
Synergistic Effect of the Heteronuclear Double Sites in C9N4 on the Electrochemical Reduction of CO2 to CO

Abstract
:1. Introduction
2. Results and Discussion
2.1. The Structure and Stability of TM-B@C9N4
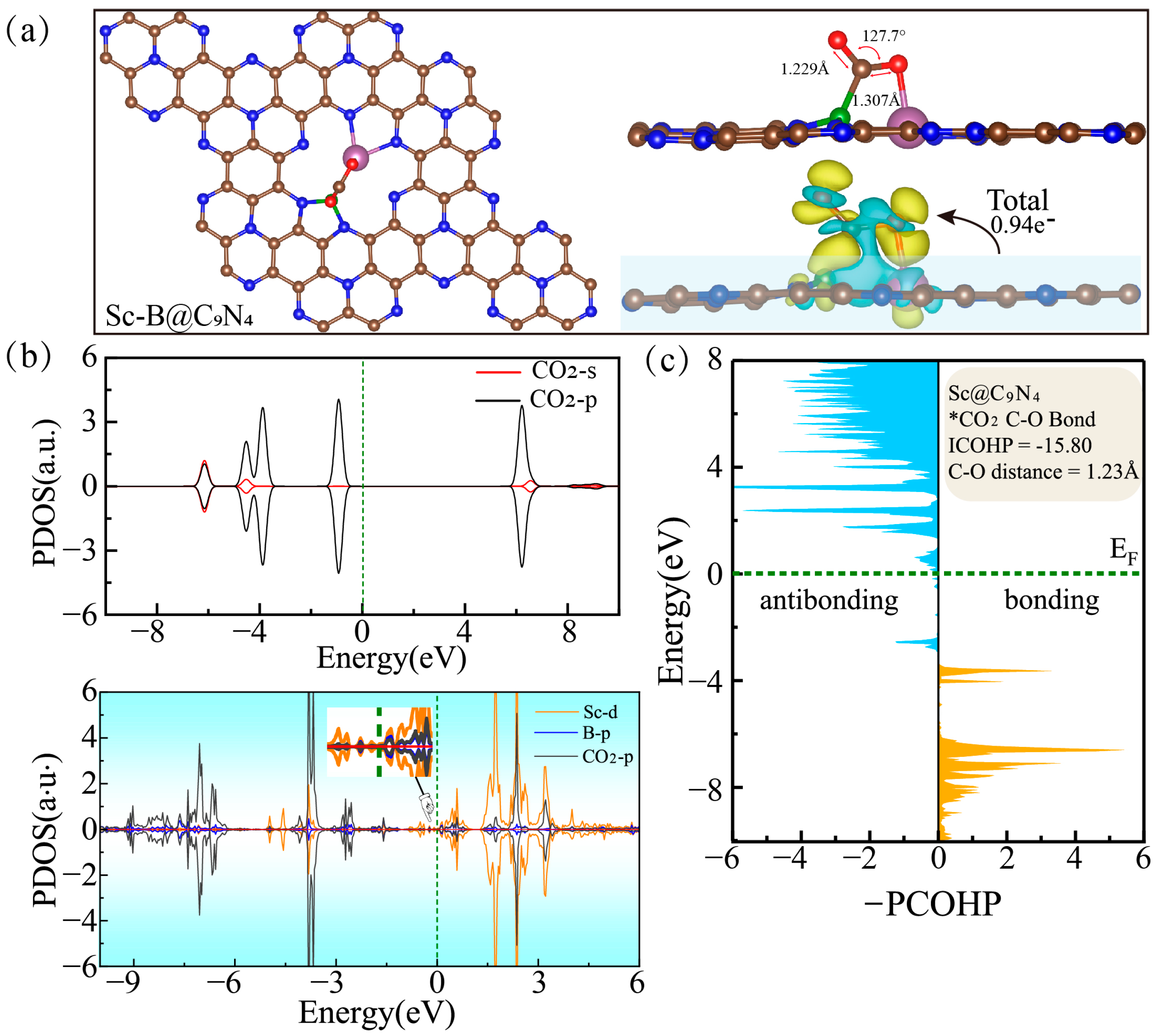
2.2. Adsorption and Activation of CO2
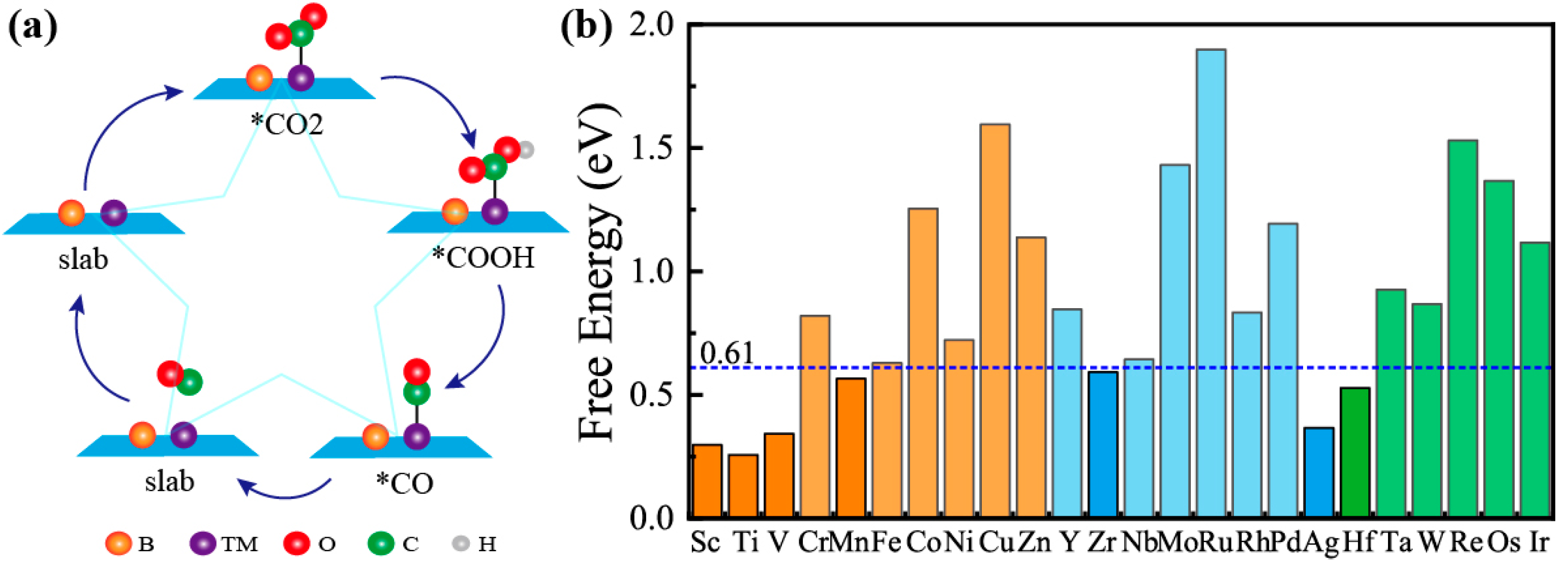
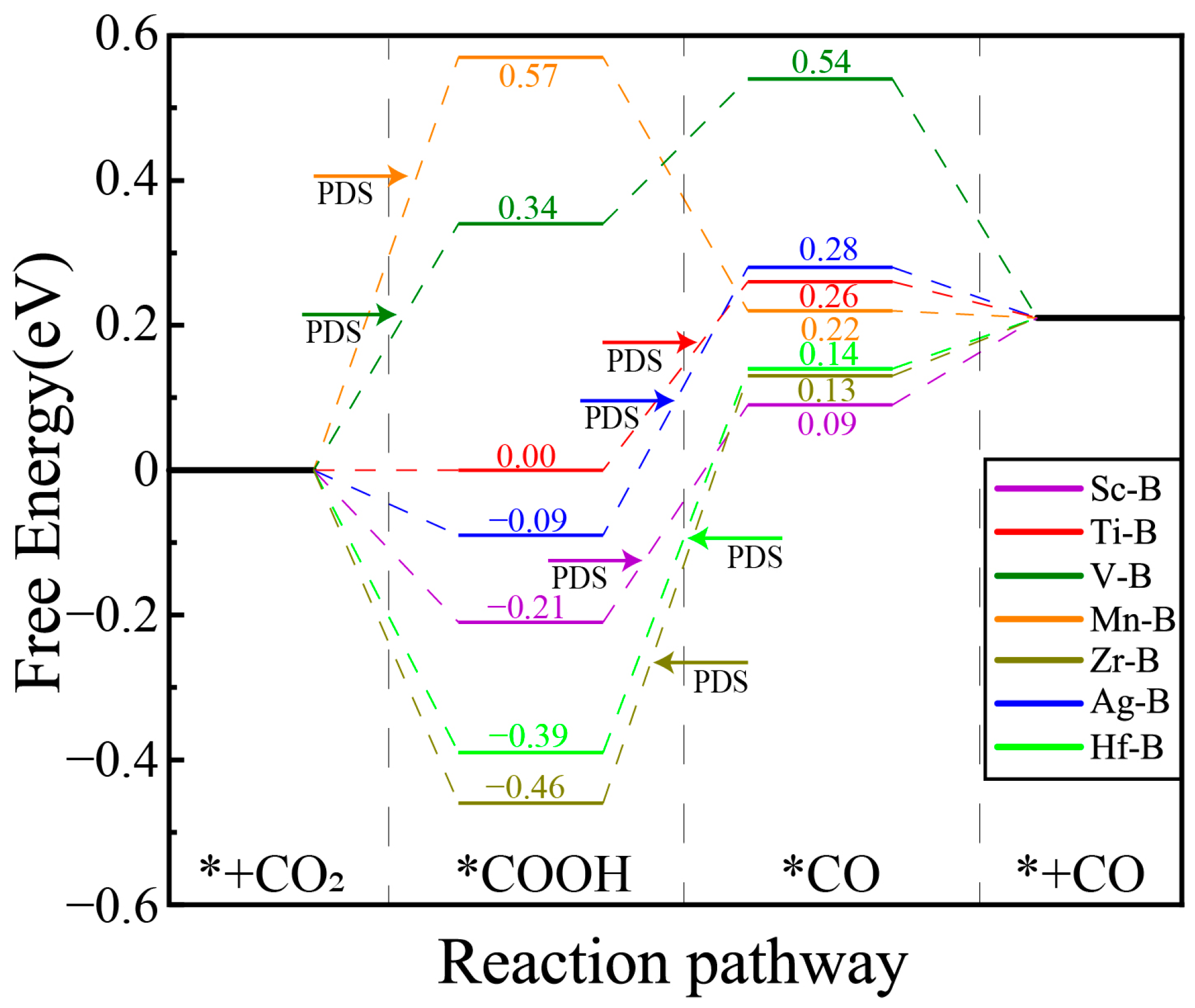
2.3. The Mechanism and Free Energy of CO2 Reduction to CO
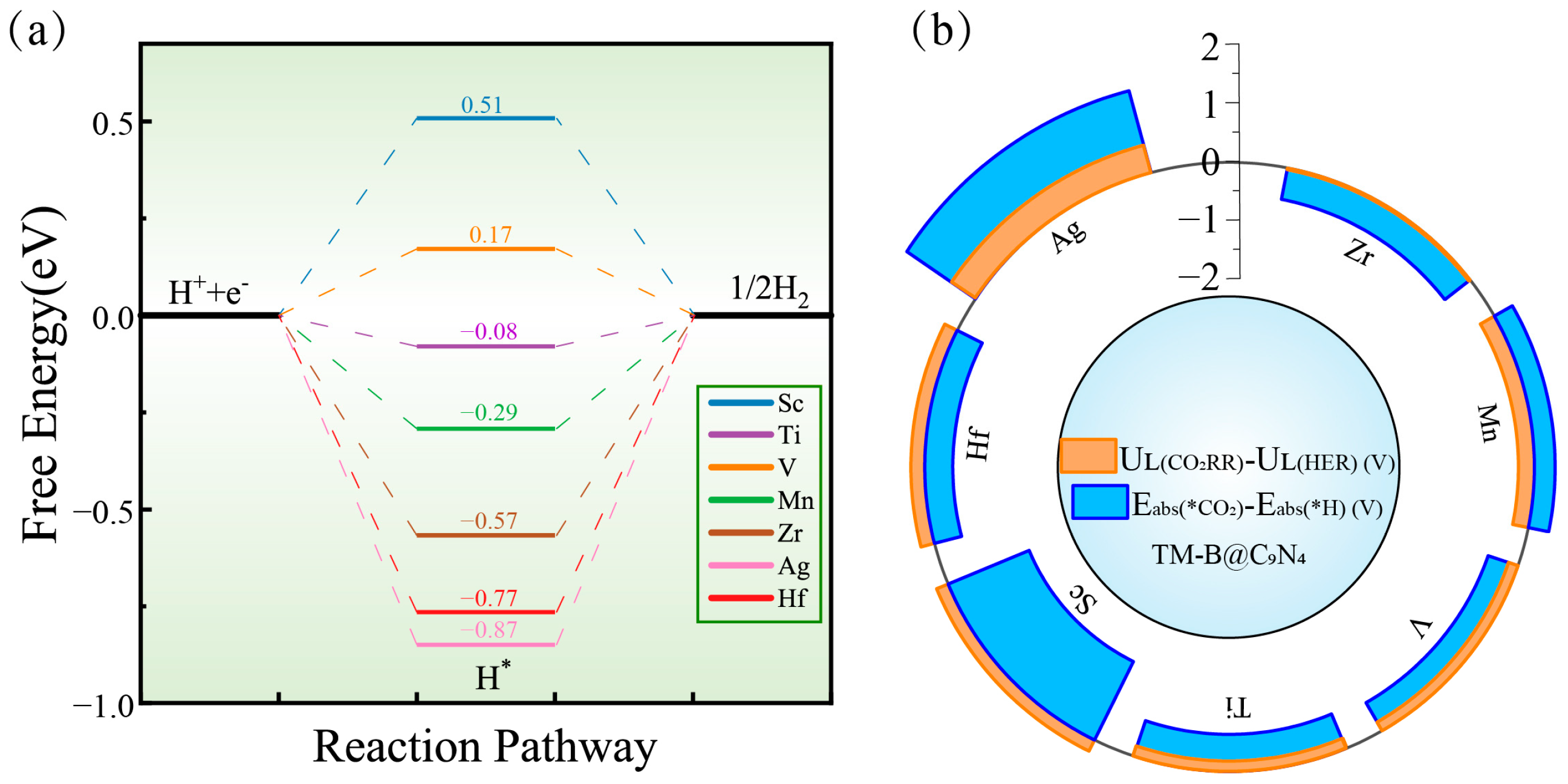
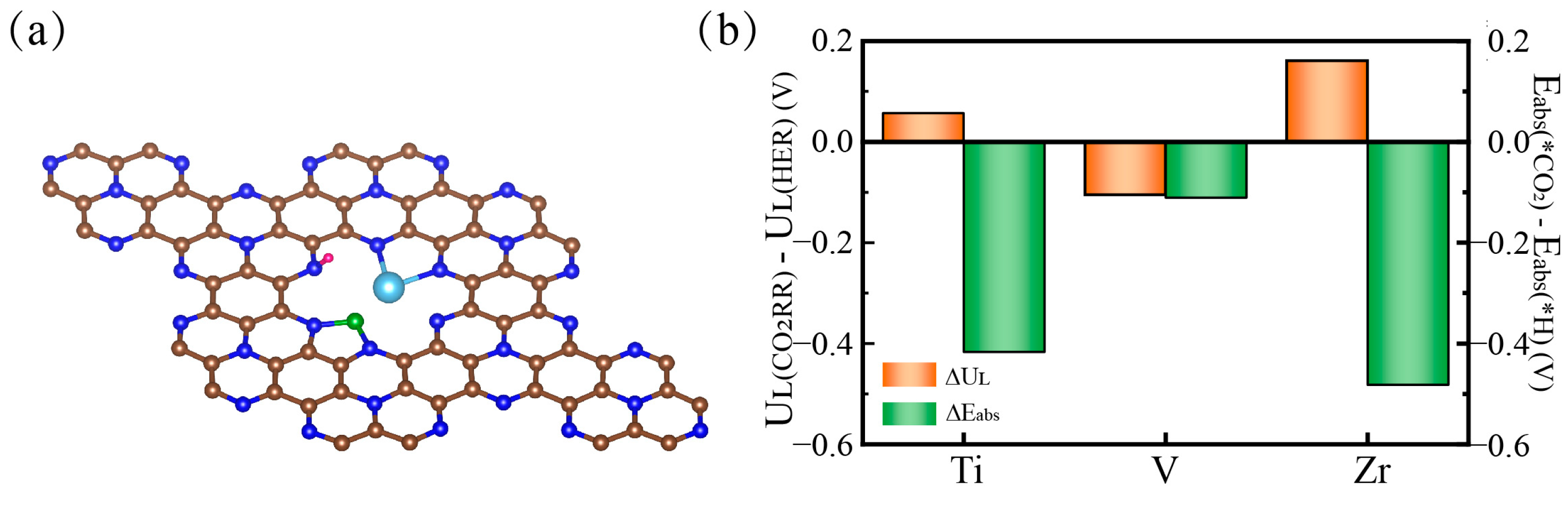
2.4. Competitive HER and Catalyst Improvements
- (1)
- HER is the only side reaction in the CO2RR process;
- (2)
- The rate of HER and CO2RR do not depend mainly on proton and electron transfer;
- (3)
- Boltzmann distribution can be used to estimate the selectivity of CO2RR relative to HER.
3. Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudd, J.A.; Hernandez-Aldave, S.; Kazimierska, E.; Hamdy, L.B.; Bain, O.J.E.; Barron, A.R.; Andreoli, E. Investigation into the Re-Arrangement of Copper Foams Pre-and Post-CO2 Electrocatalysis. Chemistry 2021, 3, 687–703. [Google Scholar] [CrossRef]
- Chu, S.; Cui, Y.; Liu, N. The Path towards Sustainable Energy. Nat. Mater. 2017, 16, 16–22. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Zhu, D.D.; Liu, J.L.; Qiao, S.Z. Recent Advances in Inorganic Heterogeneous Electrocatalysts for Reduction of Carbon Dioxide. Adv. Mater. 2016, 28, 3423–3452. [Google Scholar] [CrossRef]
