
Transition Metal-Promoted LDH-Derived CoCeMgAlO Mixed Oxides as Active Catalysts for Methane Total Oxidation

 ,
,  ,
, 

Abstract

1. Introduction
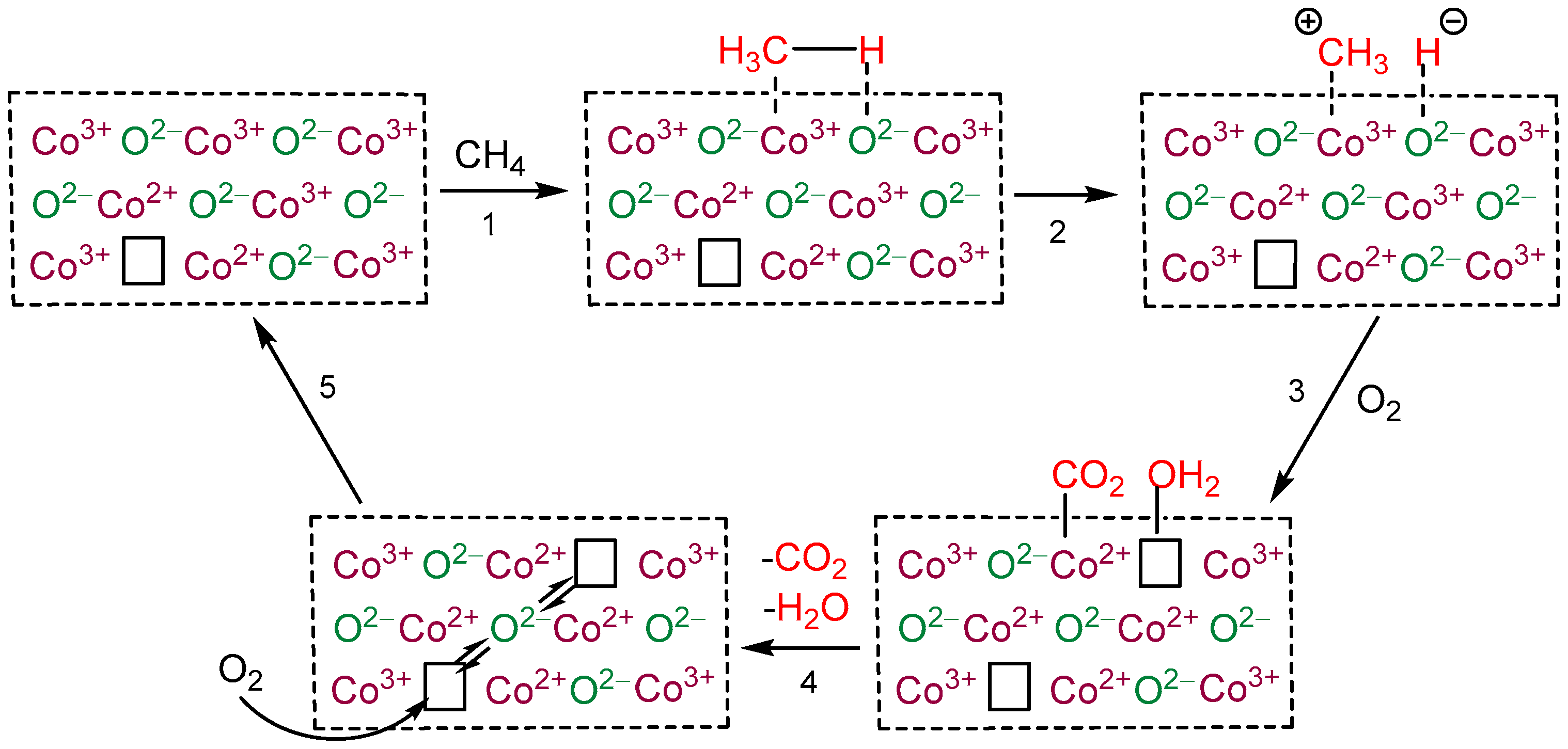
2. Results and Discussion
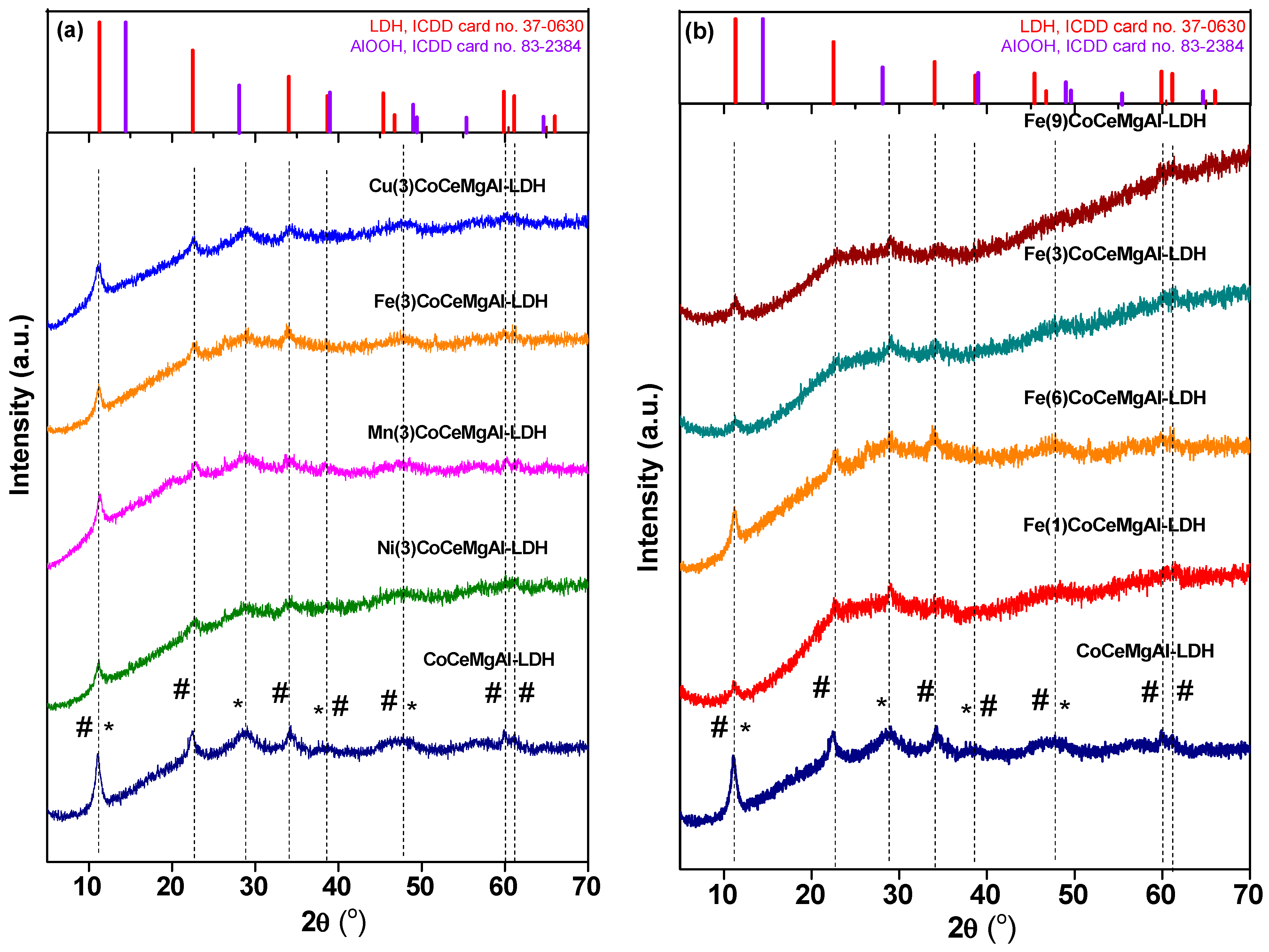
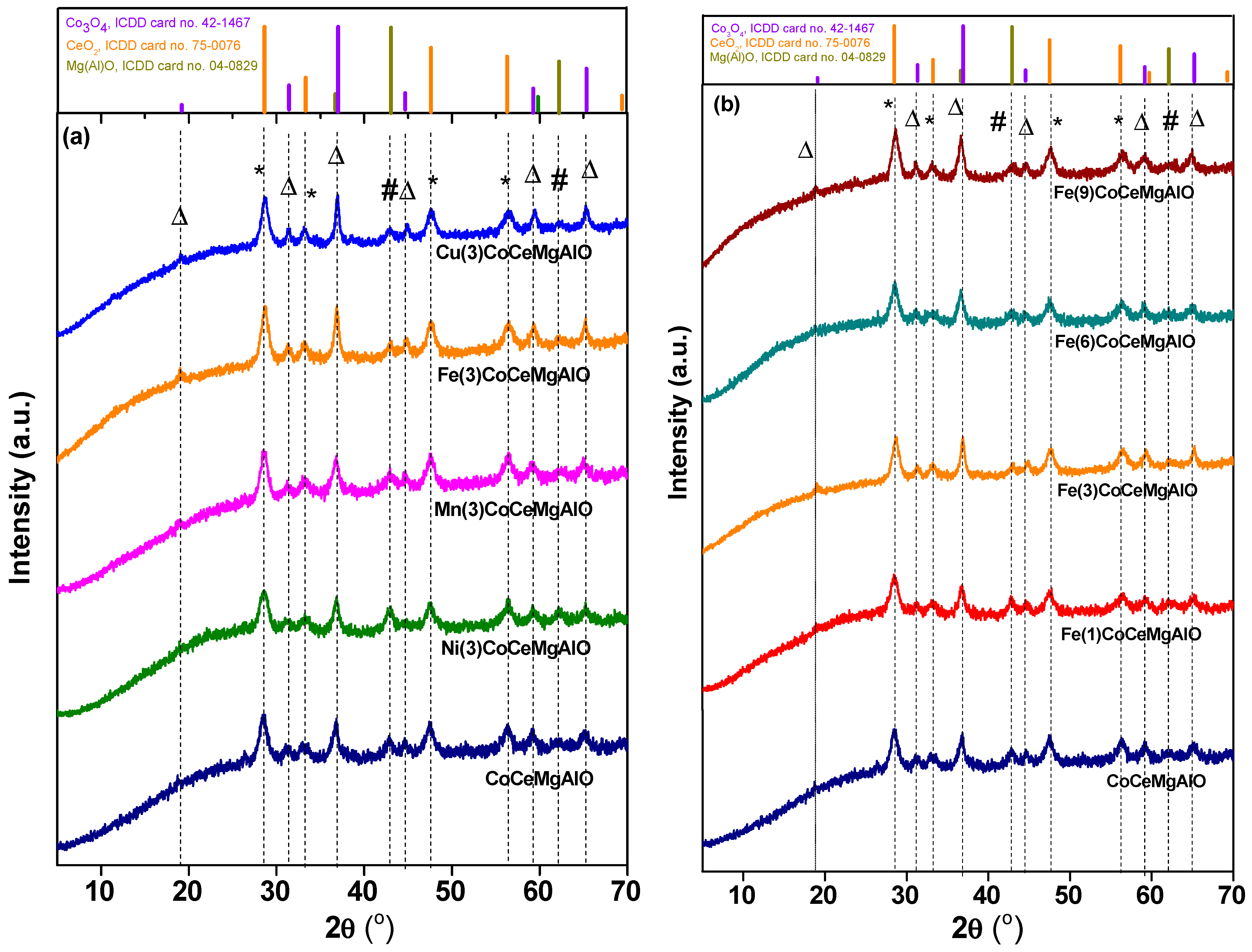
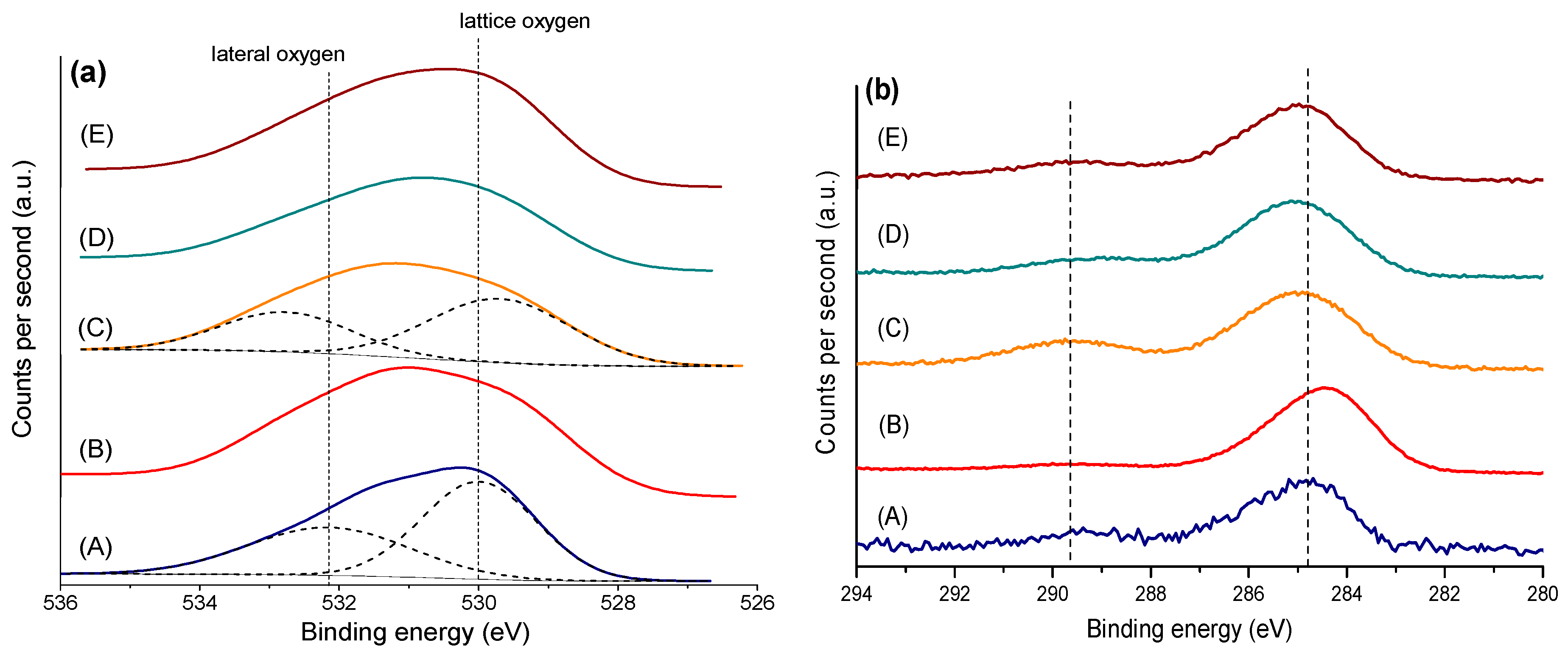
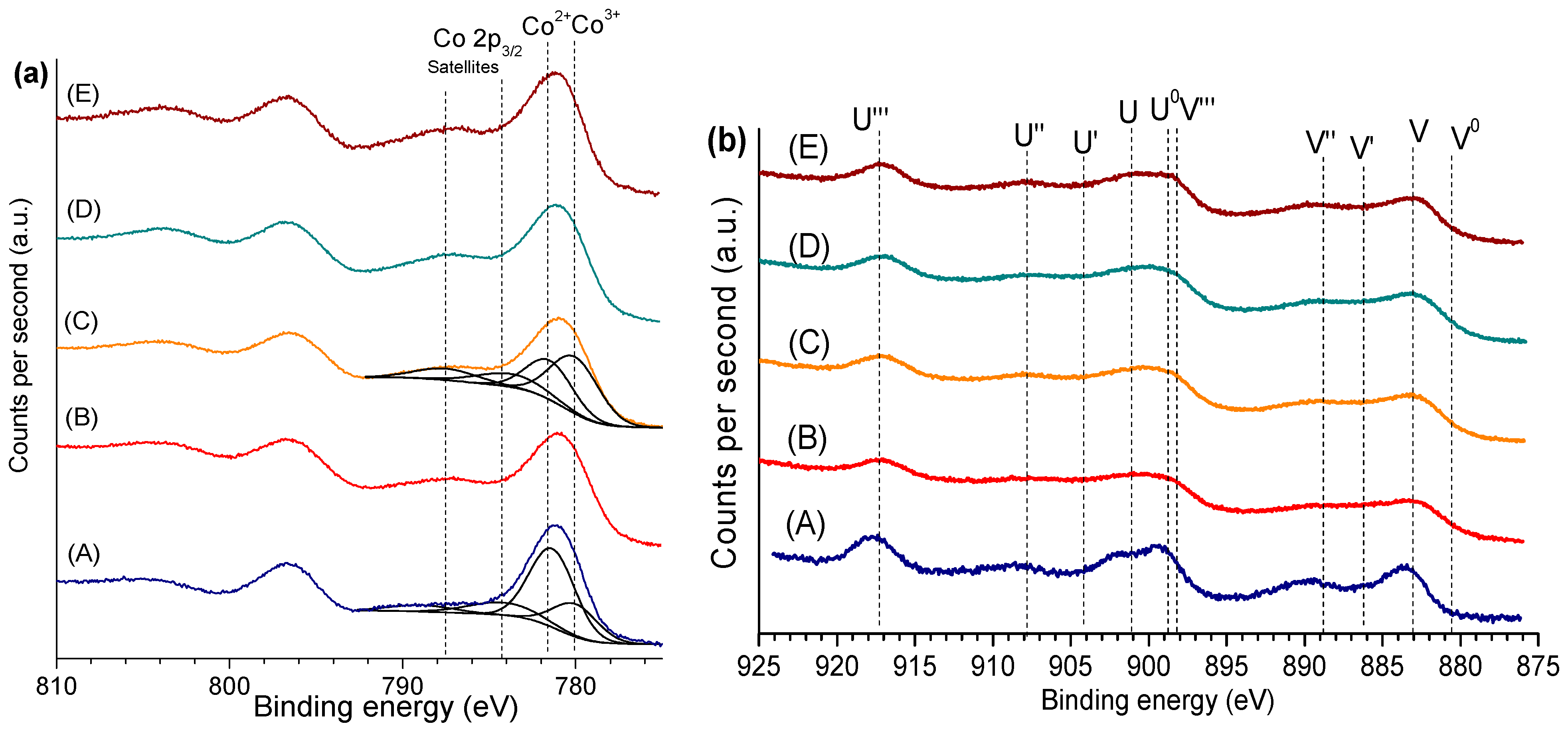
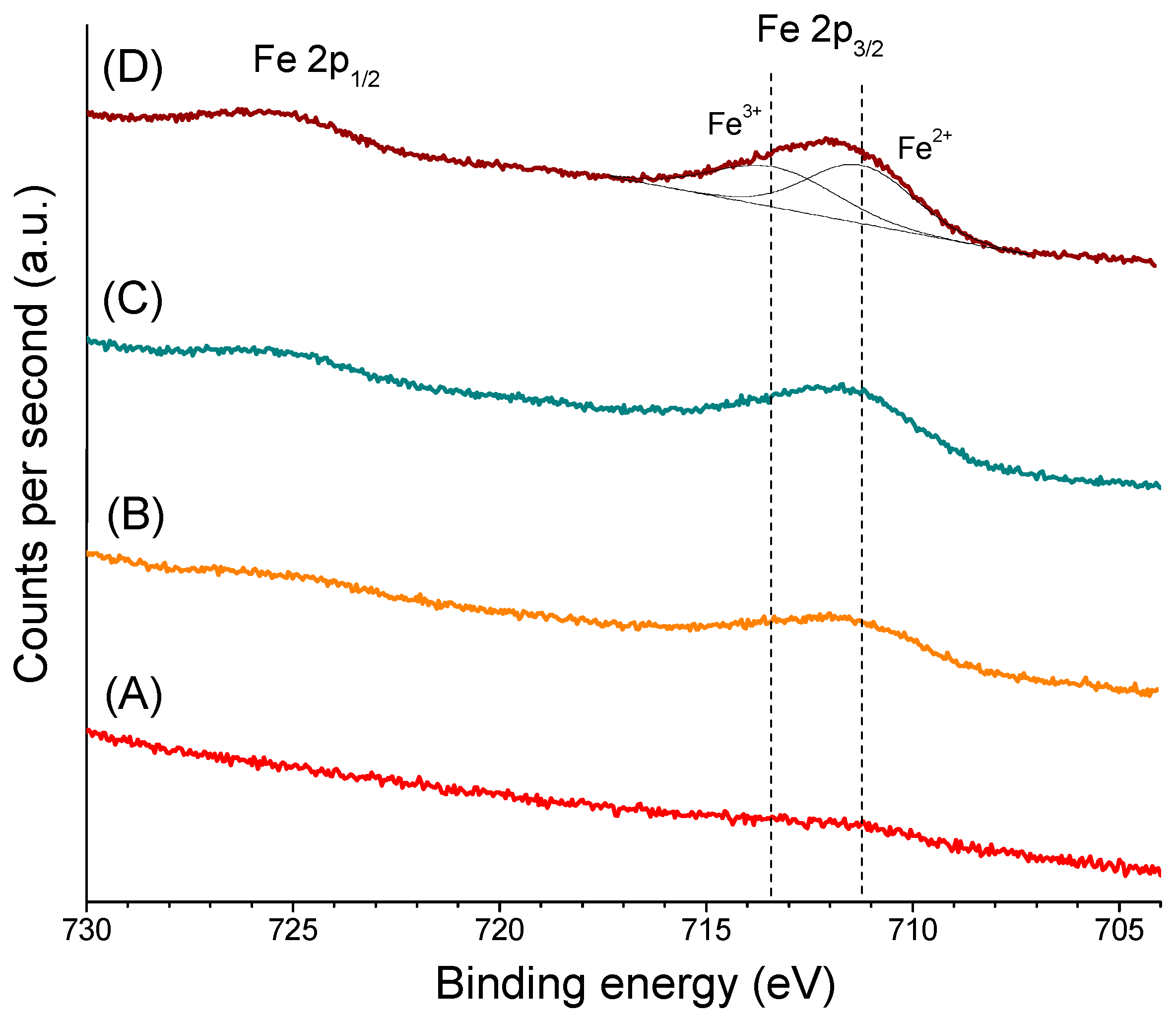
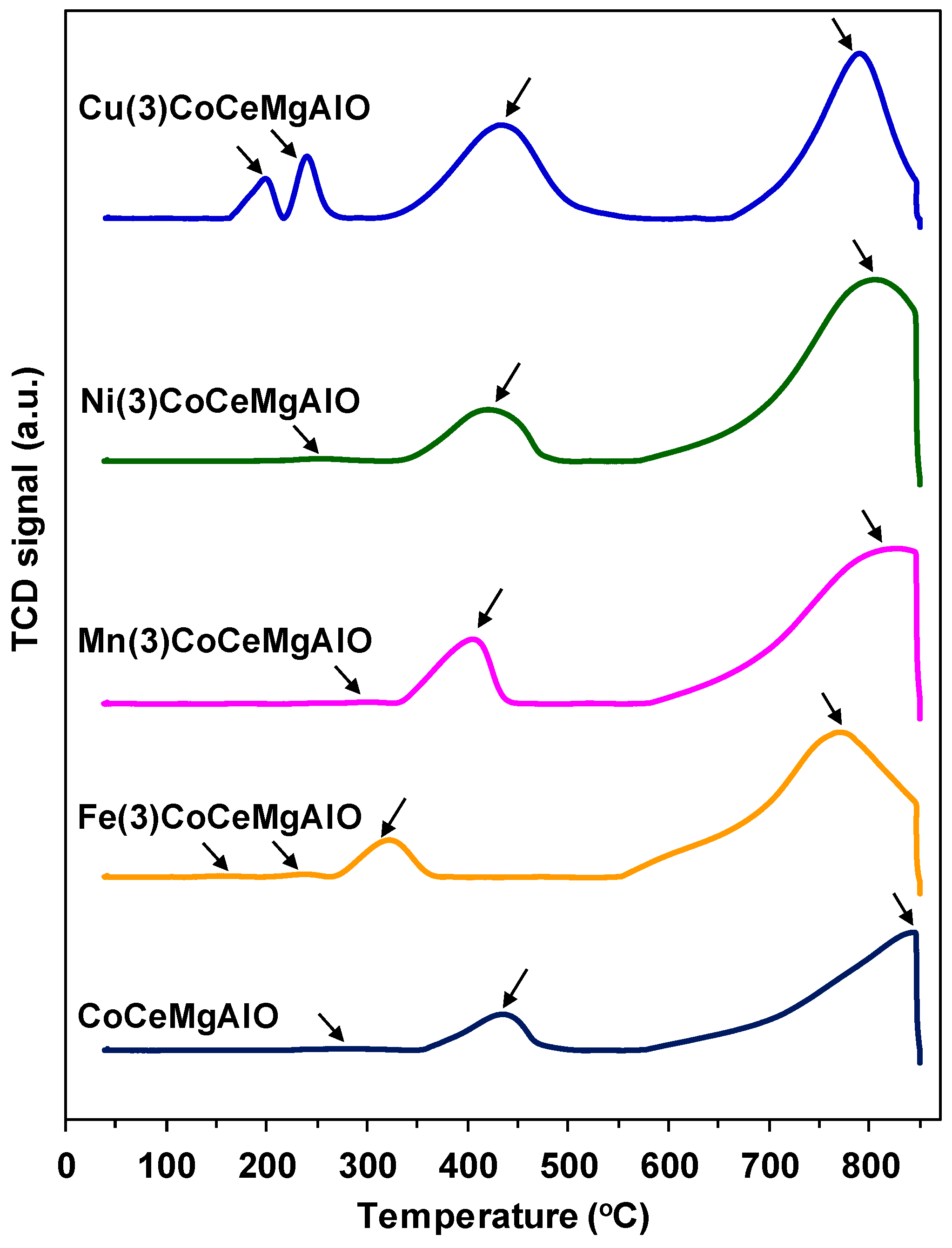
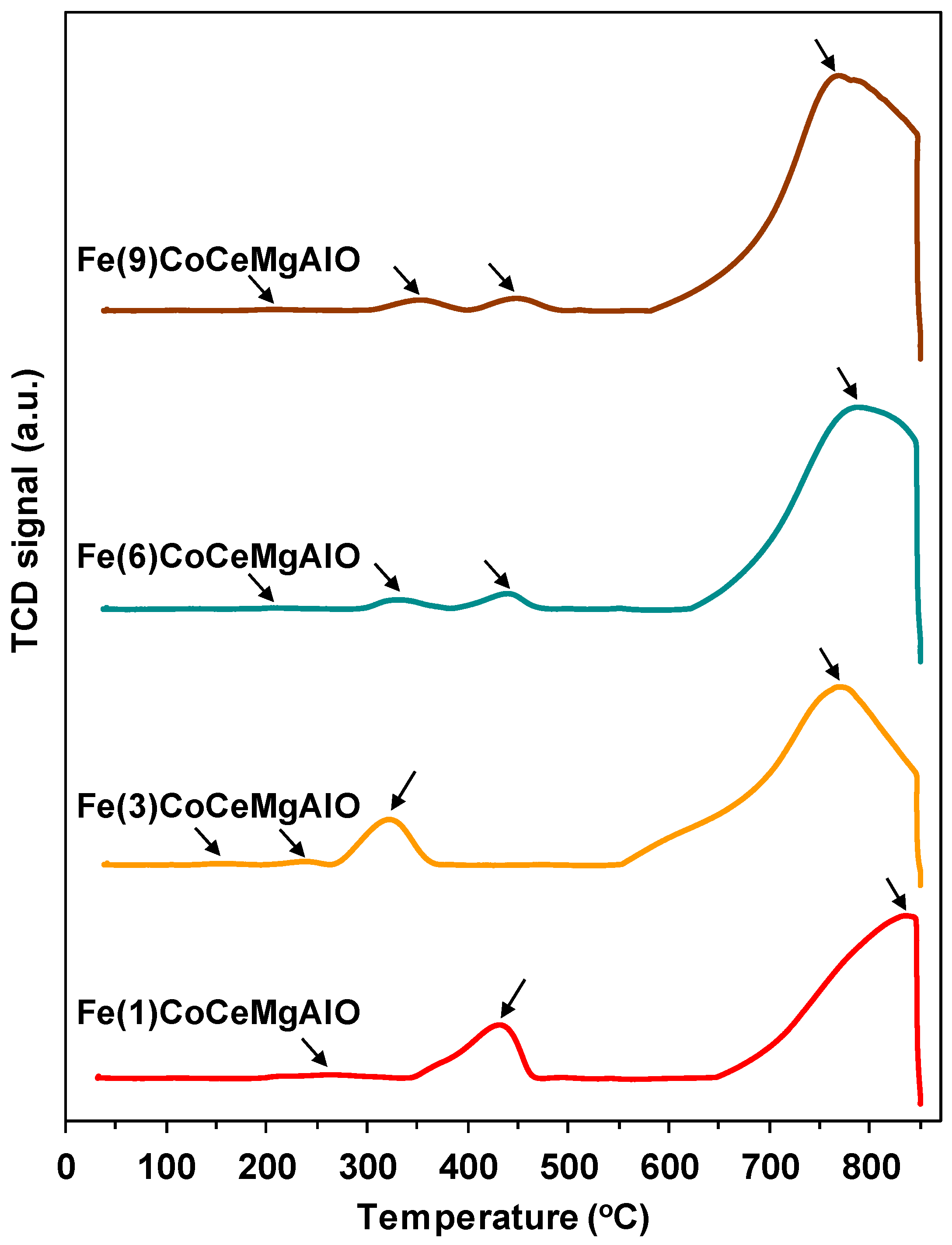
2.1. Catalysts Characterization
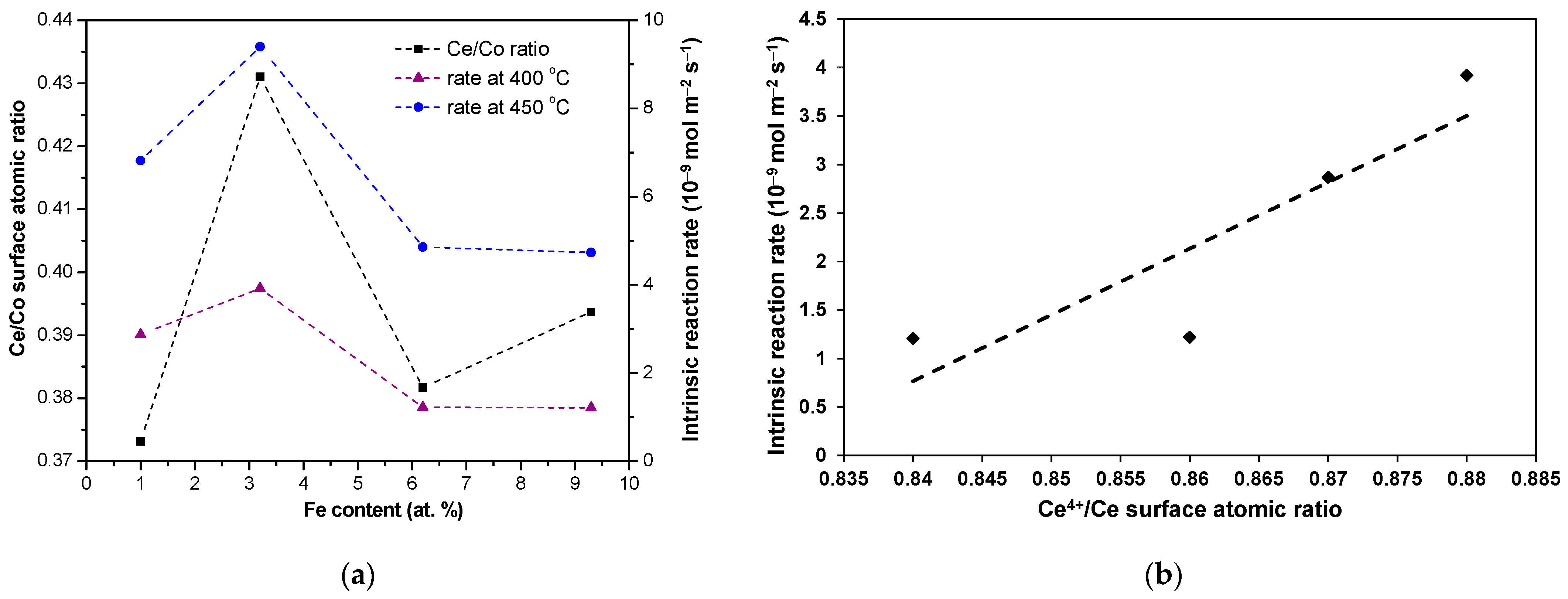
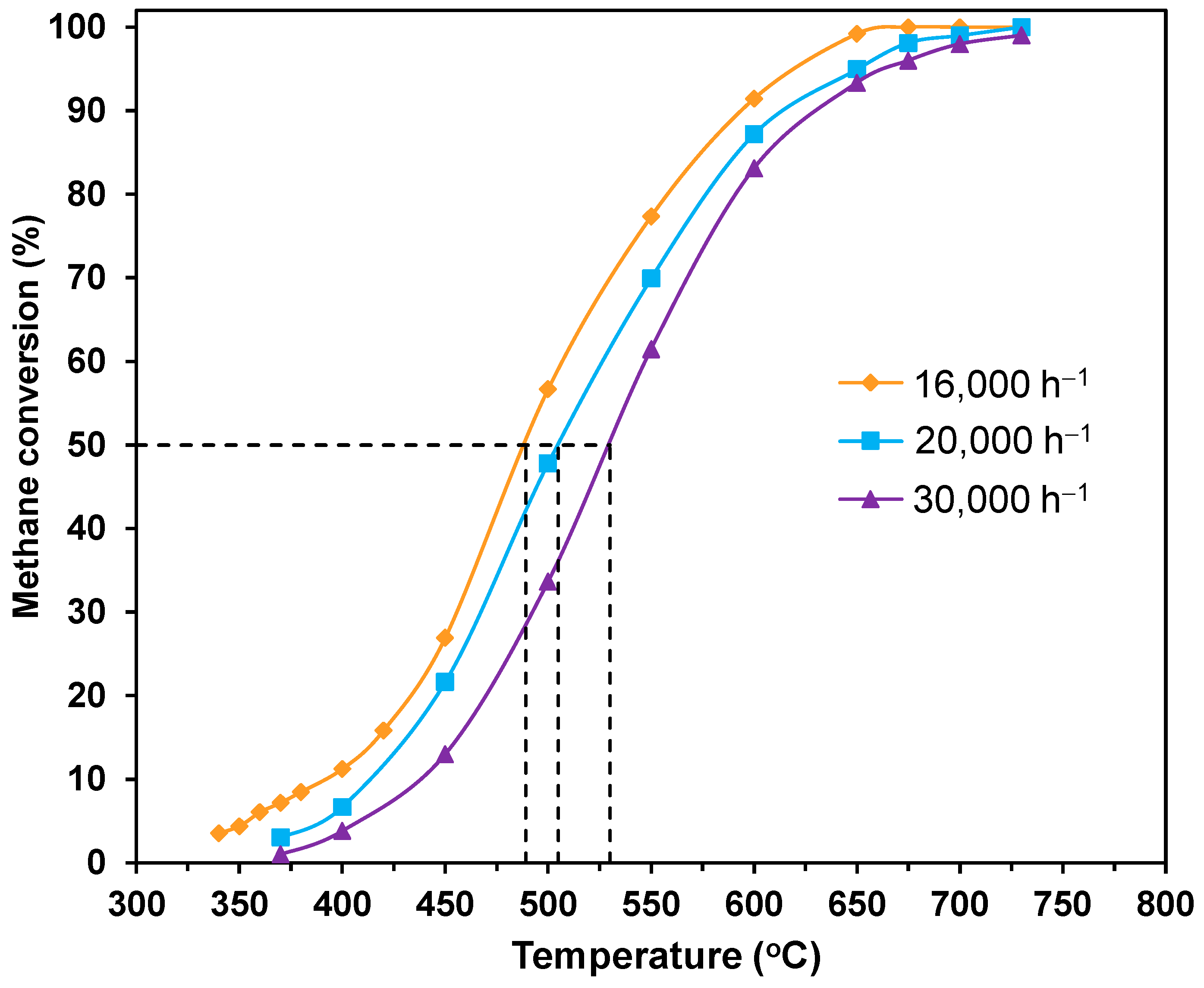
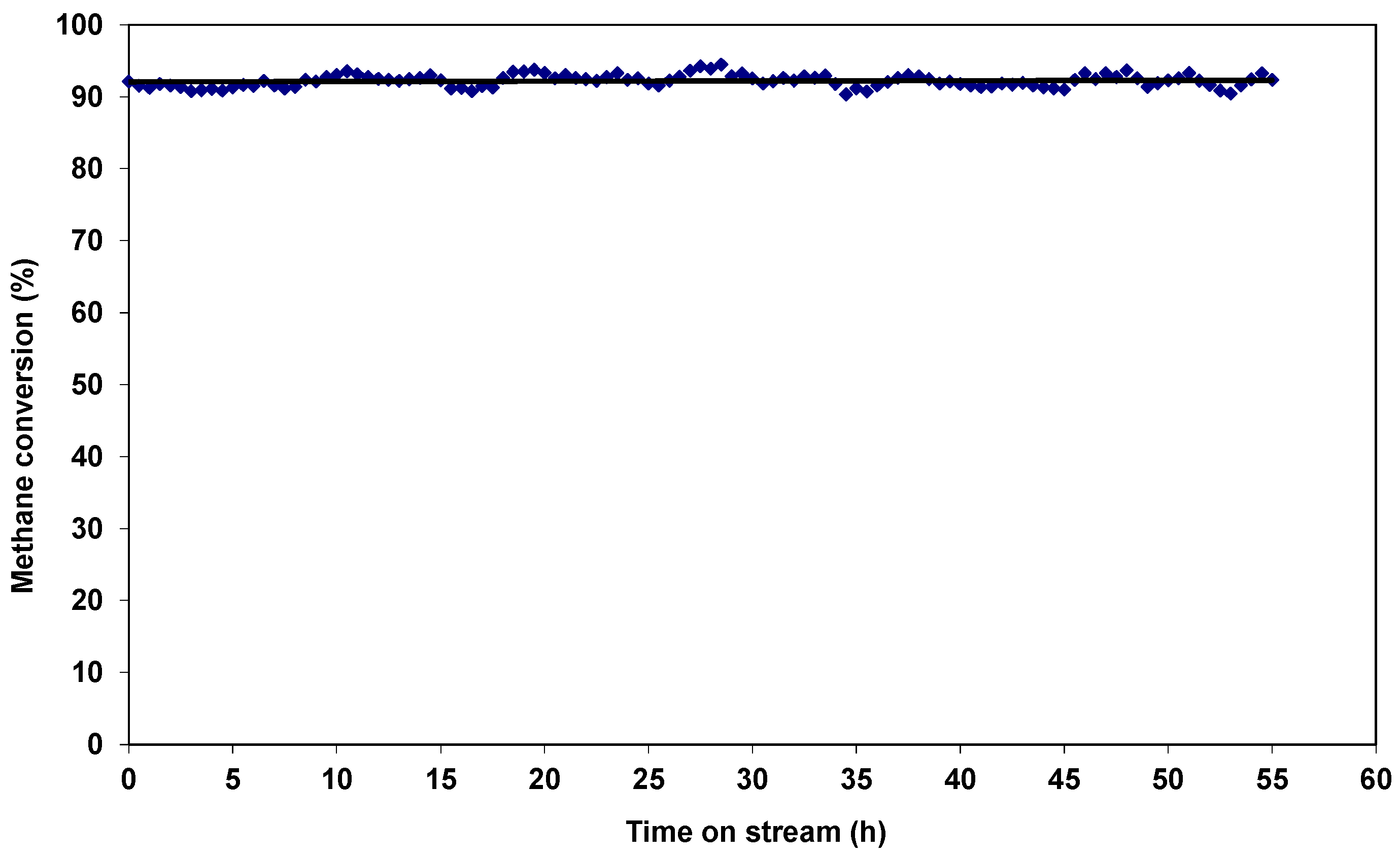
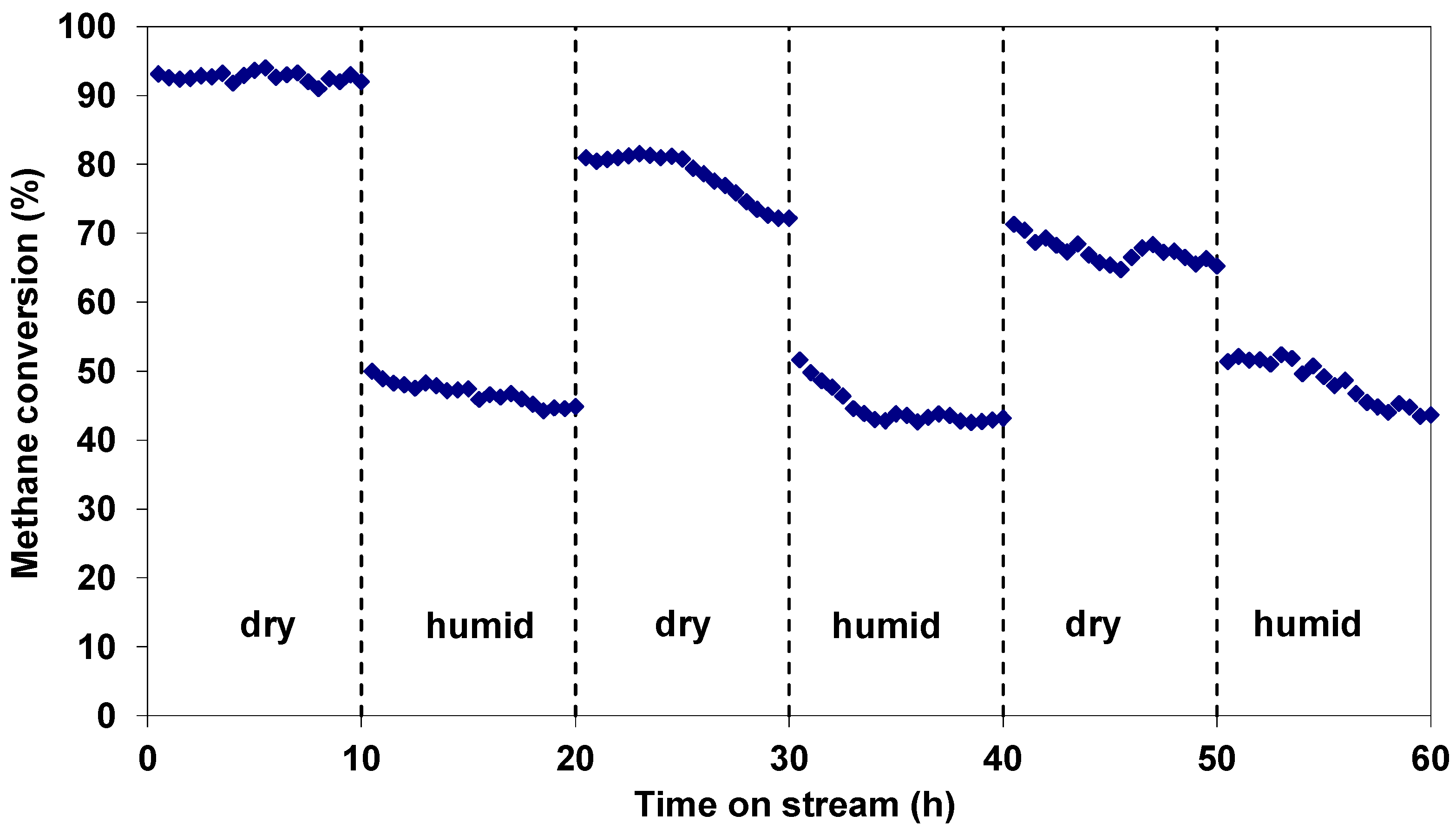
2.2. Catalytic Properties
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Catalysts Characterization
3.3. Catalytic Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kuśtrowski, P.; Rokicińska, A.; Kondratowicz, T. Abatement of Volatile Organic Compounds Emission as a Target for Various Human Activities Including Energy Production. Adv. Inorg. Chem. 2018, 72, 385–419. [Google Scholar] [CrossRef]
- Spivey, J.J. Complete Catalytic Oxidation of Volatile Organics. Ind. Eng. Chem. Res. 1987, 26, 2165–2180. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N. Tropospheric Air Pollution: Ozone, Airborne Toxics, Polycyclic Aromatic Hydrocarbons, and Particles. Science 1997, 276, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Sicard, P.; Agathokleous, E.; De Marco, A.; Paoletti, E.; Calatayud, V. Urban Population Exposure to Air Pollution in Europe over the Last Decades. Environ. Sci. Eur. 2021, 33, 28. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.S.; Razzak, S.A.; Hossain, M.M. Catalytic Oxidation of Volatile Organic Compounds (VOCs)—A Review. Atmos. Environ. 2016, 140, 117–134. [Google Scholar] [CrossRef]
- Moro-Oka, Y.; Morikawa, Y.; Ozaki, A. Regularity in the Catalytic Properties of Metal Oxides in Hydrocarbon Oxidation. J. Catal. 1967, 7, 23–32. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic Combustion of Methane over Palladium-Based Catalysts. Catal. Rev. Sci. Eng. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Martinez-Ortega, F.; Batiot-Dupeyrat, C.; Valderrama, G.; Tatibouët, J.M. Methane Catalytic Combustion on La-Based Perovskite Catalysts. Comptes Rendus De L’academie Des Sci.—Ser. IIc Chem. 2001, 4, 49–55. [Google Scholar] [CrossRef]
- Pengpanich, S.; Meeyoo, V.; Rirksomboon, T.; Bunyakiat, K. Catalytic Oxidation of Methane over CeO2-ZrO2 Mixed Oxide Solid Solution Catalysts Prepared via Urea Hydrolysis. Appl. Catal. A Gen. 2002, 234, 221–233. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for Combustion of Methane and Lower Alkanes. Appl. Catal. A Gen. 2002, 234, 1–23. [Google Scholar] [CrossRef]
- Liotta, L.F. Catalytic Oxidation of Volatile Organic Compounds on Supported Noble Metals. Appl. Catal. B 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent Advances in Catalysts for Methane Combustion. Catal. Surv. Asia 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Dalla Betta, R.A. Catalytic Combustion Gas Turbine Systems: The Preferred Technology for Low Emissions Electric Power Production and Co-Generation. Catal. Today 1997, 35, 129–135. [Google Scholar] [CrossRef]
- Spivey, J.J.; Butt, J.B. Literature Review: Deactivation of Catalysts in the Oxidation of Volatile Organic Compounds. Catal. Today 1992, 11, 465–500. [Google Scholar] [CrossRef]
