
Synthesis of Star Isotactic Polypropylene via Styryldichlorosilane/Hydrogen Consecutive Chain Transfer Reaction


Abstract

1. Introduction
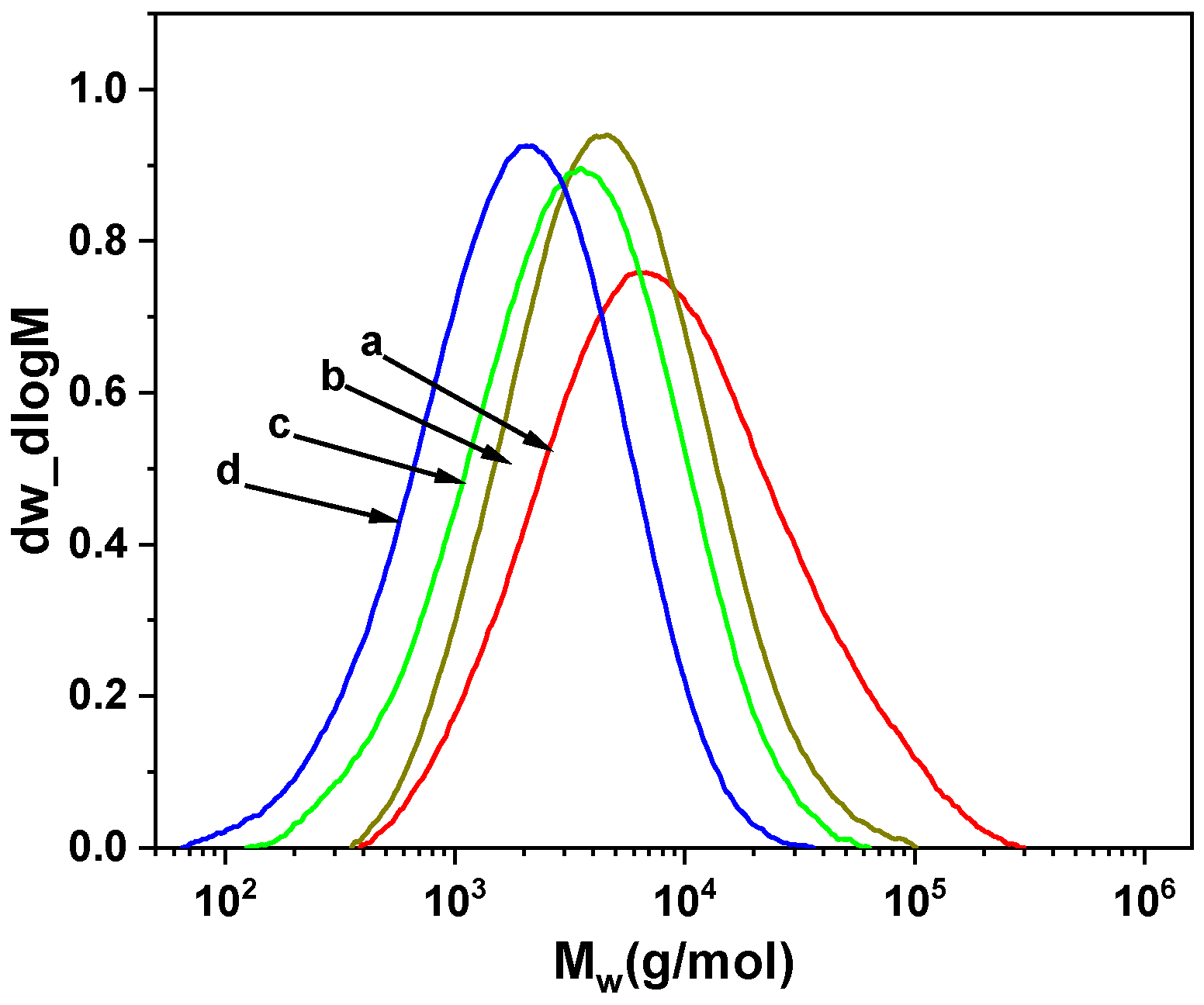
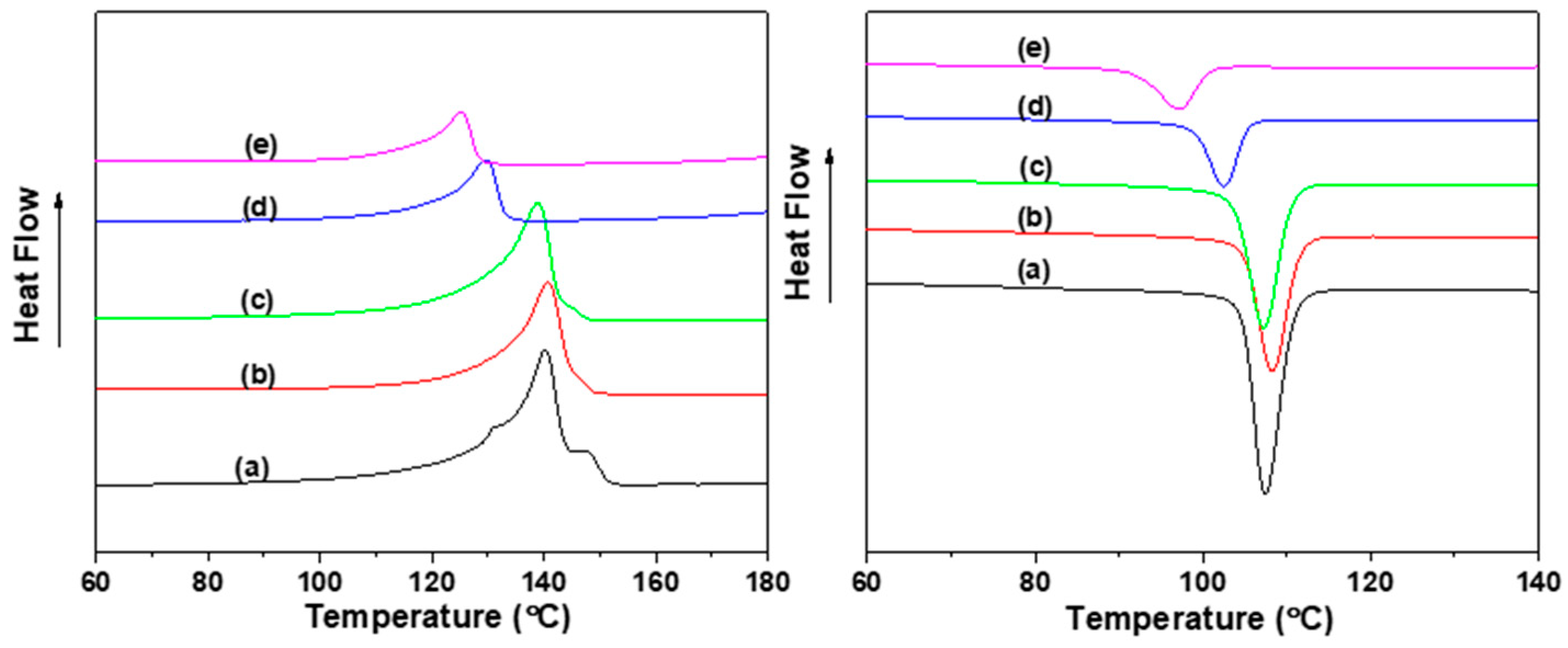
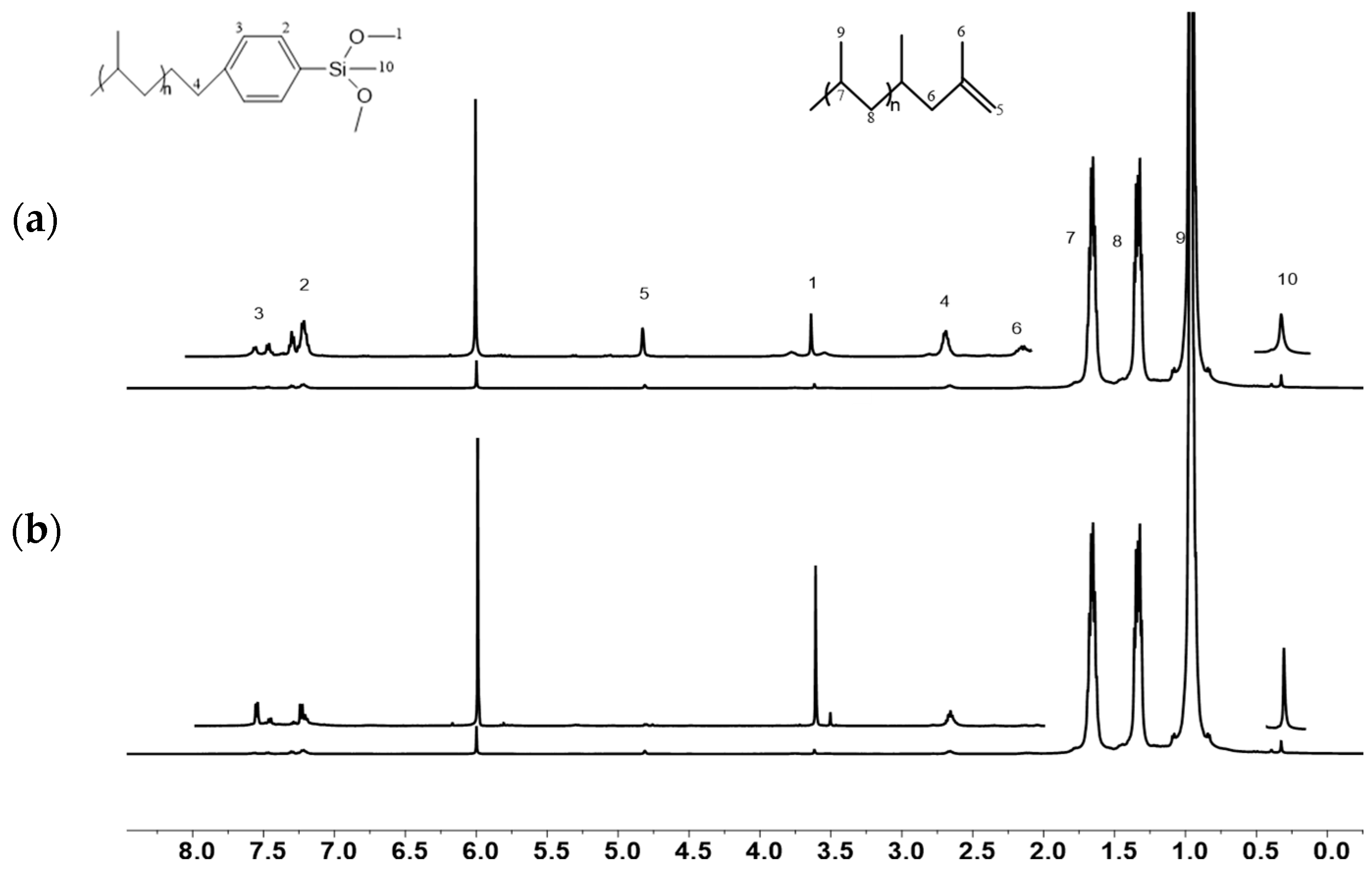
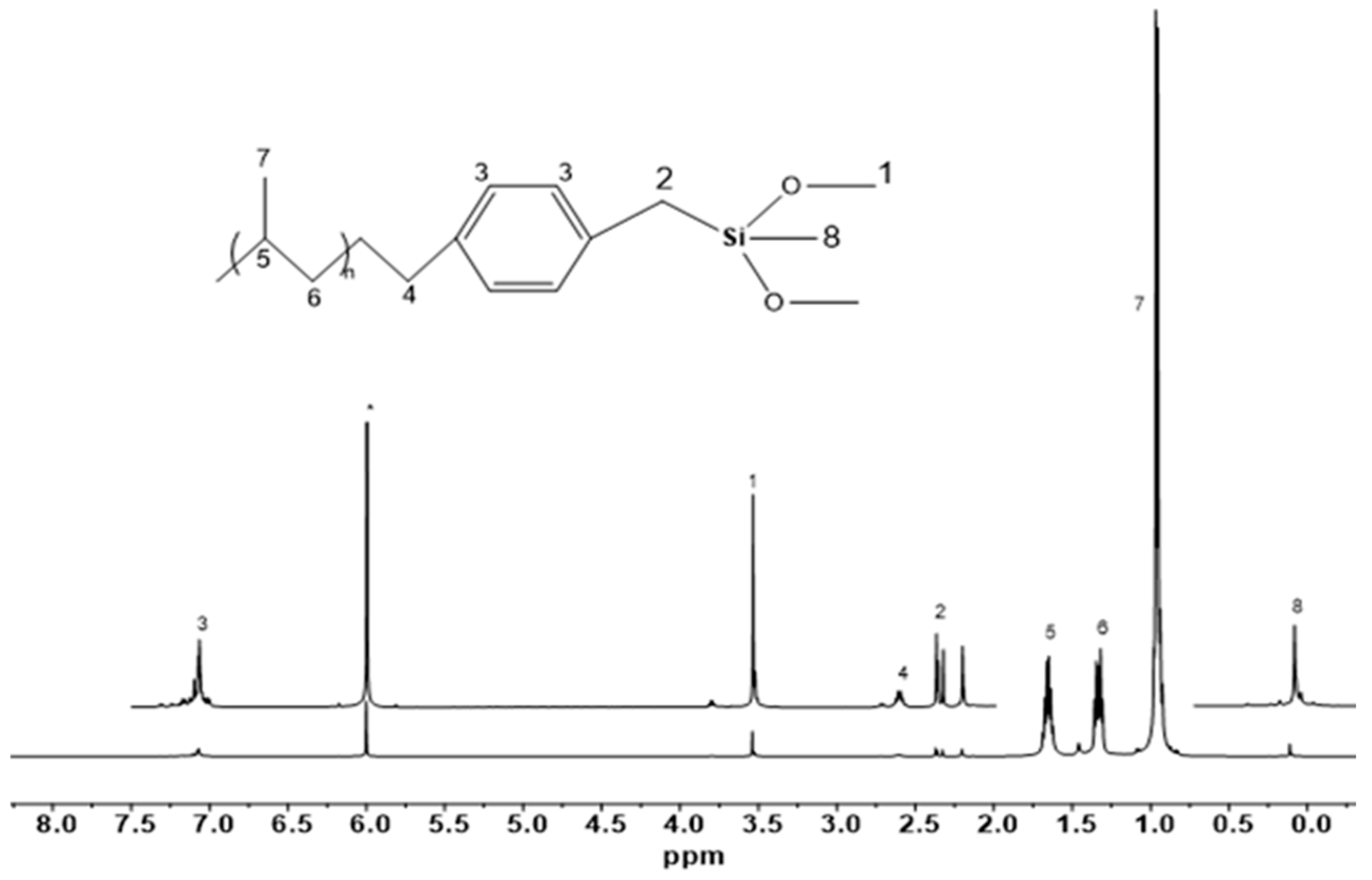
2. Results and Discussion
2.1. Propylene Polymerization
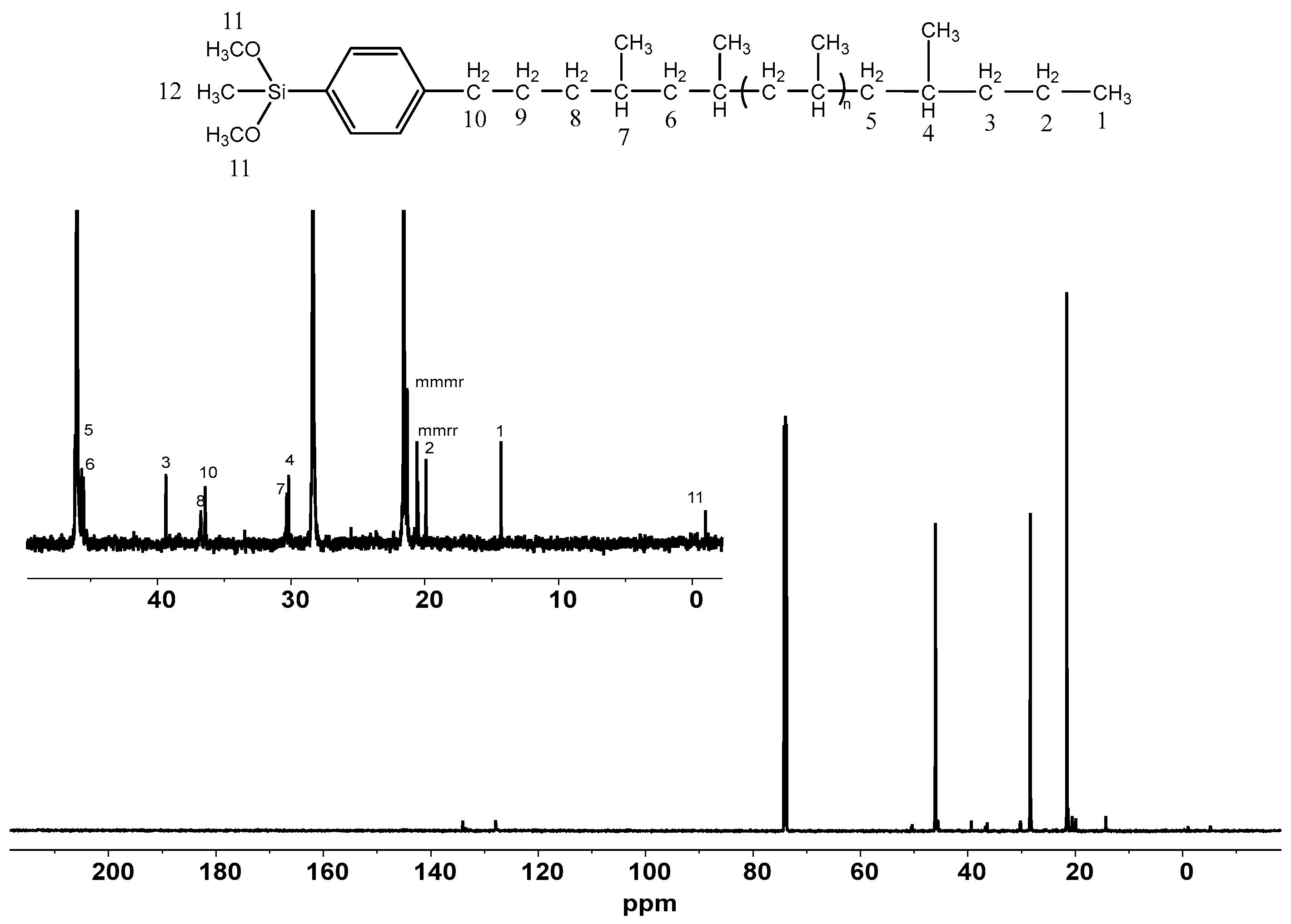
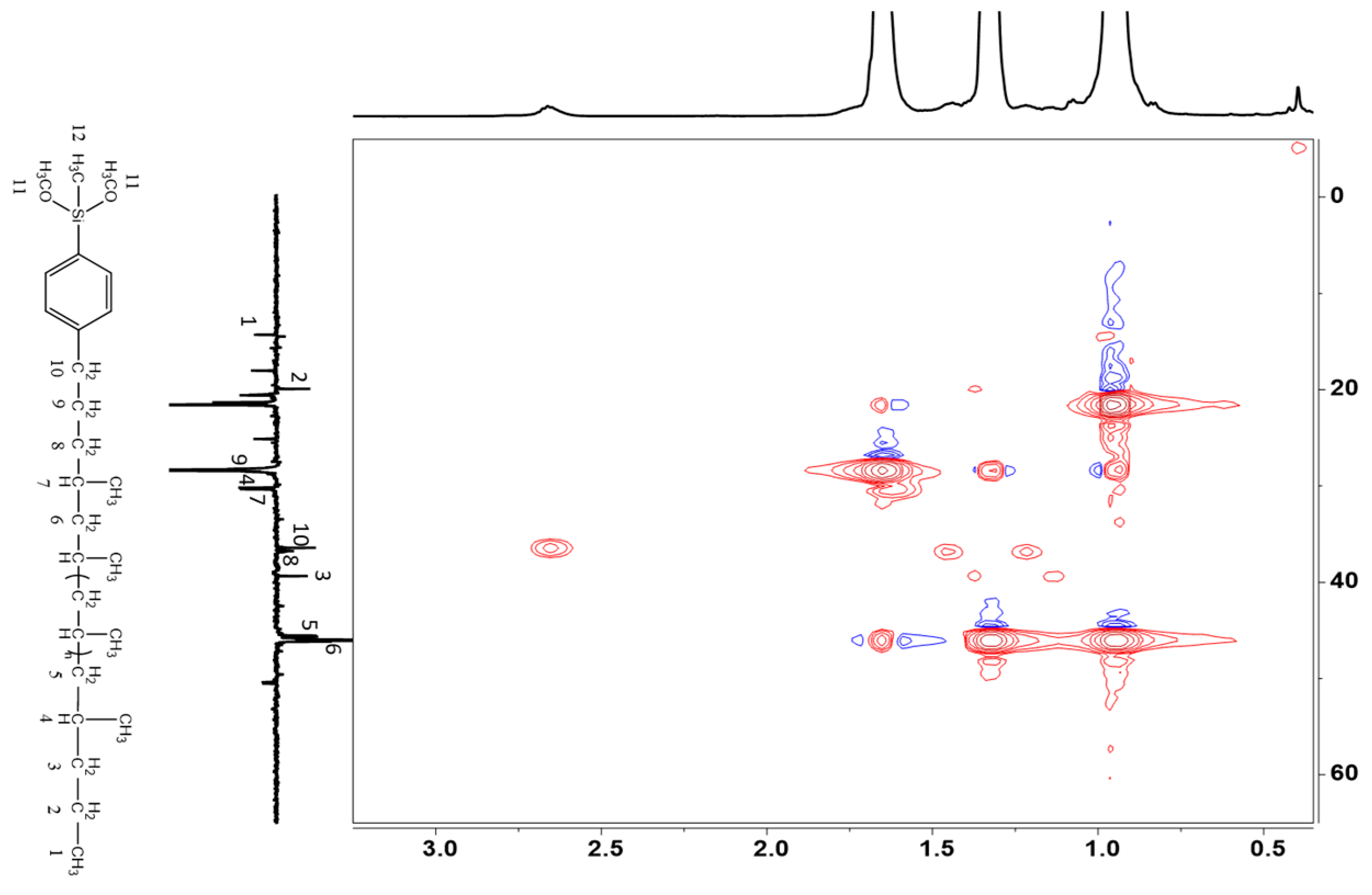
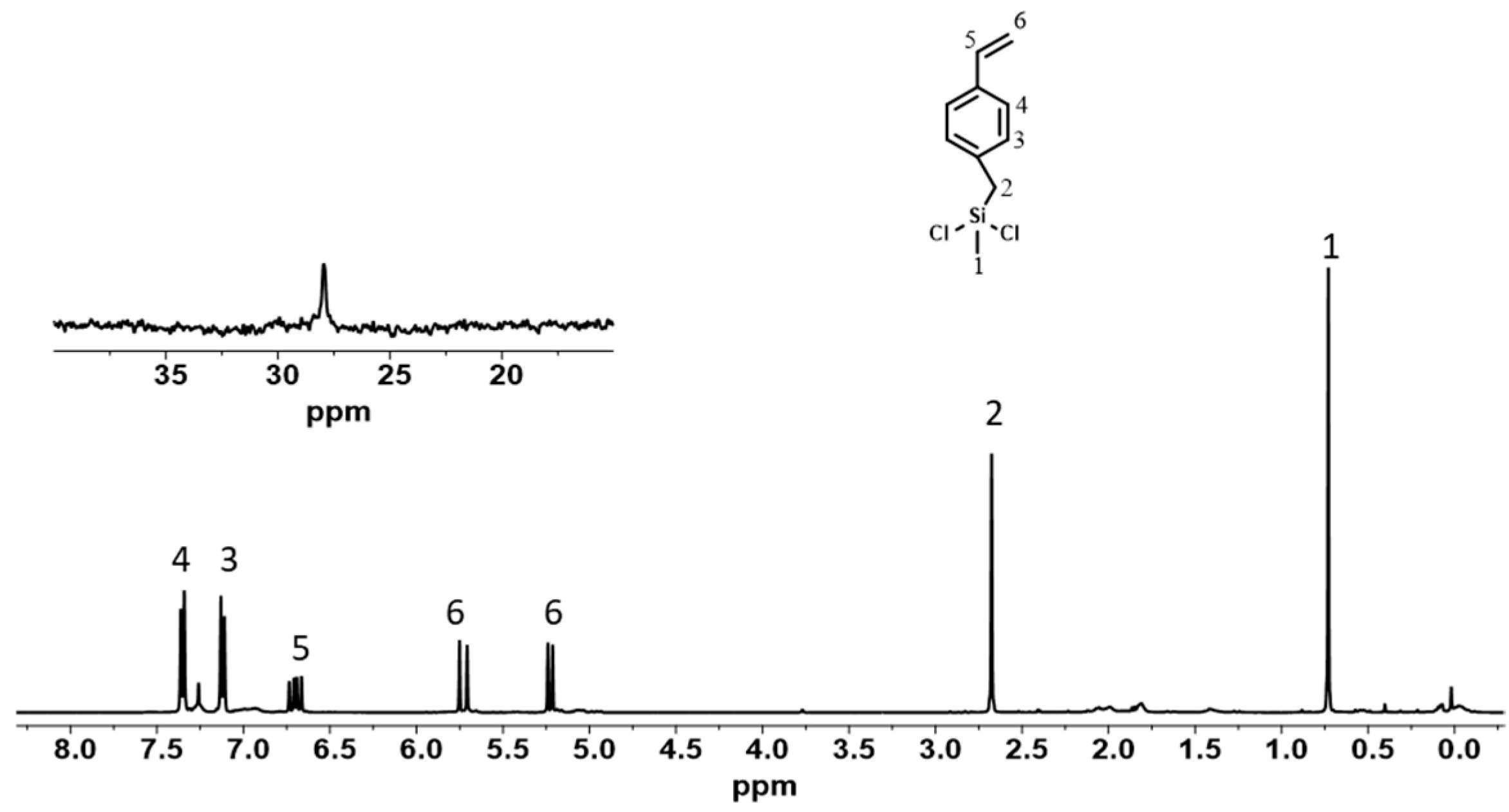
2.2. End Group Analysis
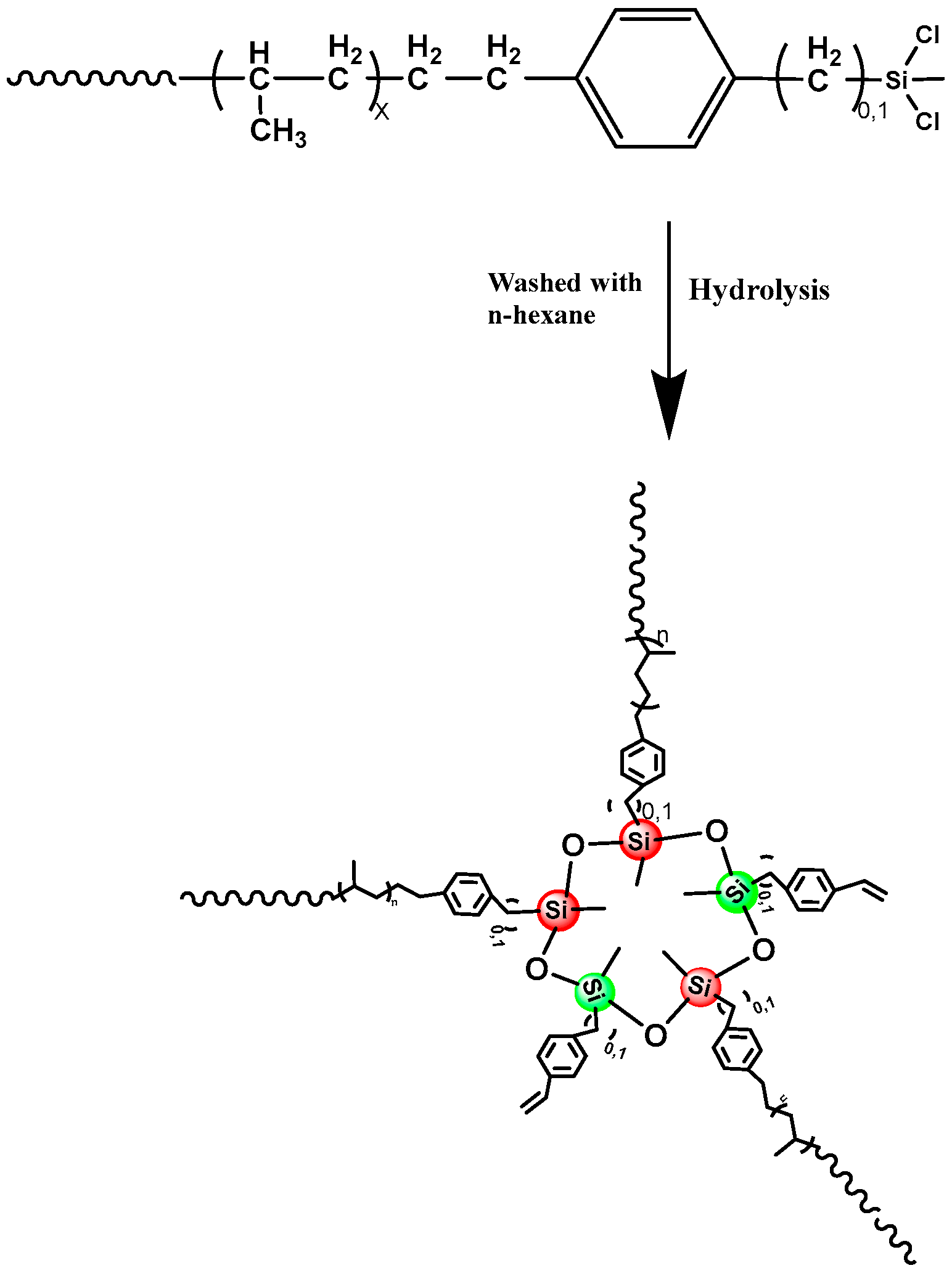
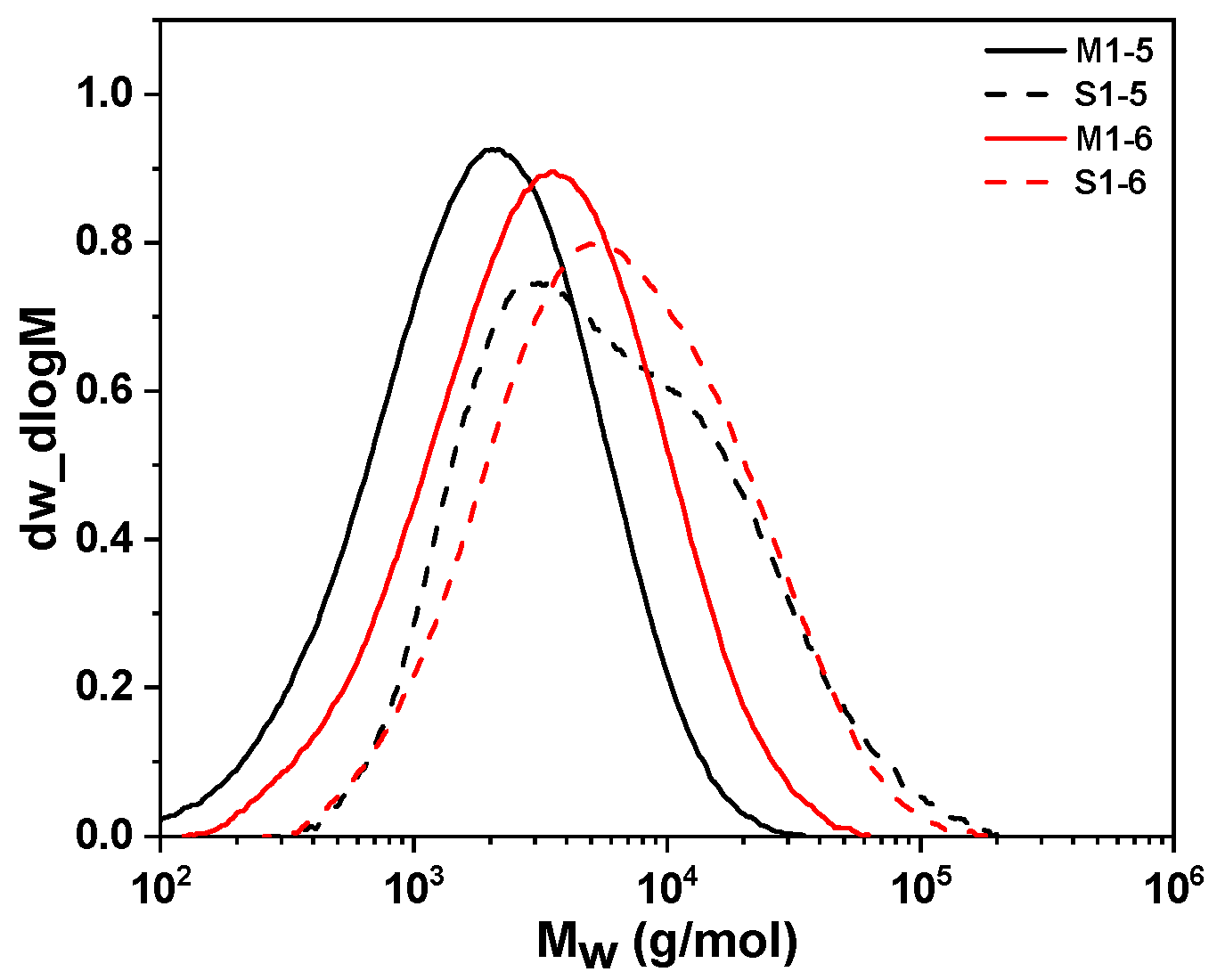
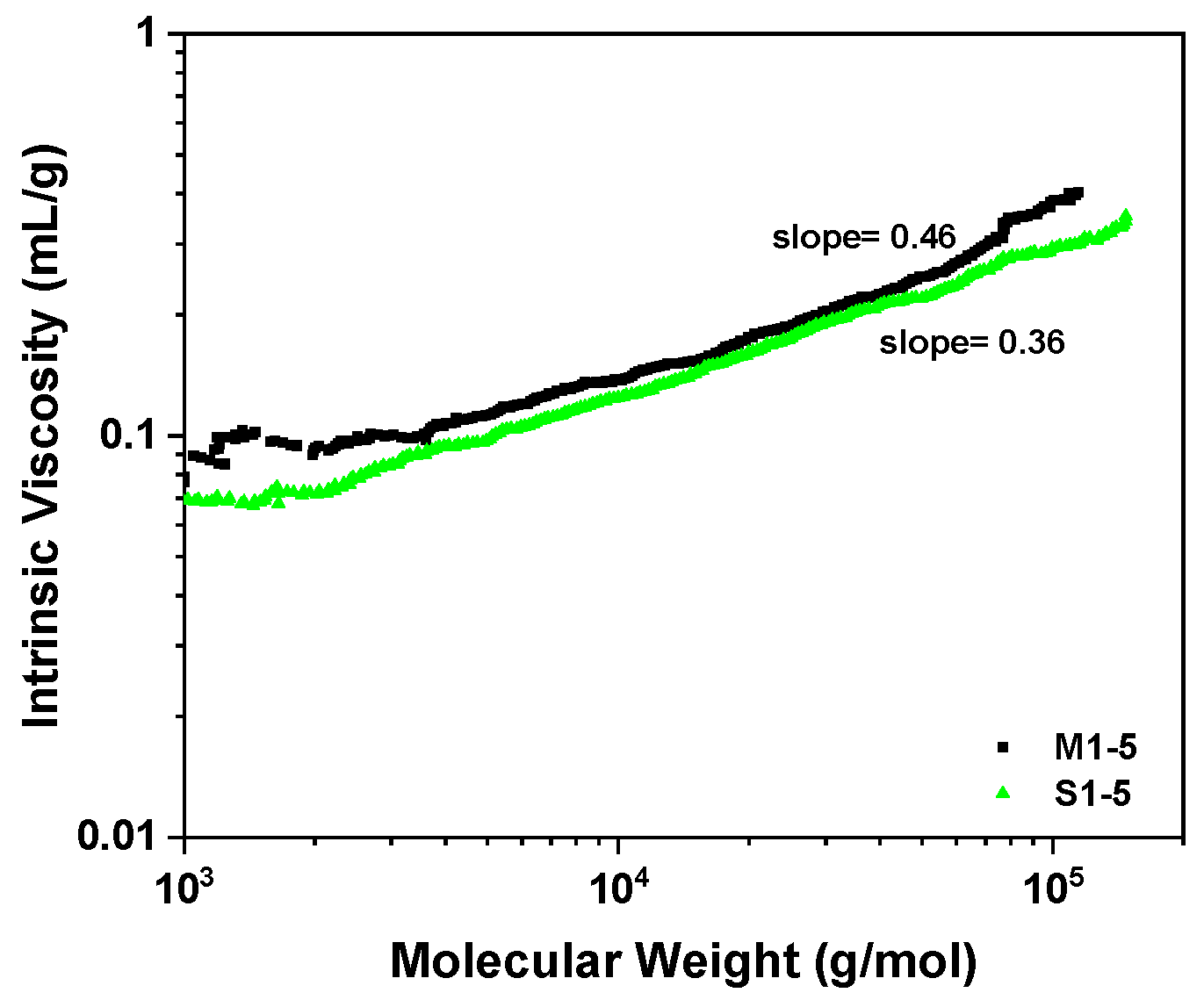
2.3. Fabricating i-PP Star Polymer with Hydrolytic Condensation Mechanism
3. Materials and Methods
3.1. Preparation of the Chain Transfer Agent
3.2. Synthesis of Methoxysilane-Terminated iPP
3.3. Synthesis of Star iPP
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Natta, G. Une Nouvelle Classe de Polymeres d’α-Olefines Ayant Une Régularité de Structure Exceptionnelle. J. Polym. Sci. 1955, 16, 143–154. [Google Scholar] [CrossRef]
- Natta, G.; Mazzanti, G.; Longi, P.; Bernardini, F. Isotactic Polymers of Silicon-Containing Vinyl Monomers. J. Polym. Sci. 1958, 31, 181–183. [Google Scholar] [CrossRef]