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Roldan Cuenya, B. Rational Catalyst and Electrolyte Design for CO2 Electroreduction towards Multicarbon Products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Ross, M.B.; De Luna, P.; Li, Y.; Dinh, C.-T.; Kim, D.; Yang, P.; Sargent, E.H. Designing Materials for Electrochemical Carbon Dioxide Recycling. Nat. Catal. 2019, 2, 648–658. [Google Scholar] [CrossRef]
- Kornienko, N.; Zhao, Y.; Kley, C.S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C.J.; Yaghi, O.M.; Yang, P. Metal-Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. [Google Scholar] [CrossRef]
- Ma, W.; Xie, S.; Zhang, X.-G.; Sun, F.; Kang, J.; Jiang, Z.; Zhang, Q.; Wu, D.-Y.; Wang, Y. Promoting Electrocatalytic CO2 Reduction to Formate via Sulfur-Boosting Water Activation on Indium Surfaces. Nat. Commun. 2019, 10, 892. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T. Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide and Methane at an Immobilized Cobalt Protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Wei, Z.; Liu, Q.; Wang, C.; Ma, J. Electrochemical CO2 Reduction over Nitrogen-Doped SnO2 Crystal Surfaces. J. Energy Chem. 2019, 33, 22–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Mo, Y.; Cao, Z. Rational