catalyzed Dehydrogenation Enhanced by Charge Transfer from MoOx Promoter. ACS Catal. 2025, 15, 1563–1575. [Google Scholar] [CrossRef]
- Mokrane, T.; Boudjahem, A.-G.; Bettahar, M. Benzene hydrogenation over alumina-supported nickel nanoparticles prepared by polyol method. RSC Adv. 2016, 6, 59858–59864. [Google Scholar] [CrossRef]
- Zhou, G.; Li, T.; Chen, J.; Deng, L.; Xie, H. Nano-Pd/CeO2 catalysts for hydrogen storage by reversible benzene hydrogenation/dehydrogenation reactions. Int. J. Hydrogen Energy 2021, 46, 14540–14555. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, W.; Zhao, Y.; Wang, X.; Chen, S.; Jia, A.; Xie, H. Reversible hydrogenation and dehydrogenation of benzene for hydrogen storage on highly dispersed Pd/γ-Al2O3 catalyst. J. Ind. Eng. Chem. 2024, 134, 561–573. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Kultin, D.Y.; Lebedeva, O.K.; Tkachenko, O.; He, T.; Kustov, L.M. Influence of steric factors on the reaction of hydrogenation of aromatic hydrocarbons in hydrogen storage systems. Int. J. Hydrogen Energy 2025. [Google Scholar] [CrossRef]
- Johnson, R.; Hu, P.; Pugh, J.; Haridasan, R.K.; Searles, K. Benzene hydrogenation utilizing organometallic early transition metal precursors. Catal. Sci. Technol. 2024, 15, 41–45. [Google Scholar] [CrossRef]
- Ishii, T.; Kitamura, Y.; Hasegawa, S.; Sasaki, C.; Ozaki, J.-I. Benzene hydrogenation activities of Ni catalyst supported on N- and B-doped carbons. Diam. Relat. Mater. 2021, 119, 108550. [Google Scholar] [CrossRef]
- Lindfors, L.P.; Salmi, T.; Smeds, S. Kinetics of toluene hydrogenation on Ni/Al2O3 catalyst. Chem. Eng. Sci. 1993, 48, 3813–3828. [Google Scholar] [CrossRef]
- Janiszewska, E.; Kot, M.; Zieliński, M. Modification of silica with NH4+ agents to prepare an acidic support for iridium hydrogenation catalyst. Microporous Mesoporous Mater. 2018, 255, 94–102. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, H.; Hu, W.; Zheng, J.; Zhang, N.; Yu, C.; Ye, H.; Yang, Z.; Chen, B.H. Nickel Hydroxide–Cobalt Hydroxide Nanoparticle Supported Ruthenium–Nickel–Cobalt Islands as an Efficient Nanocatalyst for the Hydrogenation Reaction. ChemCatChem 2018, 10, 1998–2002. [Google Scholar] [CrossRef]
- Medina-Mendoza, A.K.; Cortés-Jácome, M.A.; Toledo-Antonio, J.A.; Angeles-Chávez, C.; López-Salinas, E.; Cuauhtémoc-López, I.; Barrera, M.C.; Escobar, J.; Navarrete, J.; Hernández, I. Highly dispersed uniformly sized Pt nanoparticles on mesoporous Al-SBA-15 by solid state impregnation. Appl. Catal. B Environ. 2011. [Google Scholar] [CrossRef]
- Montesinos-Castellanos, A.; Zepeda, T. High hydrogenation performance of the mesoporous NiMo/Al(Ti, Zr)–HMS catalysts. Microporous Mesoporous Mater. 2008, 113, 146–162. [Google Scholar] [CrossRef]
- Jorschick, H.; Preuster, P.; Dürr, S.; Seidel, A.; Müller, K.; Bösmann, A.; Wasserscheid, P. Hydrogen storage using a hot pressure swing reactor. Energy Environ. Sci. 2017, 10, 1652–1659. [Google Scholar] [CrossRef]