- Clark, K.J.; Powell, T. Polymers of Halogen-Substituted 1-Olefins. Polymer 1965, 6, 531–534. [Google Scholar] [CrossRef]
- Chung, T.C. Synthesis of Polyalcohols via Ziegler-Natta Polymerization. Macromolecules 1988, 21, 865–869. [Google Scholar] [CrossRef]
- Galli, P.; Vecellio, G. Polyolefins: The Most Promising Large-Volume Materials for the 21st Century. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 396–415. [Google Scholar] [CrossRef]
- Corradini, P. The Discovery of Isotactic Polypropylene and Its Impact on Pure and Applied Science. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 391–395. [Google Scholar] [CrossRef]
- Chung, T.C. Functionalization of Polyolefins; Academic Press: Cambridge, MA, USA, 2002; ISBN 0121746518. [Google Scholar]
- Boffa, L.S.; Novak, B.M. Copolymerization of Polar Monomers with Olefins Using Transition-Metal Complexes. Chem. Rev. J. 2000, 100, 1479–1494. [Google Scholar] [CrossRef]
- Kesti, M.R.; Coates, G.W.; Waymouth, R.M. Homogeneous Ziegler-Natta Polymerization of Functionalized Monomers Catalyzed by Cationic Group IV Metallocenes. J. Am. Chem. Soc. 1992, 114, 9679–9680. [Google Scholar] [CrossRef]
- Aaltonen, P.; Loefgren, B. Synthesis of Functional Polyethylenes with Soluble Metallocene/Methylaluminoxane Catalyst. Macromolecules 1995, 28, 5353–5357. [Google Scholar] [CrossRef]