Design of Main Group Metal-Embedded Nitrogen-Doped Carbon Materials as Frustrated Lewis Pair Catalysts for CO2 Hydrogenation to Formic Acid. ACS Appl. Mater. Interfaces 2021, 14, 1002–1014. [Google Scholar] [CrossRef]
- Li, Y.; Su, H.; Chan, S.H.; Sun, Q. CO2 Electroreduction Performance of Transition Metal Dimers Supported on Graphene: A Theoretical Study. ACS Catal. 2015, 5, 6658–6664. [Google Scholar] [CrossRef]
- Lin, L.; Liu, T.; Xiao, J.; Li, H.; Wei, P.; Gao, D.; Nan, B.; Si, R.; Wang, G.; Bao, X. Enhancing CO2 Electroreduction to Methane with a Cobalt Phthalocyanine and Zinc-Nitrogen-Carbon Tandem Catalyst. Angew. Chem. Int. Ed. 2020, 59, 22408–22413. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Kenis, P.J.A. Electrochemical Conversion of CO2 to Useful Chemicals: Current Status, Remaining Challenges, and Future Opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar]
- Ganesh, I. Applications of Nanomaterials; Elsevier: Amsterdam, The Netherlands, 2018; pp. 83–131. [Google Scholar]
- Ren, W.; Tan, X.; Yang, W.; Jia, C.; Xu, S.; Wang, K.; Smith, S.C.; Zhao, C. Isolated Diatomic Ni-Fe Metal-Nitrogen Sites for Synergistic Electroreduction of CO2. Angew. Chem. Int. Ed. 2019, 58, 6972–6976. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, X.; Kang, Y.; Ye, C.; Jin, R.; Yan, H.; Lin, R.; Yang, J.; Xu, Q.; Wang, Y. A Supported Pd2 Dual-Atom Site Catalyst for Efficient Electrochemical CO2 Reduction. Angew. Chem. 2021, 133, 13500–13505. [Google Scholar] [CrossRef]