- Craciun, R.; Nentwick, B.; Hadjiivanov, K.; Knözinger, H. Structure and Redox Properties of MnOx/Yttrium-Stabilized Zirconia (YSZ) Catalyst and Its Used in CO and CH4 Oxidation. Appl. Catal. A Gen. 2003, 243, 67–79. [Google Scholar] [CrossRef]
- Morales, M.; Barbero, B.; Cadus, L. Total Oxidation of Ethanol and Propane over Mn-Cu Mixed Oxide Catalysts. Appl. Catal. B 2006, 67, 229–236. [Google Scholar] [CrossRef]
- Bahlawane, N. Kinetics of Methane Combustion over CVD-Made Cobalt Oxide Catalysts. Appl. Catal. B 2006, 67, 168–176. [Google Scholar] [CrossRef]
- Waqas, M.; El Kasmi, A.; Wang, Y.; Mountapmbeme Kouotou, P.; Tian, Z.Y. CVD Synthesis of Cu-Doped Cobalt Spinel Thin Film Catalysts for Kinetic Study of Propene Oxidation. Colloids Surf. A Physicochem. Eng. Asp. 2018, 556, 195–200. [Google Scholar] [CrossRef]
- Ma, Z. Cobalt Oxide Catalysts for Environmental Remediation. Curr. Catal. 2014, 3, 15–26. [Google Scholar] [CrossRef]
- Li, S.; Mo, S.; Wang, D.; Wu, X.; Chen, Y. Synergistic Effect for Promoted Benzene Oxidation over Monolithic CoMnAlO Catalysts Derived from in Situ Supported LDH Film. Catal. Today 2019, 332, 132–138. [Google Scholar] [CrossRef]
- Barbosa, A.L.; Herguido, J.; Santamaria, J. Methane Combustion over Unsupported Iron Oxide Catalysts. Catal. Today 2001, 64, 43–50. [Google Scholar] [CrossRef]
- Soltan, W.B.; Sun, J.; Wang, W.; Song, Z.; Zhao, X.; Mao, Y.; Zhang, Z. Discovering the Key Role of MnO2 and CeO2 Particles in the Fe2O3 Catalysts for Enhancing the Catalytic Oxidation of VOC: Synergistic Effect of the Lattice Oxygen Species and Surface-Adsorbed Oxygen. Sci. Total Environ. 2022, 819, 152844. [Google Scholar] [CrossRef]
- Zedan, A.F.; AlJaber, A.S. Combustion Synthesis of Non-Precious CuO-CeO2 Nanocrystalline Catalysts with Enhanced Catalytic Activity for Methane Oxidation. Materials 2019, 16, 878. [Google Scholar] [CrossRef]
- Al-Aani, H.M.S.; Iro, E.; Chirra, P.; Fechete, I.; Badea, M.; Negrilă, C.; Popescu, I.; Olea, M.; Marcu, I.C. CuxCeMgAlO Mixed Oxide Catalysts Derived from Multicationic LDH Precursors for Methane Total Oxidation. Appl. Catal. A Gen. 2019, 586, 4–12. [Google Scholar] [CrossRef]
- Darda, S.; Pachatouridou, E.; Lappas, A.; Iliopoulou, E. Effect of Preparation Method of Co-Ce Catalysts on CH4 Combustion. Catalysts 2019, 9, 219. [Google Scholar] [CrossRef]
- Stoian, M.; Rogé, V.; Lazar, L.; Maurer, T.; Védrine, J.C.; Marcu, I.C.; Fechete, I. Total Oxidation of Methane on Oxide and Mixed Oxide Ceria-containing Catalysts. Catalysts 2021, 11, 427. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, J.; Xie, S.; Wang, Z.; Dai, H. Catalytic Removal of Volatile Organic Compounds Using Ordered Porous Transition Metal Oxide and Supported Noble Metal Catalysts. Cuihua Xuebao/Chin. J. Catal. 2016, 37, 1193–1205. [Google Scholar] [CrossRef]
- Marcu, I.-C.; Urdă, A.; Popescu, I.; Hulea, V. Layered Double Hydroxides-Based Materials as Oxidation Catalysts. In Sustainable Nanosystems Development, Properties, and Applications; Putz, M.V., Mirica, M.C., Eds.; IGI Global: Hershey, PA, USA, 2017; pp. 59–121. [Google Scholar]
- Li, W.B.; Wang, J.X.; Gong, H. Catalytic Combustion of VOCs on Non-Noble Metal Catalysts. Catal. Today 2010, 148, 81–87. [Google Scholar] [CrossRef]
- Rǎciulete, M.; Layrac, G.; Tichit, D.; Marcu, I.C. Comparison of CuxZnAlO Mixed Oxide Catalysts Derived from Multicationic and Hybrid LDH Precursors for Methane Total Oxidation. Appl. Catal. A Gen. 2014, 477, 195–204. [Google Scholar] [CrossRef]