- Shi, L.; Zhou, Y.; Qi, S.; Smith, K.J.; Tan, X.; Yan, J.; Yi, C. Pt Catalysts Supported on H2 and O2 Plasma-Treated Al2O3 for Hydrogenation and Dehydrogenation of the Liquid Organic Hydrogen Carrier Pair Dibenzyltoluene and Perhydrodibenzyltoluene. ACS Catal. 2020, 10, 10661–10671. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Zhao, L.; Smith, K.J. Kinetics of H2 recovery from dodecahydro-N-ethylcarbazole over a supported Pd catalyst. Appl. Catal. A Gen. 2009, 362, 155–162. [Google Scholar] [CrossRef]
- Yu, H.; Yang, X.; Jiang, X.; Wu, Y.; Chen, S.; Lin, W.; Wu, Y.; Xie, L.; Li, X.; Zheng, J. LaNi5.5 particles for reversible hydrogen storage in N-ethylcarbazole. Nano Energy 2021, 80, 105476. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, F.; Li, H.; Liu, D. High-performance hydrogenation of N-propylcarbazole over Ru-Ni alloy catalyst by efficient overall hydrogenation transition. J. Alloy. Compd. 2025, 1013, 178619. [Google Scholar] [CrossRef]
- Ojo, A.U.; Shakya, D.M.; Stetzler, J.; Gbadamosi, M.; Masudur, R.M.; Acharya, N.; Thornburg, N.; Tengco, J.; Balijepalli, S.K.; Monnier, J.R.; et al. The enhanced reactivity of graphitic supports for Pd catalyzed toluene hydrogenation. J. Catal. 2025, 445, 116029. [Google Scholar] [CrossRef]
- Na, G.-J.; Hwang, J.; Park, H.; Ryoo, R. Enhanced dispersion of Pt and Ni nanoparticles on ammonia-treated siliceous MFI zeolites for toluene hydrogenation. Appl. Catal. A Gen. 2024, 692, 120096. [Google Scholar] [CrossRef]
- Oda, A.; Ogawa, N.; Yamamoto, Y.; Sawabe, K.; Satsuma, A. Atom-to-nm Scale Engineering of PtCo Alloy Catalysts over Supported Co Nanoparticles for Advanced Toluene Hydrogenation Efficiency in Hydrogen Storage Applications. ACS Catal. 2025, 15, 3191–3202. [Google Scholar] [CrossRef]
- Wang, E.; Cheng, K.; Liu, Z.; Kong, X.; Wang, W.; Duan, A.; Wang, X. Engineering the pore structure of hierarchical silica via simple mechanical shear for the enhanced selective hydrogenation of naphthalene. Fuel 2025, 388, 134507. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, E.; Mei, J.; Wang, A.; Zou, Y.; Wang, C.; Shang, H.; Gong, Y.; Duan, A.; Xu, C.; et al. Hierarchical porous Pt/Hβ catalyst with controllable acidity for efficient hydrogenation of naphthalene. Chem. Eng. J. 2024, 481, 148763. [Google Scholar] [CrossRef]
- Ali, A.; Rohini, A.K.; Noh, Y.S.; Moon, D.J.; Lee, H.J. Hydrogenation of dibenzyltoluene and the catalytic performance of Pt/Al2 O3with various Pt loadings for hydrogen production from perhydro-dibenzyltoluene. Int. J. Energy Res. 2021, 46, 6672–6688. [Google Scholar] [CrossRef]
- Zhu, T.; Liu, L.; Zhao, Y.; Gao, M.; Dong, Y.; Xia, D.; Huang, P.; Cheng, H.; Yang, M. La promoted Ni0-Niδ+ synergistic interaction for rapid and deep hydrogenation of liquid organic hydrogen carriers. Chem. Eng. J. 2024, 493, 152354. [Google Scholar] [CrossRef]