- Zhang, C.; He, Q.; Chu, W.; Zhao, Y. Transition Metals Doped Borophene-Graphene Heterostructure for Robust Polysulfide Anchoring: A First Principle Study. Appl. Surf. Sci. 2020, 534, 147575. [Google Scholar] [CrossRef]
- Duplock, E.J.; Scheffler, M.; Lindan, P.J.D. Hallmark of Perfect Graphene. Phys. Rev. Lett. 2004, 92, 225502. [Google Scholar] [CrossRef]
- Zhao, Q.; Crespo-Otero, R.; Di Tommaso, D. The Role of Copper in Enhancing the Performance of Heteronuclear Diatomic Catalysts for the Electrochemical CO2 Conversion to C1 Chemicals. J. Energy Chem. 2023, 85, 490–500. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shahrokhi, M.; Shapeev, A.V.; Rabczuk, T.; Zhuang, X. Prediction of C7N6 and C9N4: Stable and Strong Porous Carbon-Nitride Nanosheets with Attractive Electronic and Optical Properties. J. Mater. Chem. C 2019, 7, 10908–10917. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, X.; Ma, X.; Liu, J.; Li, Y.; Zhao, M. Tunable Topological Electronic States in the Honeycomb-Kagome Lattices of Nitrogen/Oxygen-Doped Graphene Nanomeshes. Nanoscale Adv. 2022, 4, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-L.; Hu, H.-J.; Wei, S.-H. Transition Metal Anchored on C9N4 as a Single-Atom Catalyst for CO2 Hydrogenation: A First-Principles Study. Chin. Phys. B 2022, 31, 107306. [Google Scholar] [CrossRef]