- Lamonier, J.F.; Boutoundou, A.B.; Gennequin, C.; Pérez-Zurita, M.J.; Siffert, S.; Aboukais, A. Catalytic Removal of Toluene in Air over Co-Mn-Al Nano-Oxides Synthesized by Hydrotalcite Route. Catal. Lett. 2007, 118, 165–172. [Google Scholar] [CrossRef]
- Kameda, T.; Takeuchi, H.; Yoshioka, T. Hybrid Inorganic/Organic Composites of Mg-Al Layered Double Hydroxides Intercalated with Citrate, Malate, and Tartrate Prepared by Co-Precipitation. Mater. Res. Bull. 2009, 44, 840–845. [Google Scholar] [CrossRef]
- Quiros, D.C.; Smith, J.; Thiruvengadam, A.; Huai, T.; Hu, S. Greenhouse Gas Emissions from Heavy-Duty Natural Gas, Hybrid, and Conventional Diesel on-Road Trucks during Freight Transport. Atmos. Environ. 2017, 168, 36–45. [Google Scholar] [CrossRef]
- Lopez, M.; Sherwood, O.A.; Dlugokencky, E.J.; Kessler, R.; Giroux, L.; Worthy, D.E.J. Isotopic Signatures of Anthropogenic CH4 Sources in Alberta, Canada. Atmos. Environ. 2017, 164, 280–288. [Google Scholar] [CrossRef]
- O’Malley, A.; Hodnett, B.K. The Influence of Volatile Organic Compound Structure on Conditions Required for Total Oxidation. Catal. Today 1999, 54, 31–38. [Google Scholar] [CrossRef]
- He, L.; Fan, Y.; Bellettre, J.; Yue, J.; Luo, L. A Review on Catalytic Methane Combustion at Low Temperatures: Catalysts, Mechanisms, Reaction Conditions and Reactor Designs. Renew. Sustain. Energy Rev. 2020, 119, 109589. [Google Scholar]
- Stoian, M.; Maurer, T.; Lamri, S.; Fechete, I. Techniques of Preparation of Thin Films: Catalytic Combustion. Catalysts 2021, 11, 1530. [Google Scholar] [CrossRef]
- Tanasoi, S.; Mitran, G.; Tanchoux, N.; Cacciaguerra, T.; Fajula, F.; Sndulescu, I.; Tichit, D.; Marcu, I.C. Transition Metal-Containing Mixed Oxides Catalysts Derived from LDH Precursors for Short-Chain Hydrocarbons Oxidation. Appl. Catal. A Gen. 2011, 395, 78–86. [Google Scholar] [CrossRef]
- Tanasoi, S.; Tanchoux, N.; Urdǎ, A.; Tichit, D.; Sǎndulescu, I.; Fajula, F.; Marcu, I.C. New Cu-Based Mixed Oxides Obtained from LDH Precursors, Catalysts for Methane Total Oxidation. Appl. Catal. A Gen. 2009, 363, 135–142. [Google Scholar] [CrossRef]
- Urdǎ, A.; Popescu, I.; Cacciaguerra, T.; Tanchoux, N.; Tichit, D.; Marcu, I.C. Total Oxidation of Methane over Rare Earth Cation-Containing Mixed Oxides Derived from LDH Precursors. Appl. Catal. A Gen. 2013, 464–465, 20–27. [Google Scholar] [CrossRef]
- Stoian, M.C.; Romaniţan, C.; Crăciun, G.; Culiţă, D.C.; Papa, F.; Badea, M.; Negrilă, C.; Popescu, I.; Marcu, I.C. Multicationic LDH-Derived Co(x)CeMgAlO Mixed Oxide Catalysts for the Total Oxidation of Methane. Appl. Catal. A Gen. 2023, 650, 119001. [Google Scholar] [CrossRef]
- Zhang, S.; Shan, J.J.; Zhu, Y.; Frenkel, A.I.; Patlolla, A.; Huang, W.; Yoon, S.J.; Wang, L.; Yoshida, H.; Takeda, S.; et al. WGS Catalysis and in Situ Studies of CoO1−x, PtCon/Co3O4, and PtmCoM′/CoO1−x Nanorod Catalysts. J. Am. Chem. Soc. 2013, 135, 8283–8293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yu, Y.; Zhou, H.; Huang, W.; Pu, Z. Combustion of Lean Methane over Co3O4 Catalysts Prepared with Different Cobalt Precursors. RSC Adv. 2020, 10, 4490–4498. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Oxidation of Lean Methane over Cobalt Catalysts Supported on Ceria/Alumina. Appl. Catal. A Gen. 2020, 591, 117381. [Google Scholar] [CrossRef]
- Al-Aani, H.M.S.; Trandafir, M.M.; Fechete, I.; Leonat, L.N.; Badea, M.; Negrilă, C.; Popescu, I.; Florea, M.; Marcu, I.C. Highly Active Transition Metal-Promoted Cucemgalo Mixed Oxide Catalysts Obtained from Multicationic Ldh Precursors for the Total Oxidation of Methane. Catalysts 2020, 10, 613. [Google Scholar] [CrossRef]