- Zhang, M.; Song, Q.; He, Z.; Wang, Q.; Wang, L.; Zhang, X.; Li, G. Tuning the mesopore-acid-metal balance in Pd/HY for efficient deep hydrogenation saturation of naphthalene. Int. J. Hydrogen Energy 2022, 47, 20881–20893. [Google Scholar] [CrossRef]
- Brückner, N.; Obesser, K.; Bösmann, A.; Teichmann, D.; Arlt, W.; Dungs, J.; Wasserscheid, P. Evaluation of Industrially Applied Heat-Transfer Fluids as Liquid Organic Hydrogen Carrier Systems. ChemSusChem 2013, 7, 229–235. [Google Scholar] [CrossRef]
- Do, G.; Preuster, P.; Aslam, R.; Bösmann, A.; Müller, K.; Arlt, W.; Wasserscheid, P. Hydrogenation of the liquid organic hydrogen carrier compound dibenzyltoluene—Reaction pathway determination by 1H NMR spectroscopy. React. Chem. Eng. 2016, 1, 313–320. [Google Scholar] [CrossRef]
- Modisha, P.; Bessarabov, D. Stress tolerance assessment of dibenzyltoluene-based liquid organic hydrogen carriers. Sustain. Energy Fuels 2020, 4, 4662–4670. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, T.; Xia, M.; Zhu, Y.; Ke, H.; Yang, M.; Cheng, H.; Dong, Y. Identifying Noble Metal Catalysts for the Hydrogenation and Dehydrogenation of Dibenzyltoluene: A Combined Theoretical–Experimental Study. Inorg. Chem. 2023, 62, 17390–17400. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Ren, G.; Wei, H.; Ju, X.; Ding, Q.; Xi, Y.; Shang, H.; Ma, L.; Lin, X. A new insight into the kinetics of dibenzyltoluene hydrogenation over ruthenium catalyst for hydrogen storage. React. Kinet. Catal. Lett. 2024, 137, 1683–1699. [Google Scholar] [CrossRef]
- Ye, X.; An, Y.; Xu, G. Kinetics of 9-ethylcarbazole hydrogenation over Raney-Ni catalyst for hydrogen storage. J. Alloy. Compd. 2011, 509, 152–156. [Google Scholar] [CrossRef]
- Liu, H.; Xue, J.; Yu, P.; Zhang, Y.; Wang, J.; Che, D. Hydrogenation of N-ethylcarbazole with Hydrogen-Methane mixtures for hydrogen storage. Fuel 2022, 331, 125920. [Google Scholar] [CrossRef]
- Wu, Y.; Yu, H.; Guo, Y.; Zhang, Y.; Jiang, X.; Sun, B.; Fu, K.; Chen, J.; Qi, Y.; Zheng, J.; et al. Promoting hydrogen absorption of liquid organic hydrogen carriers by solid metal hydrides. J. Mater. Chem. A 2019, 7, 16677–16684. [Google Scholar] [CrossRef]
- Chiyoda’s Hydrogen Supply Chain Business. Available online: https://www.chiyodacorp.com/en/service/spera-hydrogen/innovations/ (accessed on 9 April 2025).
- Hynertech Corporation. Available online: https://www.hynertech.com/en/ (accessed on 9 April 2025).
- Wei, D.; Shi, X.; Qu, R.; Junge, K.; Junge, H.; Beller, M. Toward a Hydrogen Economy: Development of Heterogeneous Catalysts for Chemical Hydrogen Storage and Release Reactions. ACS Energy Lett. 2022, 7, 3734–3752. [Google Scholar] [CrossRef]
- Kim, T.W.; Jeong, H.; Baik, J.H.; Suh, Y.-W. State-of-the-art Catalysts for Hydrogen Storage in Liquid Organic Hydrogen Carriers. Chem. Lett. 2022, 51, 239–255. [Google Scholar] [CrossRef]
- Gong, X.; Li, L.; Shi, R.; Zhang, R.; Jiang, Z.; Fang, T. Novel Liquid Organic Hydrogen Carriers with High Hydrogen Performance: NPhCZ/18H-NPhCZ. ACS Sustain. Chem. Eng. 2023, 11, 3085–3092. [Google Scholar] [CrossRef]