- Fu, J.; Dong, J.; Si, R.; Sun, K.; Zhang, J.; Li, M.; Yu, N.; Zhang, B.; Humphrey, M.G.; Fu, Q.; et al. Synergistic Effects for Enhanced Catalysis in a Dual Single-Atom Catalyst. ACS Catal. 2021, 11, 1952–1961. [Google Scholar] [CrossRef]
- Zhang, L.; Si, R.; Liu, H.; Chen, N.; Wang, Q.; Adair, K.; Wang, Z.; Chen, J.; Song, Z.; Li, J. Atomic Layer Deposited Pt-Ru Dual-Metal Dimers and Identifying Their Active Sites for Hydrogen Evolution Reaction. Nat. Commun. 2019, 10, 4936. [Google Scholar] [CrossRef]
- Zheng, T.; Liu, C.; Guo, C.; Zhang, M.; Li, X.; Jiang, Q.; Xue, W.; Li, H.; Li, A.; Pao, C.-W.; et al. Copper-Catalysed Exclusive CO2 to Pure Formic Acid Conversion via Single-Atom Alloying. Nat. Nanotechnol. 2021, 16, 1386–1393. [Google Scholar] [CrossRef]
- Légaré, M.-A.; Bélanger-Chabot, G.; Dewhurst, R.D.; Welz, E.; Krummenacher, I.; Engels, B.; Braunschweig, H. Nitrogen Fixation and Reduction at Boron. Science 2018, 359, 896–900. [Google Scholar] [CrossRef]
- Yu, X.; Han, P.; Wei, Z.; Huang, L.; Gu, Z.; Peng, S.; Ma, J.; Zheng, G. Boron-Doped Graphene for Electrocatalytic N2 Reduction. Joule 2018, 2, 1610–1622. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Song, Y.; Zhang, J.; Zeng, L.; Wang, C.; Lin, W. Cooperative Copper Centres in a Metal-Organic Framework for Selective Conversion of CO2 to Ethanol. Nat. Catal. 2019, 2, 709–717. [Google Scholar] [CrossRef]