- Xue, L.; Yuan, C.; Wu, S.; Huang, Z.; Yan, Z.; Streiff, S.; Xu, H.; Shen, W. Preparation of Mn-Doped Co3O4 Catalysts by an Eco-Friendly Solid-State Method for Catalytic Combustion of Low-Concentration Methane. Catalysts 2023, 13, 529. [Google Scholar] [CrossRef]
- Singh, S.A.; Madras, G.; Sreedhar, I. Transition Metal (Ni, Cu and Fe) Substituted Co3O4–ZrO2 Catalysts for Lean Methane Combustion. Top. Catal. 2021, 64, 243–255. [Google Scholar] [CrossRef]
- Suarez Anzorena, R.; Mazan, M.O.; Soldati, A.; Larrondo, S.A. The Effect of Incorporation of Iron in Cerium Oxide Structure on Reducibility and Catalytic Performance for Methane Oxidation in Diluted Streams. Ceram. Int. 2019, 45, 19757–19765. [Google Scholar] [CrossRef]
- Shafaei, A.; Irankhah, A. Evaluation of Co-Fe, Cu-Fe, Ni-Al and Ni-Fe Mixed Oxides in Catalytic Combustion of Methane: Comparison Study and Investigating the Effect of Preparation Method. Mol. Catal. 2023, 538, 112989. [Google Scholar] [CrossRef]
- Li, D.; Li, K.; Xu, R.; Wang, H.; Tian, D.; Wei, Y.; Zhu, X.; Zeng, C.; Zeng, L. Ce1-XFexO2-δ Catalysts for Catalytic Methane Combustion: Role of Oxygen Vacancy and Structural Dependence. Catal. Today 2018, 318, 73–85. [Google Scholar] [CrossRef]
- Li, H.; Lu, G.; Qiao, D.; Wang, Y.; Guo, Y.; Guo, Y. Catalytic Methane Combustion over Co3O4/CeO2 Composite Oxides Prepared by Modified Citrate Sol-Gel Method. Catal. Lett. 2011, 141, 452–458. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Thommes, M. Progress in the Physisorption Characterization of Nanoporous Gas Storage Materials. Engineering 2018, 4, 559–566. [Google Scholar]
- Zhang, H.; Zhang, G.; Bi, X.; Chen, X. Facile Assembly of a Hierarchical Core@shell Fe3O4@CuMgAl-LDH (Layered Double Hydroxide) Magnetic Nanocatalyst for the Hydroxylation of Phenol. J. Mater. Chem. A Mater. 2013, 1, 5934–5942. [Google Scholar] [CrossRef]
- Savova, B.; Filkova, D.; Crişan, D.; Crişan, M.; Rǎileanu, M.; Drǎgan, N.; Galtayries, A.; Védrine, J.C. Neodymium Doped Alkaline-Earth Oxide Catalysts for Propane Oxidative Dehydrogenation. Part I. Catalyst Characterisation. Appl. Catal. A Gen. 2009, 359, 47–54. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Shi, J.; Yuan, Y.; Li, Y.; Zhao, W.; Shi, J. One-Pot Pyrolytic Synthesis of Mesoporous MCo2O4(4.5) (M = Mn, Ni, Fe, Cu) Spinels and Its High Efficient Catalytic Properties for CO Oxidation at Low Temperature. J. Mol. Catal. A Chem. 2014, 390, 97–104. [Google Scholar] [CrossRef]
- Shilina, M.I.; Rostovshchikova, T.N.; Nikolaev, S.A.; Udalova, O.V. Polynuclear Co-Oxo Cations in the Catalytic Oxidation of CO on Co-Modified ZSM-5 Zeolites. Mater. Chem. Phys. 2019, 223, 287–298. [Google Scholar] [CrossRef]
- Burroughs, P.; Hamnett, A.; Orchard, A.F.; Thornton, G. 1686 J.C.S. Dalton. J. Chem. Soc. Dalton Trans. 1976, 17, 1686–1698. [Google Scholar]
- Bêche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS Investigation of Cerium Oxides and Mixed Cerium Oxide (Ce XTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Yu, X.; Li, G. XPS Study of Cerium Conversion Coating on the Anodized 2024 Aluminum Alloy. J. Alloys Compd. 2004, 364, 193–198. [Google Scholar] [CrossRef]