- Kim, T.W.; Jo, Y.; Jeong, K.; Yook, H.; Han, J.W.; Jang, J.H.; Han, G.B.; Park, J.H.; Suh, Y.-W. Tuning the isomer composition is a key to overcome the performance limits of commercial benzyltoluene as liquid organic hydrogen carrier. J. Energy Storage 2023, 60, 106676. [Google Scholar] [CrossRef]
- Schörner, M.; Solymosi, T.; Razcka, T.; Nathrath, P.; Johner, N.P.; Zimmermann, T.; Mandel, K.; Wasserscheid, P.; Wintzheimer, S.; Schühle, P. Inductively heatable catalytic materials for the dehydrogenation of the liquid organic hydrogen carrier (LOHC) perhydro dibenzyltoluene. Catal. Sci. Technol. 2024, 14, 4450–4457. [Google Scholar] [CrossRef]
- Sun, J.; Shang, H.; Miao, C.; Yang, J.; Liao, Y. Microwave enhanced hydrogen production from liquid organic hydrogen carriers: A review. Chem. Eng. Process.-Process. Intensif. 2023, 190, 109432. [Google Scholar] [CrossRef]
- Wang, N.; Qiu, J.; Wu, J.; Yuan, X.; You, K.; Luo, H. Microwave assisted synthesis of Sn-modified MgAlO as support for platinum catalyst in cyclohexane dehydrogenation to cyclohexene. Appl. Catal. A Gen. 2016, 516, 9–16. [Google Scholar] [CrossRef]
- Ichikawa, T.; Matsuo, T.; Tachikawa, T.; Yamada, T.; Yoshimura, T.; Yoshimura, M.; Takagi, Y.; Sawama, Y.; Sugiyama, J.-I.; Monguchi, Y.; et al. Microwave-Mediated Site-Selective Heating of Spherical-Carbon-Bead-Supported Platinum for the Continuous, Efficient Catalytic Dehydrogenative Aromatization of Saturated Cyclic Hydrocarbons. ACS Sustain. Chem. Eng. 2019, 7, 3052–3061. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclohexane |  | Benzene | 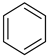 |
| Melting point | 5 °C | Melting point | 6.5 °C |
| Boiling point | 80 °C | Boiling point | 80 °C |
| Flash point | −18 °C | Flash point | −11 °C |
| Dehydrogenation pressure | 1–5 bar | Hydrogenation pressure | 1–50 bar |
| Dehydrogenation temperature | 280–350 °C | Hydrogenation temperature | 120–250 °C |
| Methylcyclohexane |  | Toluene |  |
| Melting point | −126 °C | Melting point | −95 °C |
| Boiling point | 101 °C | Boiling point | 111 °C |
| Flash point | −3 °C | Flash point | 6 °C |
| Dehydrogenation pressure | 1–5 bar | Hydrogenation pressure | 1–50 bar |
| Dehydrogenation temperature | 300–400 °C | Hydrogenation temperature | 60–200 °C |
| Decalin |  | Naphthalene |  |
| Melting point | −37 °C | Melting point | 79 °C |
| Boiling point | 189 °C | Boiling point | 218 °C |
| Flash point | 57 °C | Flash point | 80 °C |
| Dehydrogenation pressure | 1–5 bar | Hydrogenation pressure | 20–70 bar |
| Dehydrogenation temperature | 270–350 °C | Hydrogenation temperature | 150–330 °C |
| Perhydrodibenzyltoluene | 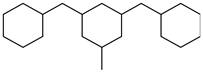 | Dibenzyltoluene | 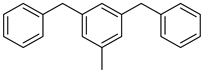 |
| Melting point | −34 °C | Melting point | −30 °C |
| Boiling point | 354 °C | Boiling point | 390 °C |
| Flash point | - | Flash point | 190 °C |
| Dehydrogenation pressure | 1–5 bar | Hydrogenation pressure | 10–50 bar |