- Jiao, J.; Lin, R.; Liu, S.; Cheong, W.-C.; Zhang, C.; Chen, Z.; Pan, Y.; Tang, J.; Wu, K.; Hung, S.-F.; et al. Copper Atom-Pair Catalyst Anchored on Alloy Nanowires for Selective and Efficient Electrochemical Reduction of CO2. Nat. Chem. 2019, 11, 222–228. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, H.; An, W.; Wang, Y.; Feng, Y.; Men, Y. Synergy of a Metallic NiCo Dimer Anchored on a C2N-Graphene Matrix Promotes the Electrochemical CO2 Reduction Reaction. ACS Sustain. Chem. Eng. 2019, 7, 19113–19121. [Google Scholar] [CrossRef]
- Ding, C.; Feng, C.; Mei, Y.; Liu, F.; Wang, H.; Dupuis, M.; Li, C. Carbon Nitride Embedded with Transition Metals for Selective Electrocatalytic CO2 Reduction. Appl. Catal. B Environ. 2020, 268, 118391. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Meaning and Functional Form of the Electron Localization Function. Acta Phys.-Chim. Sin. 2011, 27, 2786–2792. [Google Scholar]
- Pittalis, S.; Varsano, D.; Delgado, A.; Rozzi, C.A. Bonds, Lone Pairs, and Shells Probed by Means of On-Top Dynamical Correlations. Eur. Phys. J. B 2018, 91, 187. [Google Scholar] [CrossRef]