- Schofield, P.F.; Henderson, C.M.B.; Redfern, S.A.T.; van der Laan, G. Cu 2p Absorption Spectroscopy as a Probe for the Site Occupancy of (ZnxCu1−x)WO4 Solid Solution. Phys. Chem. Miner. 1993, 20, 375–381. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Cai, T.; Huang, H.; Deng, W.; Dai, Q.; Liu, W.; Wang, X. Catalytic Combustion of 1,2-Dichlorobenzene at Low Temperature over Mn-Modified Co3O4 Catalysts. Appl. Catal. B 2015, 166–167, 393–405. [Google Scholar] [CrossRef]
- Emamdoust, A.; La Parola, V.; Pantaleo, G.; Testa, M.L.; Farjami Shayesteh, S.; Venezia, A.M. Partial Oxidation of Methane over SiO2 Supported Ni and NiCe Catalysts. J. Energy Chem. 2020, 47, 1–9. [Google Scholar] [CrossRef]
- Zhu, X.; Tao, H.; Li, M. Co-Precipitation Synthesis of Nickel Cobalt Hexacyanoferrate for Binder-Free High-Performance Supercapacitor Electrodes. Int. J. Hydrogen Energy 2020, 45, 14452–14460. [Google Scholar] [CrossRef]
- Luo, D.; Liu, S.; Liu, J.; Zhao, J.; Miao, C.; Ren, J. Catalytic Combustion of Toluene over Cobalt Oxides Supported on Graphitic Carbon Nitride (CoOx/g-C3N4) Catalyst. Ind. Eng. Chem. Res. 2018, 57, 11920–11928. [Google Scholar] [CrossRef]
- Sohn, H.; Celik, G.; Gunduz, S.; Dogu, D.; Zhang, S.; Shan, J.; Tao, F.F.; Ozkan, U.S. Oxygen Mobility in Pre-Reduced Nano- and Macro-Ceria with Co Loading: An AP-XPS, In-Situ DRIFTS and TPR Study. Catal. Lett. 2017, 147, 2863–2876. [Google Scholar] [CrossRef]
- Beckers, J.; Rothenberg, G. Redox Properties of Doped and Supported Copper–Ceria Catalysts. Dalton Trans. 2008, 6573–6578. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. On the Beneficial Effect of MgO Promoter on the Performance of Co3O4/Al2O3 Catalysts for Combustion of Dilute Methane. Appl. Catal. A Gen. 2019, 582, 117099. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; González-Velasco, J.R.; López-Fonseca, R. Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials 2019, 12, 3174. [Google Scholar] [CrossRef] [PubMed]
- Choya, A.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Comparative Study of Strategies for Enhancing the Performance of Co3O4/Al2O3 Catalysts for Lean Methane Combustion. Catalysts 2020, 10, 757. [Google Scholar] [CrossRef]
- Choya, A.; Gudyka, S.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; Kotarba, A.; López-Fonseca, R. Design, Characterization and Evaluation of Ce-Modified Cobalt Catalysts Supported on Alpha Alumina in the Abatement of Methane Emissions from Natural Gas Engines. Appl. Catal. A Gen. 2021, 617, 118105. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, Y.; Fu, J.; Kyzas, G.Z.; Billah, S.M.R.; An, S. Synthesis, Characterization, and Catalytic Evaluation of Co3O4/γ-Al2O3 as Methane Combustion Catalysts: Significance of Co Species and the Redox Cycle. Appl. Catal. B 2015, 168–169, 42–50. [Google Scholar] [CrossRef]
- Jackson, S.D.; Brandreth, B.J.; Winstanley, D. Carbon Monoxide Hydrogenation over Group VIII Metals Supported on Tungsten and Molybdenum Trioxides. Appl. Catal. 1986, 27, 325–333. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takeguchi, T.; Kikuchi, R.; Eguchi, K. Influence of Preparation Method and Additive for Cu-Mn Spinel Oxide Catalyst on Water Gas Shift Reaction of Reformed Fuels. Appl. Catal. A Gen. 2005, 279, 59–66. [Google Scholar] [CrossRef]
- Huang, Y.H.; Wang, S.F.; Tsai, A.P.; Kameoka, S. Reduction Behaviors and Catalytic Properties for Methanol Steam Reforming of Cu-Based Spinel Compounds CuX2O4 (X=Fe, Mn, Al, La). Ceram. Int. 2014, 40, 4541–4551. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Gong, M.; Jiang, H.; Gao, X.; Ma, W.; Luo, J.; Ji, H.; Ge, J.; Jia, S.; et al. Emergent Superconductivity in Topological-Kagome-Magnet/Metal Heterostructures. Nat. Commun. 2023, 14, 6998. [Google Scholar] [CrossRef]