| Dehydrogenation temperature | 260–320 °C | Hydrogenation temperature | 140–300 °C |
| Perhydro-N-Ethylcarbazole | 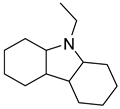 | N-ethylcarbazole | 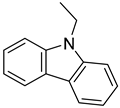 |
| Melting point | −85 °C | Melting point | 70 °C |
| Boiling point | - | Boiling point | 348 °C |
| Flash point | - | Flash point | 186 °C |
| Dehydrogenation pressure | 1–5 bar | Hydrogenation pressure | 50–70 bar |
| Dehydrogenation temperature | 170–270 °C | Hydrogenation temperature | 140–180 °C |
| Catalyst | Reactor | T (C°) | p (Bar) | WHSV a (h−1) | Conv. (%) | Sel. (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Cyclohexane | ||||||||
| 10%Pt/ACC b | P.S. c | 330 | 1 | 51 | - | 35 | 100 | [35] |
| Pt-Rh/ACC b | P.S. c | 328 | 1 | 78 | - | 25–35 | 100 | [35] |
| 17%Ni,3%Cu/SiO2 | flow through | 350 | 1 | 54 | 1.6 | 94.9 | 99.5 | [37] |
| 20%Ni/ACC b | P.S. c | 300 | 1 | 8.5 | - | 21.9 | 98.8 | [38] |
| 0.5%Pt/ACC b | P.S. c | 300 | 1 | 0.22 | - | 0.4 | 100 | [38] |
| 20%Ni,0.5%Pt/ACC b | P.S. c | 300 | 1 | 13.1 | - | 31.1 | 99.7 | [38] |
| Methylcyclohexane | ||||||||
| 0.4%Pt/active carbon | flow through | 300 | 1 | 1.4 | 2.5 | 95 | 100 | [40] |
| 1%Pt/CeO2 | flow through | 350 | 1 | 3.5 | 7.7 | 78 | - | [41] |
| 1%Pt/CeO2-SiO2 | flow through | 310 | 1 | 1.0 | 1.9 | 97 | 100 | [42] |
| 52%Ni,13%Sn + 17% SiO2 | flow through | 350 | 1 | 2.9 | 6.2 | 92 | 99 | [43] |
| 17%Ni/TiO2 | flow through | 375 | 1 | 0.84 | 1.9 | 86.5 | 96.5 | [44] |
| 1.2%Pt,0.4%Zn/self-pillared silicate-1 | flow through | 400 | 1 | 41.5 | 100 | 80 | - | [45] |
| 1%Pt/N-Ti3C2Tx | flow through | 375 | 1 | 3.6 | 7.7 | 93 | 100 | [46] |
| 0.4%Pt,0.6%Fe/silicalite-1 | flow through | 350 | 1 | 12.9 | 90 | 28 | 100 | [47] |
| 0.4%Pt,2.8%Sn encap-sulated on silicate-1 | flow through | 300 | 1 | 2.5 | 1.56 | 88 | 100 | [48] |
| Decalin | ||||||||
| 5%Pt/C (impreg.) | batch | 270 | 1 | 0.45 (4 h) e | - | 69 | 68 | [49] |
| 8%Ni,2%Cu/ACC b | P.S. c | 350 | 1 | 9.0 | - | - | - | [50] |
| Perhydrodibenzyltoluene | ||||||||
| 0.3%Pt/Al2O3 | batch | 310 | 1 | - | - | 85 | 100 | [51] |
| 5%Pt/Al2O3 | flow through | 300 | 1 | 10.8 | 10 d | 5.0 | 100 | [52] |
| 5%Pt/CeO2 | flow through | 300 | 1 | 25.2 | 10 d | 37 | 100 | [52] |
| 5%Pt/Al2O3 | batch | 270 | 1 | - | - | 58 | 100 | [53] |
| 1%La,5%Pt/Al2O3 | batch | 270 | 1 | - | - | 65 | 100 | [53] |
| 5%Pt/Al2O3 | batch | 300 | 1 | 12.5 | - | 48 | - | [54] |
| Perhydro-N-Ethylcarbazole | ||||||||
| 2.5% Pd/graphene-oxide | batch | 170 | 1 | 21.1 (12 h) e | - | 100 | 85 | [55] |
| 5% Pd/C | batch | 180 | 1 | - | - | 99.9 | 98 | [56] |
| 2.5%Pd + 2.5%Ni/Al2O3 f | batch | 180 | 1 | - | - | 98 | 100 | [57] |
| 1% Pd/Al2O3 | flow through | 180 | 1 | 2.3 | 2.26 c | 100 | 92 | [58] |
| Catalyst | Reactor Type | T (C°) | p (bar) | W/F a (h−1) | Initial Conv. (%) | Initial Sel. (%) | TOS b (h) | Number of Recycling | Decrease of Reaction Rate (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyclohexane | |||||||||||