- Wilen, C.-E.; Nasman, J.H. Polar Activation in Copolymerization of Propylene and 6-Tert-Butyl-[2-(1,1-Dimethylhept-6-Enyl)]-4-Methylphenol over a Racemic [1,1′-(Dimethylsilylene)Bis(.Eta.5-4,5,6,7-Tetrahydro-1-Indenyl)]Zirconium Dichloride/Methylalumoxane Catalyst System. Macromolecules 1994, 27, 4051–4057. [Google Scholar] [CrossRef]
- Rix, F.C.; Brookhart, M.; White, P.S. Mechanistic Studies of the Palladium (II)-Catalyzed Copolymerization of Ethylene with Carbon Monoxide. J. Am. Chem. Soc. 1996, 118, 4746–4764. [Google Scholar]
- Gao, H.; Matyjaszewski, K. Synthesis of Functional Polymers with Controlled Architecture by CRP of Monomers in the Presence of Cross-Linkers: From Stars to Gels. Prog. Polym. Sci. 2009, 34, 317–350. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with Complex Architecture by Living Anionic Polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef] [PubMed]
- Goh, T.K.; Coventry, K.D.; Blencowe, A.; Qiao, G.G. Rheology of Core Cross-Linked Star Polymers. Polymer 2008, 49, 5095–5104. [Google Scholar] [CrossRef]
- Snijkers, F.; Cho, H.Y.; Nese, A.; Matyjaszewski, K.; Pyckhout-Hintzen, W.; Vlassopoulos, D. Effects of Core Microstructure on Structure and Dynamics of Star Polymer Melts: From Polymeric to Colloidal Response. Macromolecules 2014, 47, 5347–5356. [Google Scholar]