- Mosallanejad, S.; Dlugogorski, B.Z.; Kennedy, E.M.; Stockenhuber, M. On the Chemistry of Iron Oxide Supported on γ-Alumina and Silica Catalysts. ACS Omega 2018, 3, 5362–5374. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Cheng, X.; Ma, C.; Qin, Y. Investigation of SO2 Tolerance of Ce-Modified Activated Semi-Coke Based Catalysts for the NO + CO Reaction. RSC Adv. 2017, 7, 53631–53642. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Oxidation of Residual Methane from VNG Vehicles over Co3O4-Based Catalysts: Comparison among Bulk, Al2O3-Supported and Ce-Doped Catalysts. Appl. Catal. B 2018, 237, 844–854. [Google Scholar] [CrossRef]
- Arnone, S.; Bagnasco, G.; Busca, G.; Lisi, L.; Russo, G.; Turco, M. Catalytic Combustion of Methane over Transition Metal Oxides. Stud. Surf. Sci. Catal. 1998, 119, 65–71. [Google Scholar]
- Ercolino, G.; Stelmachowski, P.; Kotarba, A.; Specchia, S. Reactivity of Mixed Iron–Cobalt Spinels in the Lean Methane Combustion. Top. Catal. 2017, 60, 1370–1379. [Google Scholar] [CrossRef]
- Zasada, F.; Janas, J.; Piskorz, W.; Gorczyńska, M.; Sojka, Z. Total Oxidation of Lean Methane over Cobalt Spinel Nanocubes Controlled by the Self-Adjusted Redox State of the Catalyst: Experimental and Theoretical Account for Interplay between the Langmuir-Hinshelwood and Mars-Van Krevelen Mechanisms. ACS Catal. 2017, 7, 2853–2867. [Google Scholar] [CrossRef]
- Zasada, F.; Gryboś, J.; Budiyanto, E.; Janas, J.; Sojka, Z. Oxygen Species Stabilized on the Cobalt Spinel Nano-Octahedra at Various Reaction Conditions and Their Role in Catalytic CO and CH4 Oxidation, N2O Decomposition and Oxygen Isotopic Exchange. J. Catal. 2019, 371, 224–235. [Google Scholar] [CrossRef]
- Cheng, J.; Yu, J.; Wang, X.; Li, L.; Li, J.; Hao, Z. Novel CH4 Combustion Catalysts Derived from Cu-Co/X-Al (X = Fe, Mn, La, Ce) Hydrotalcite-like Compounds. Energy Fuels 2008, 22, 2131–2137. [Google Scholar] [CrossRef]
- Machej, T.; Serwicka, E.M.; Zimowska, M.; Dula, R.; Michalik-Zym, A.; Napruszewska, B.; Rojek, W.; Socha, R. Cu/Mn-Based Mixed Oxides Derived from Hydrotalcite-like Precursors as Catalysts for Methane Combustion. Appl. Catal. A Gen. 2014, 474, 87–94. [Google Scholar] [CrossRef]
- Jiang, Z.; Hao, Z.; Yu, J.; Hou, H.; Hu, C.; Su, J. Catalytic Combustion of Methane on Novel Catalysts Derived from Cu-Mg/Al-Hydrotalcites. Catal. Lett. 2005, 99, 157–163. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, J.; Cheng, J.; Xiao, T.; Jones, M.O.; Hao, Z.; Edwards, P.P. Catalytic Combustion of Methane over Mixed Oxides Derived from Co-Mg/Al Ternary Hydrotalcites. Fuel Proc. Technol. 2010, 91, 97–102. [Google Scholar] [CrossRef]
- Tao, F.F.; Shan, J.J.; Nguyen, L.; Wang, Z.; Zhang, S.; Zhang, L.; Wu, Z.; Huang, W.; Zeng, S.; Hu, P. Understanding Complete Oxidation of Methane on Spinel Oxides at a Molecular Level. Nat. Commun. 2015, 6, 7798. [Google Scholar] [CrossRef]
- Dai, Y.; Kumar, V.P.; Zhu, C.; Wang, H.; Smith, K.J.; Wolf, M.O.; MacLachlan, M.J. Bowtie-Shaped NiCo2O4 Catalysts for Low-Temperature Methane Combustion. Adv. Funct. Mater. 2019, 29, 1807519. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Surface Area (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size a (nm) | CeO2 Lattice Parameter (nm) | CeO2 Crystallite Size (nm) | Periclase Lattice Parameter (nm) | Periclase Crystallite Size (nm) | Co3O4 Lattice Parameter (nm) | Co3O4 Crystallite Size (nm) |