| 3%Ni/Beta (Si/Al > 300) | flow through | 280 | 1 | 4 | 1.5 | 66 | 93 | 60 | - | 6 | [39] |
| 9% Ni/Beta (Si/Al > 300) | flow through | 300 | 1 | 13.3 | 4.7 | 61 | 96 | 150 | - | 16 | [39] |
| Methylcyclohexane | |||||||||||
| 0.4%Pt/active carbon | flow through | 300 | 1 | 2.5 | 1.4 | 98 | 100 | 52 | - | 0 | [40] |
| 1%Pt/CeO2 | flow through | 350 | 1 | 7.7 | 3.5 | 83 | - | 72 | - | 35 | [42] |
| 52%Ni, 13%Sn, 17%SiO2 | flow through | 350 | 1 | 18.5 | 6.2 | 64 | 100 | 100 | - | 19 | [43] |
| 17%Ni/TiO2 | flow through | 375 | 1 | 1.9 | 0.9 | 98 | 90 | 6 | - | 6 | [44] |
| 1.2%Pt, 0.4%Zn/self-pillared silicalite-1 | flow through | 400 | 1 | 100 | 41.5 | 80 | 100 | 100 | - | 0 | [45] |
| 9.5%Pt, 0.6%Fe/silicalite-1 | flow through | 350 | 1 | 2.2 | 314 | 100 | 99 | 72 | - | 6 | [47] |
| 0.4%Pt, 2.8%Sn encapsulated on silicalite-1 | flow through | 350 | 1 | 7.8 | 5.1 | 37 | 100 | 32 | - | 46 | [48] |
| Decalin | |||||||||||
| 8%Ni, 2%Cu/ ACC c | P.S. d | 350 | 1 | - | 3.6 | - | - | 3 | - | 0 | [50] |
| Perhydrodibenzyltoluene | |||||||||||
| 5%Pt/Al2O3 | flow through | 300 | 1 | 6.5 | 1.3 | 45 | - | 24 | - | 16 | [54] |
| Perhydro-N-Ethylcarbazole | |||||||||||
| 2.5%Pd/graph-ene oxide | batch | 180 | 1 | - | 5.1 | 100 | 84 | - | 5 | 15 | [55] |
| 5%Pd/C | batch | 180 | 1 | - | 24 | 99 | 99 | - | 4 | 8 | [56] |
| 2.5%Pd, 2.5%Ni/Al2O3 | batch | 200 | 1 | - | 27 | 100 | 100 | - | 5 | 0 | [57] |
| 1%Pd/Al2O3 | flow through | 180 | 1 | 2.9 e | 2.3 | 100 | 92 | 200 | - | 0 | [58] |
| Catalyst | Reactor | T (°C) | p (bar) | Rate of H2 Consumption (mmol min−1) | WHSV a (h−1) | Conv. (%) | Sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Benzene | ||||||||
| 0.5%Ni/Al2O3 | flow through | 180 | 1 | 0.28 | 0.96 | 45 | 100 | [67] |
| 3%Pd/CeO2 | flow through | 200 | 1 | 0.79 | 1.23 | 99.5 | 100 | [68] |
| 1%Pd/Al2O3 | flow through | 200 | 1 | 0.97 | 1.48 | 97.5 | 100 | [69] |
| 10%Ni/ N-doped carbon | flow through | 145 | 1 | 284 | 15.3 | - | 100 | [72] |
| Toluene | ||||||||
| 60%Ni/Al2O3 | flow through | 150 | 1 | - | - | 100 | 100 | [73] |
| 1%Ir/SiO2 | flow through | 125 | 1 | 3.75 | 7.03 | 83 | 100 | [74] |
| 3.7%Ru/C | autoclave | 60 | 45 | 20 (1 h) b | - | 10.2 | 100 | [75] |
| 3.7%Ru,9.3%Ni/C | autoclave | 60 | 45 | 518 (1 h) b | - | 100 | 100 | [75] |
| 3.7%Ru,9.2%Co/C | autoclave | 60 | 45 | 547 (1 h) b | - | 100 | 100 | [75] |
| 3.7%Ru,4.6%Ni, 4.6%Co/C | autoclave | 60 | 45 | 683 (1 h) b | - | 100 | 100 | [75] |
| Naphthalene | ||||||||
| 1%Pt/Al-SBA-15 | autoclave | 290 | 70 | 26 (1 h) b | - | 100 | 100 | [76] |
| 2.9%Ni,9%Mo/HMS | autoclave | 325 | 65 | - | - | 90 | 43.8 | [77] |
| 2.9%Ni,9%Mo/Al-HMS | autoclave | 325 | 65 | - | - | 100 | 75.7 | [77] |
| Dibenzyltoluene | ||||||||
| 0.3%Pt/Al2O3 | autoclave | 270 | 30 | 24 (15 min) b | - | 100 | 100 | [78] |
| 3%Pt/Al2O3 | autoclave | 140 | 40 | 17 (10 min) b | - | 100 | 100 | [79] |
| 3%Pt/Al2O3-H2 plasma | autoclave | 140 | 40 | 19 (10 min) b | - | 100 | 100 | [79] |