- Chung, T. Functional Polyolefins for Energy Applications. Macromolecules 2013, 46, 6671–6698. [Google Scholar] [CrossRef]
- Liu, P.; Landry, E.; Ye, Z.; Joly, H.; Wang, W.-J.; Li, B.-G. “Arm-First” Synthesis of Core-Cross-Linked Multiarm Star Polyethylenes by Coupling Palladium-Catalyzed Ethylene “Living” Polymerization with Atom-Transfer Radical Polymerization. Macromolecules 2011, 44, 4125–4139. [Google Scholar] [CrossRef]
- Zhang, K.; Ye, Z.; Subramanian, R. A Trinuclear Pd−Diimine Catalyst for “Core-First” Synthesis of Three-Arm Star Polyethylenes via Ethylene “Living” Polymerization. Macromolecules 2009, 42, 2313–2316. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Xu, Z.; Bu, W.; Liu, C.; Dong, J.-Y.; Hu, Y. Synthesis of Low Dispersity Star-like Polyethylene: A Combination of Click Chemistry and a Sol–Gel Process. Polym. Chem. 2014, 5, 3963–3967. [Google Scholar] [CrossRef]
- Huang, H.; Niu, H.; Dong, J.-Y. Synthesis of Star Isotactic Polypropylene Using Click Chemistry. Macromolecules 2010, 43, 8331–8335. [Google Scholar]
- Liu, X.; Niu, H.; Li, Y.; Dong, J.-Y. New Effort to Synthesize Star Isotactic Polypropylene. Polym. Chem. 2018, 9, 3347–3354. [Google Scholar] [CrossRef]