|---|---|---|---|---|---|---|---|---|---|
| CoCeMgAlO | 92.7 | 0.22 | 22.1 | 0.5416 | 8.0 | 0.4247 | 14.0 | 0.8078 | 13.9 |
| Cu(3)CoCeMgAlO | 40.1 | 0.21 | 13.8 | 0.5408 | 9.0 | 0.4220 | 9.4 | 0.8077 | 17.9 |
| Mn(3)CoCeMgAlO | 71.4 | 0.41 | 22.7 | 0.5403 | 8.6 | 0.4196 | 9.0 | 0.8099 | 9.1 |
| Fe(3)CoCeMgAlO | 62.8 (58.8) b | 0.28 (0.28) b | 17.6 (23.5) b | 0.5408 | 8.6 (9.8) b | 0.4203 | 8.9 (10.0) b | 0.8107 | 15.9 (18.3) b |
| Ni(3)CoCeMgAlO | 67.6 | 0.31 | 14.3 | 0.5409 | 8.0 | 0.4216 | 10.3 | 0.8087 | 13.1 |
| Fe(1)CoCeMgAlO | 74.9 | 0.36 | 17.6 and 11 c | 0.5420 | 7.8 | 0.4205 | 8.1 | 0.8097 | 12.4 |
| Fe(6)CoCeMgAlO | 77.3 | 0.39 | 17.5 | 0.5415 | 8.0 | 0.4213 | 8.0 | 0.8123 | 13.0 |
| Fe(9)CoCeMgAlO | 72.3 | 0.32 | 17.8 | 0.5411 | 8.7 | 0.4251 | 8.7 | 0.8138 | 15.2 |
| Mixed Oxide | Atomic Content (%) a | Atomic Ratio b | ||||||
|---|---|---|---|---|---|---|---|---|
| M | Co | Ce | Mg | Al | Mg/Al | Co/Ce | Co/M | |
| CoCeMgAlO | - | 39.8 | 9.2 | 37.6 | 13.4 | 2.8 | 4.3 | - |
| Cu(3)CoCeMgAlO | 3.0 | 41.8 | 9.9 | 33.2 | 12.1 | 2.7 | 4.2 | 13.9 |
| Mn(3)CoCeMgAlO | 3.0 | 40.0 | 9.4 | 35.0 | 12.6 | 2.8 | 4.2 | 13.3 |
| Fe(3)CoCeMgAlO | 2.6 | 40.5 | 9.3 | 34.6 | 13.0 | 2.7 | 4.3 | 15.6 |
| Ni(3)CoCeMgAlO | 3.6 | 41.1 | 9.7 | 34.1 | 11.5 | 3.0 | 4.2 | 11.4 |
| Fe(1)CoCeMgAlO | 0.6 | 39.5 | 9.1 | 37.7 | 13.1 | 2.9 | 4.3 | 65.8 |
| Fe(6)CoCeMgAlO | 5.7 | 42.6 | 9.6 | 31.2 | 10.9 | 2.9 | 4.4 | 7.5 |
| Fe(9)CoCeMgAlO | 8.2 | 39.6 | 9.3 | 32.0 | 10.9 | 2.9 | 4.2 | 4.8 |
| Mixed Oxide | Atomic Content (%) a | Atomic Ratio | ||||||
|---|---|---|---|---|---|---|---|---|
| M | Co | Ce | Mg | Al | Mg/Al | Co/Ce | Co/M | |
| CoCeMgAlO | - | |||||||
| Cu(3)CoCeMgAlO | 3.1 | 41.4 | 10.0 | 33.4 | 12.1 | 2.8 | 4.1 | 13.3 |
| Mn(3)CoCeMgAlO | 3.1 | 41.5 | 10.3 | 33.1 | 12.0 | 2.8 | 4.0 | 13.4 |
| Fe(3)CoCeMgAlO | 3.2 | 42.0 | 10.8 | 31.9 | 12.1 | 2.6 | 3.9 | 13.1 |
| Ni(3)CoCeMgAlO | 3.0 | 40.2 | 10.1 | 34.9 | 11.8 | 3.0 | 4.0 | 13.4 |
| Fe(1)CoCeMgAlO | 1.0 | 41.1 | 10.1 | 35.2 | 12.6 | 2.8 | 4.1 | 41.1 |
| Fe(6)CoCeMgAlO | 6.2 | 41.1 | 10.1 | 31.4 | 11.2 | 2.8 | 4.1 | 6.6 |
| Fe(9)CoCeMgAlO | 9.3 | 40.7 | 10.2 | 29.3 | 10.5 | 2.8 | 4.0 | 4.4 |
| Sample | Surface Atomic Content (at.%) | Surface Atomic Content with Respect to Cations (at.%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ce | Mg | Al | Co | M | O | C | Co | M | Ce | |
| CoCeMgAlO | 3.2 | 15.9 | 5.7 | 9.2 | - | 54.6 | 11.5 | 27.2 | - | 9.3 |
| Cu(3)CoCeMgAlO | 2.7 | 15.7 | 4.7 | 8.0 | 1.5 | 52.9 | 14.4 | 24.6 | 4.7 | 8.2 |
| Mn(3)CoCeMgAlO | 2.8 | 15.1 | 7.6 | 8.5 | 0.6 | 52.1 | 13.2 | 24.6 | 1.8 | 8.1 |
| Fe(3)CoCeMgAlO | 3.8 | 10.5 | 2.5 | 8.8 | 0.8 | 55.0 | 18.6 | 33.3 | 3.0 | 14.4 |
| Ni(3)CoCeMgAlO | 3.0 | 14.9 | 5.6 | 9.9 | 1.1 | 50.9 | 14.5 | 28.7 | 3.3 | 8.7 |
| Fe(1)CoCeMgAlO | 2.5 | 9.6 | 3.0 | 6.7 | 0.2 | 42.4 | 35.6 | 30.4 | 0.9 | 11.4 |
| Fe(6)CoCeMgAlO | 3.7 | 10.6 | 2.6 | 9.7 | 1.4 | 52.4 | 19.6 | 34.6 | 5.0 | 13.2 |
| Fe(9)CoCeMgAlO | 4.1 | 11.0 | 3.2 | 10.4 | 1.9 | 52.0 | 17.4 | 34.0 | 6.2 | 13.4 |
| Fe(3)CoCeMgAlO-used | 4.7 | 10.8 | 3.1 | 6.1 | 0.6 | 52.9 | 21.8 | 24.1 | 2.4 | 18.6 |
| Sample | Surface Atomic Ratios | |||||
|---|---|---|---|---|---|---|
| Co/Ce | Mg/Al | Co/M | Ce(IV)/Ce | Co(III)/Co(II) | M(n + 1) /M(n) b | |
| CoCeMgAlO | 2.9 | 2.8 | - | 0.94 | 0.3 | - |
| Cu(3)CoCeMgAlO | 3.0 | 3.3 | 5.2 | 0.85 | 2.5 | 0.48 |
| Mn(3)CoCeMgAlO | 3.0 | 2.0 | 13.7 | 0.80 | 1.9 | 0.94 |
| Fe(3)CoCeMgAlO | 2.3 | 4.2 | 11.0 | 0.88 | 1.5 | 0.81 |