| 3%Pt/Al2O3-O2 plasma | autoclave | 140 | 40 | 23 (10 min) b | - | 100 | 100 | [79] |
| N-ethylcarbazole | ||||||||
| 5%Ru/Al2O3 | autoclave | 150 | 70 | 3.0 (1 h) b | - | 100 | 95 | [80] |
| LiNi5.5 | autoclave | 180 | 70 | - | - | 97 | 100 | [81] |
| 2.5%Ru,2.5%Ni/Al2O3 c | autoclave | 160 | 70 | 8.5 (2 h) b | - | 100 | 100 | [82] |
| Catalyst | Reactor Type | T (°C) | p (bar) | W/F a (h−1) | Initial Conv. (%) | Initial Sel. (%) | TOS b (h) | Number of Recycling | Decrease of Reaction Rate (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Benzene | |||||||||||
| Nb2(μ2-CSiMe3)2 (CH2SiMe3)4 | autoclave | 120 | 27.6 | - | 149 | 80 | 100 | - | 2 | 28 | [71] |
| Toluene | |||||||||||
| 3.7%Ru 4.6%Ni, 4.6%Co/C | autoclave | 60 | 45 | - | 94 (1 h) | 100 | 100 | - | 5 | 4 | [75] |
| 1.3wt%Pd/ graphite nanopla-telets | flow through | 200 | 1 | - | 287 | - | - | 24 | - | 16.2 | [83] |
| 1.5wt%Pd/ Timrex (graphite) | flow through | 200 | 1 | - | 127 | - | - | 24 | - | 14.6 | [83] |
| 0.039wt%Pd, 6wt%Co/ ZrO2 | flow through | 120 | 1 | 36.1 | 6 | - | - | 24 | - | 0 | [85] |
| Naphtalene | |||||||||||
| 0.8wt%Pt-Al2O3-NH2/SiO2 | flow through | 260 | 40 | 10 | - | 100 | 96.4 | 60 | - | 0 | [86] |
| 1wt%Pt/ H-Beta-75 | flow through | 220 | 40 | 10 | - | 96.7 | 79.3 | 40 | - | [87] | |
| Dibenzyltoluene | |||||||||||
| 0.3%Pt/Al2O3 | autoclave | 300 | 30 | - | 37.5 (1 h) | 96 | 100 | - | 4 | 0 | [78] |
| 3%Pt/Al2O3 | batch | 140 | 40 | - | - | 100 | 100 | - | 4 | 3.7 | [79] |
| 3%Pt/Al2O3-O2 plasma treated | batch | 140 | 40 | - | - | 100 | 100 | - | 4 | 3.7 | [79] |
| 3%Pt/Al2O3-H2 plasma treated | batch | 140 | 40 | - | - | 100 | 100 | - | 4 | 5.3 | [79] |
| 5%Pt/Al2O3 | batch | 240 | 50 | - | 100 | 100 | - | 3 | 61.3 | [88] | |
| N-Ethylcarbazole | |||||||||||
| LaNi5.5 | batch | 180 | 70 | - | - | 100 | 96.8 | - | 9 | 0 | [81] |
| Ru2.5Ni2.5/Al2O3 | batch | 150 | 60 | - | - | 93.6 | 100 | - | 5 | [82] | |
| 30%Ni, 10%La2O3, 60%Al2O3 | batch | 140 | 50 | - | - | 100 | 100 | - | 10 | 0 | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barthos, R.; Lónyi, F.; Shi, Y.; Szegedi, Á.; Vikár, A.; Solt, H.E.; Novodárszki, G. Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology. Catalysts 2025, 15, 427. https://doi.org/10.3390/catal15050427
Barthos R, Lónyi F, Shi Y, Szegedi Á, Vikár A, Solt HE, Novodárszki G. Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology. Catalysts. 2025; 15(5):427. https://doi.org/10.3390/catal15050427
Chicago/Turabian StyleBarthos, Róbert, Ferenc Lónyi, Yuting Shi, Ágnes Szegedi, Anna Vikár, Hanna E. Solt, and Gyula Novodárszki. 2025. "Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology" Catalysts 15, no. 5: 427. https://doi.org/10.3390/catal15050427
APA StyleBarthos, R., Lónyi, F., Shi, Y., Szegedi, Á., Vikár, A., Solt, H. E., & Novodárszki, G. (2025). Catalytic Aspects of Liquid Organic Hydrogen Carrier Technology. Catalysts, 15(5), 427. https://doi.org/10.3390/catal15050427