- Dong, J.Y.; Chung, T.C. Synthesis of Polyethylene Containing a Terminal p-Methylstyrene Group: Metallocene-Mediated Ethylene Polymerization with a Consecutive Chain Transfer Reaction to p-Methylstyrene and Hydrogen. Macromolecules 2002, 35, 1622–1631. [Google Scholar] [CrossRef]
- Spaleck, W.; Kueber, F.; Winter, A.; Rohrmann, J.; Bachmann, B.; Antberg, M.; Dolle, V.; Paulus, E.F. The Influence of Aromatic Substituents on the Polymerization Behavior of Bridged Zirconocene Catalysts. Organometallics 1994, 13, 954–963. [Google Scholar] [CrossRef]
- Chadwick, J.C.; Miedema, A.; Sudmeijer, O. Hydrogen Activation in Propene Polymerization with MgCl2-Supported Ziegler-Natta Catalysts: The Effect of the External Donor. Macromol. Chem. Phys. 1994, 195, 167–172. [Google Scholar] [CrossRef]
- Chadwick, J.C.; Van Kessel, G.M.M.; Sudmeijer, O. Regio- and Stereospecificity in Propene Polymerization with MgCl2-Supported Ziegler-Natta Catalysts: Effects of Hydrogen and the External Donor. Macromol. Chem. Phys. 1995, 196, 1431–1437. [Google Scholar] [CrossRef]
- Jüngling, S.; Mülhaupt, R.; Stehling, U.; Brintzinger, H.-H.; Fischer, D.; Langhauser, F. Propene Polymerization Using Homogeneous MAO-Activated Metallocene Catalysts: Me2Si(Benz[e]Indenyl)2ZrCl2/MAO vs. Me2Si(2-Me-Benz[e]Indenyl)2ZrCl2/MAO. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 1305–1317. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Talarico, G.; Caporaso, L. Highly Regioselective Transition-Metal-Catalyzed 1-Alkene Polymerizations: A Simple Method for the Detection and Precise Determination of Regioirregular Monomer Enchainments. Macromolecules 1998, 31, 2387–2390. [Google Scholar] [CrossRef]
- Lin, S.; Waymouth, R.M. Regioirregular Propene Insertion in Polypropenes Synthesized with Unbridged Bis(2-Aryl)Indenyl Zirconium Dichloride Catalysts: Implications on Activity. Macromolecules 1999, 32, 8283–8290. [Google Scholar] [CrossRef]
- Xia, X.; Ye, Z.; Morgan, S.; Lu, J. “Core-First” Synthesis of Multiarm Star Polyethylenes with a Hyperbranched Core and Linear Arms via Ethylene Multifunctional “Living” Polymerization with Hyperbranched Polyethylenes Encapsulating Multinuclear Covalently Tethered Pd-Diimine Catalysts. Macromolecules 2010, 43, 4889–4901. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [M1] or [M2] in Feed (mol/L) | H2 (MPa) | Catalyst Activity (106 g(mol Zr h)−1) | M1 or M2 in PP (mol%) | M1 or M2 Conversion (%) | Tm b (°C) | ΔHm b (J/g) | Tc b (°C) | Mn (g/mol) | PDI c |
|---|---|---|---|---|---|---|---|---|---|---|
| M1-1 | 0.031 | 0 | 2.24 | 0.26 | 9.09 | 140.72 | 78.39 | 109.48 | 6257 | 1.9 |