- Gao, Z.; Ma, H.; Yuan, S.; Ren, H.; Ge, Z.; Zhu, H.; Guo, W.; Ding, F.; Zhao, W. Benzenehexol-Based 2D MOF as High-Performance Electrocatalyst for Oxygen Reduction Reaction. Appl. Surf. Sci. 2022, 601, 154187. [Google Scholar] [CrossRef]
- Xing, G.; Liu, S.; Liu, J.-Y. Computational Evaluation of 2D Metal-Organic Frameworks with TMX4-Centers (X = O, S, and Se) for CO2 Electroreduction. Int. J. Hydrog. Energy 2023, 48, 3486–3494. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Chiu, K.-Y.; Fan, C.-H.; Chen, H.-L. Electrocatalytic Carbon Dioxide Reduction on Graphene-Supported Ni Cluster and Its Hydride: Insight from First-Principles Calculations. Appl. Surf. Sci. 2023, 629, 157418. [Google Scholar] [CrossRef]
- Hong, X.; Chan, K.; Tsai, C.; Nørskov, J.K. How Doped MoS2 Breaks Transition-Metal Scaling Relations for CO2 Electrochemical Reduction. ACS Catal. 2016, 6, 4428–4437. [Google Scholar] [CrossRef]
- Chiu, K.-Y.; Fan, C.-H.; Hsu, C.-W.; Chen, H.-L. First-Principles Insight into the Mechanistic Study of Electrochemical Cyanide Reduction Reaction on Post-Transition Metal Based Single-Atom Catalysts Anchored by Phthalocyanine Nanosheets. ACS Appl. Nano Mater. 2024, 7, 9909–9924. [Google Scholar] [CrossRef]