| Ni(3)CoCeMgAlO | 3.3 | 2.7 | 8.7 | 0.77 | 1.8 | - |
| Fe(1)CoCeMgAlO | 2.7 | 3.2 | 33.5 | 0.87 | 1.7 | n.d. c |
| Fe(6)CoCeMgAlO | 2.6 | 4.1 | 6.9 | 0.86 | 1.3 | 0.74 |
| Fe(9)CoCeMgAlO | 2.5 | 3.4 | 5.5 | 0.84 | 1.0 | 0.80 |
| Fe(3)CoCeMgAlO-used | 1.3 | 3.5 | 10.2 | 0.86 | 3.1 | 0.75 |
| Catalyst | Experimental H2 Consumption (mmol g−1) | Calculated H2 Consumption (mmol g−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| At Low Temperatures (Peak Maximum, °C) | Total Below 500 °C | At High Temperatures (Peak Maximum, °C) | Total Below 750 °C | Total Below 850 °C | Necessary for (M + Co) Reduction a | Necessary for (M + Co + Ce) Reduction a | |||
| CoCeMgAlO | 0.05 (280) | 0.91 (434) | - | 0.96 | 4.75 (845) | 2.42 | 5.71 | 7.62 | 8.28 |
| Cu(3)CoCeMgAlO | 0.29 (198) | 0.39 (239) | 2.43 (432) | 3.11 | 3.74 (790) | 3.94 | 6.85 | 7.90 | 8.59 |
| Mn(3)CoCeMgAlO | 0.03 (298) | 1.1 (404) | - | 1.13 | 5.84 (825) | 3.03 | 6.97 | 7.88 | 8.59 |
| Fe(3)CoCeMgAlO | 0.01 (157) | 0.03 (239) | 0.56 (322) | 0.60 | 6.12 (769) | 3.48 | 6.73 | 8.13 | 8.86 |
| Ni(3)CoCeMgAlO | 0.04 (254) | 1.04 (420) | - | 1.08 | 5.74 (805) | 2.89 | 6.82 | 7.87 | 8.58 |
| Fe(1)CoCeMgAlO | 0.07 (265) | 0.8 (430) | - | 0.87 | 4.03 (835) | 1.74 | 4.90 | 7.82 | 8.52 |
| Fe(6)CoCeMgAlO | 0.01 (207) | 0.11 (332) | 0.17 (440) | 0.29 | 6.07 (784) | 2.10 | 6.36 | 8.48 | 9.17 |
| Fe(9)CoCeMgAlO | 0.01 (210) | 0.13 (353) | 0.13 (448) | 0.27 | 7.04 (771) | 2.78 | 7.31 | 8.81 | 9.49 |
| Catalyst | T10 (°C) | T50 (°C) | T90 (°C) | Reaction Rate at 400 °C | Reaction Rate at 450 °C | Ea (kJ mol−1) | ||
|---|---|---|---|---|---|---|---|---|
| Specific (107 mol g−1 s−1) | Intrinsic (109 mol m−2 s−1) | Specific (107 mol g−1 s−1) | Intrinsic (109 mol m−2 s−1) | |||||
| CoCeMgAlO | 438 | 528 | 619 | 0.83 | 0.90 | 3.72 | 4.02 | 84.6 |
| Cu(3)CoCeMgAlO | 426 | 514 | 620 | 1.19 | 2.97 | 3.66 | 9.14 | 91.8 |
| Mn(3)CoCeMgAlO | 433 | 521 | 631 | 0.94 | 1.32 | 3.90 | 5.46 | 118.3 |
| Fe(3)CoCeMgAlO | 391 | 489 | 595 | 2.46 | 3.92 | 5.90 | 9.40 | 54.9 |
| Ni(3)CoCeMgAlO | 431 | 507 | 613 | 1.09 | 1.62 | 3.39 | 5.02 | 93.8 |
| Fe(1)CoCeMgAlO | 405 | 495 | 603 | 2.15 | 2.87 | 5.11 | 6.82 | 74.7 |
| Fe(6)CoCeMgAlO | 437 | 523 | 634 | 0.94 | 1.22 | 3.76 | 4.86 | 103.3 |
| Fe(9)CoCeMgAlO | 429 | 521 | 607 | 0.88 | 1.21 | 3.42 | 4.73 | 106.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoian, M.C.; Romanitan, C.; Neubauer, K.; Atia, H.; Negrilă, C.C.; Popescu, I.; Marcu, I.-C. Transition Metal-Promoted LDH-Derived CoCeMgAlO Mixed Oxides as Active Catalysts for Methane Total Oxidation. Catalysts 2024, 14, 625. https://doi.org/10.3390/catal14090625
Stoian MC, Romanitan C, Neubauer K, Atia H, Negrilă CC, Popescu I, Marcu I-C. Transition Metal-Promoted LDH-Derived CoCeMgAlO Mixed Oxides as Active Catalysts for Methane Total Oxidation. Catalysts. 2024; 14(9):625. https://doi.org/10.3390/catal14090625
Chicago/Turabian StyleStoian, Marius C., Cosmin Romanitan, Katja Neubauer, Hanan Atia, Constantin Cătălin Negrilă, Ionel Popescu, and Ioan-Cezar Marcu. 2024. "Transition Metal-Promoted LDH-Derived CoCeMgAlO Mixed Oxides as Active Catalysts for Methane Total Oxidation" Catalysts 14, no. 9: 625. https://doi.org/10.3390/catal14090625
APA StyleStoian, M. C., Romanitan, C., Neubauer, K., Atia, H., Negrilă, C. C., Popescu, I., & Marcu, I.-C. (2024). Transition Metal-Promoted LDH-Derived CoCeMgAlO Mixed Oxides as Active Catalysts for Methane Total Oxidation. Catalysts, 14(9), 625. https://doi.org/10.3390/catal14090625