| M1-2 | 0.031 | 0.005 | 4.67 | 0.34 | 19.72 | 140.21 | 92.08 | 107.47 | 6200 | 2.3 |
| M1-3 | 0.051 | 0.010 | 4.84 | 1.82 | 39.26 | 136.84 | 63.29 | 107.01 | 4454 | 1.9 |
| M1-4 | 0.103 | 0 | 0 | |||||||
| M1-5 | 0.103 | 0.015 | 1.52 | 3.45 | 15.65 | 125.23 | 63.51 | 94.30 | 3449 | 2.0 |
| M1-6 | 0.155 | 0.015 | 0.93 | 6.13 | 13.56 | 122.25 | 54.0 | 92.67 | 2105 | 2.5 |
| M2-1 | 0.029 | 0 | 1.694 | 0.65 | 13.13 | 137.02 | 54.18 | 107.38 | 5534 | 2.4 |
| M2-2 | 0.029 | 0.005 | 2.305 | 0.85 | 23.39 | 137.00 | 76.01 | 107.67 | 6026 | 2.2 |
| M2-3 | 0.048 | 0.010 | 3.393 | 1.69 | 36.97 | 130.55 | 56.81 | 103.57 | 3974 | 2.0 |
| M2-4 | 0.096 | 0 | 0 | |||||||
| M2-5 | 0.096 | 0.015 | 1.104 | 3.10 | 11.02 | 127.00 | 46.84 | 102.06 | 2768 | 1.9 |
| M2-6 | 0.145 | 0.015 | 0.57 | 4.22 | 5.16 | 120.91 | 17.4 | 99.35 | 1702 | 1.9 |
| Sample | Concentration (mol/L) | H2 (MPa) | Tm b (°C) | ΔHm b (J/g) | Tc b (°C) | Mw c (g/mol) | Mn c (g/mol) | PDI c | Mp c (g/mol) | farm | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M1 | M1-5 | 0.103 | 0.015 | 122.4 | 41.7 | 93.0 | 7374 | 2105 | 3.5 | 2789 | 1 |
| M1-6 | 0.155 | 0.015 | 120.4 | 58.5 | 91.0 | 5902 | 1929 | 3.0 | 2742 | 1 | |
| S1-5 a | 0.103 | 0.015 | 131.2 | 61.9 | 102.3 | 13,608 | 4232 | 3.2 | 11,598 | 4.2 | |
| S1-6 a | 0.155 | 0.015 | 126.9 | 73.1 | 96.1 | 16,795 | 4757 | 3.5 | 9048 | 3.3 | |