- Ickmans, K.; Clarys, P.; Nijs, J.; Meeus, M.; Aerenhouts, D.; Zinzen, E.; Aelbrecht, S.; Meersdom, G.; Lambrecht, L.; Pattyn, N. Association Between Cognitive Performance, Physical Fitness, and Physical Activity Level in Women with Chronic Fatigue Syndrome. J. Rehabil. Res. Dev. 2013, 50, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Ti-B@C9N4(-H) | V-B@C9N4(-H) | Zr-B@C9N4(-H) | |
|---|---|---|---|---|
| TM-B@C9N4 | TM | −1.544 | −1.329 | −1.982 |
| B | −0.869 | −0.938 | −0.786 | |
| H | \ | \ | \ | |
| TM-B@C9N4-H | TM | −1.456 | −1.226 | −1.794 |
| B | −0.811 | −0.776 | −0.771 | |
| H | −0.417 | −0.431 | −0.408 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, R.; Zhao, B.; Li, Z. Synergistic Effect of the Heteronuclear Double Sites in C9N4 on the Electrochemical Reduction of CO2 to CO. Catalysts 2025, 15, 370. https://doi.org/10.3390/catal15040370
Wan R, Zhao B, Li Z. Synergistic Effect of the Heteronuclear Double Sites in C9N4 on the Electrochemical Reduction of CO2 to CO. Catalysts. 2025; 15(4):370. https://doi.org/10.3390/catal15040370
Chicago/Turabian StyleWan, Rui, Bin Zhao, and Zhongyao Li. 2025. "Synergistic Effect of the Heteronuclear Double Sites in C9N4 on the Electrochemical Reduction of CO2 to CO" Catalysts 15, no. 4: 370. https://doi.org/10.3390/catal15040370
APA StyleWan, R., Zhao, B., & Li, Z. (2025). Synergistic Effect of the Heteronuclear Double Sites in C9N4 on the Electrochemical Reduction of CO2 to CO. Catalysts, 15(4), 370. https://doi.org/10.3390/catal15040370