| M2 | M2-4 | 0.045 | 0.015 | 127.0 | 32.0 | 102.0 | 4430 | 2768 | 1.9 | 2503 | 1 |
| M2-5 | 0.096 | 0.015 | 120.9 | 17.4 | 99.3 | 3321 | 1702 | 1.9 | 1300 | 1 | |
| S2-4 a | 0.045 | 0.015 | 128.1 | 32.7 | 103.0 | 8068 | 4217 | 1.9 | 6802 | 2.7 | |
| S2-5 a | 0.096 | 0.015 | 124.1 | 18.7 | 100.6 | 8660 | 4947 | 1.7 | 6459 | 4.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jar, N.; Chen, F.; Dong, J.-Y. Synthesis of Star Isotactic Polypropylene via Styryldichlorosilane/Hydrogen Consecutive Chain Transfer Reaction. Catalysts 2025, 15, 331. https://doi.org/10.3390/catal15040331
Jar N, Chen F, Dong J-Y. Synthesis of Star Isotactic Polypropylene via Styryldichlorosilane/Hydrogen Consecutive Chain Transfer Reaction. Catalysts. 2025; 15(4):331. https://doi.org/10.3390/catal15040331
Chicago/Turabian StyleJar, Naw, Fengtao Chen, and Jin-Yong Dong. 2025. "Synthesis of Star Isotactic Polypropylene via Styryldichlorosilane/Hydrogen Consecutive Chain Transfer Reaction" Catalysts 15, no. 4: 331. https://doi.org/10.3390/catal15040331
APA StyleJar, N., Chen, F., & Dong, J.-Y. (2025). Synthesis of Star Isotactic Polypropylene via Styryldichlorosilane/Hydrogen Consecutive Chain Transfer Reaction. Catalysts, 15(4), 331. https://doi.org/10.3390/catal15040331