


Mechanistic and Kinetic Analysis of Complete Methane Oxidation on a Practical PtPd/Al2O3 Catalyst

 ,
,  and
and
Abstract

1. Introduction
2. Results and Discussion
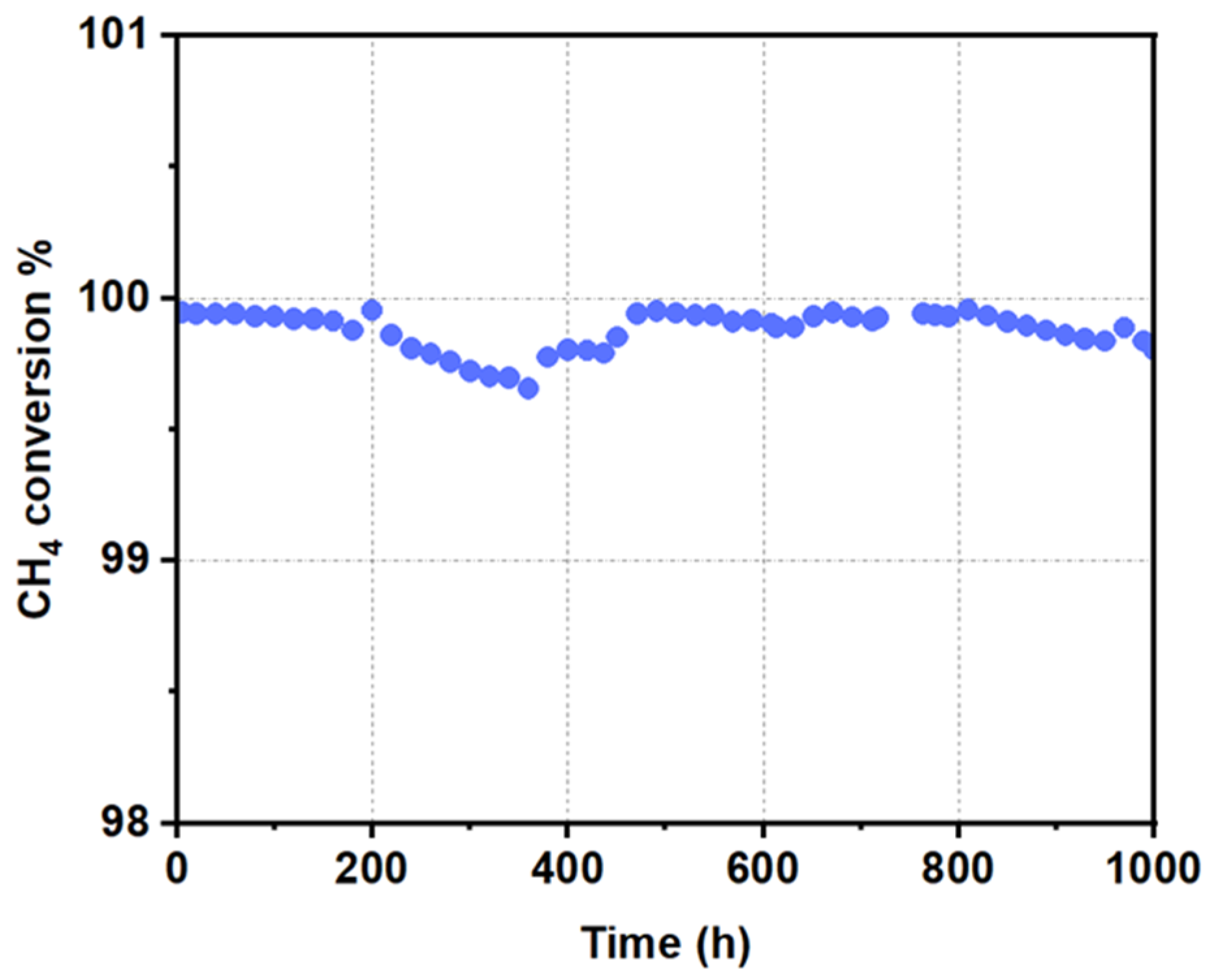
2.1. Assessment of Catalyst Stability Under CH4 Oxidation Conditions
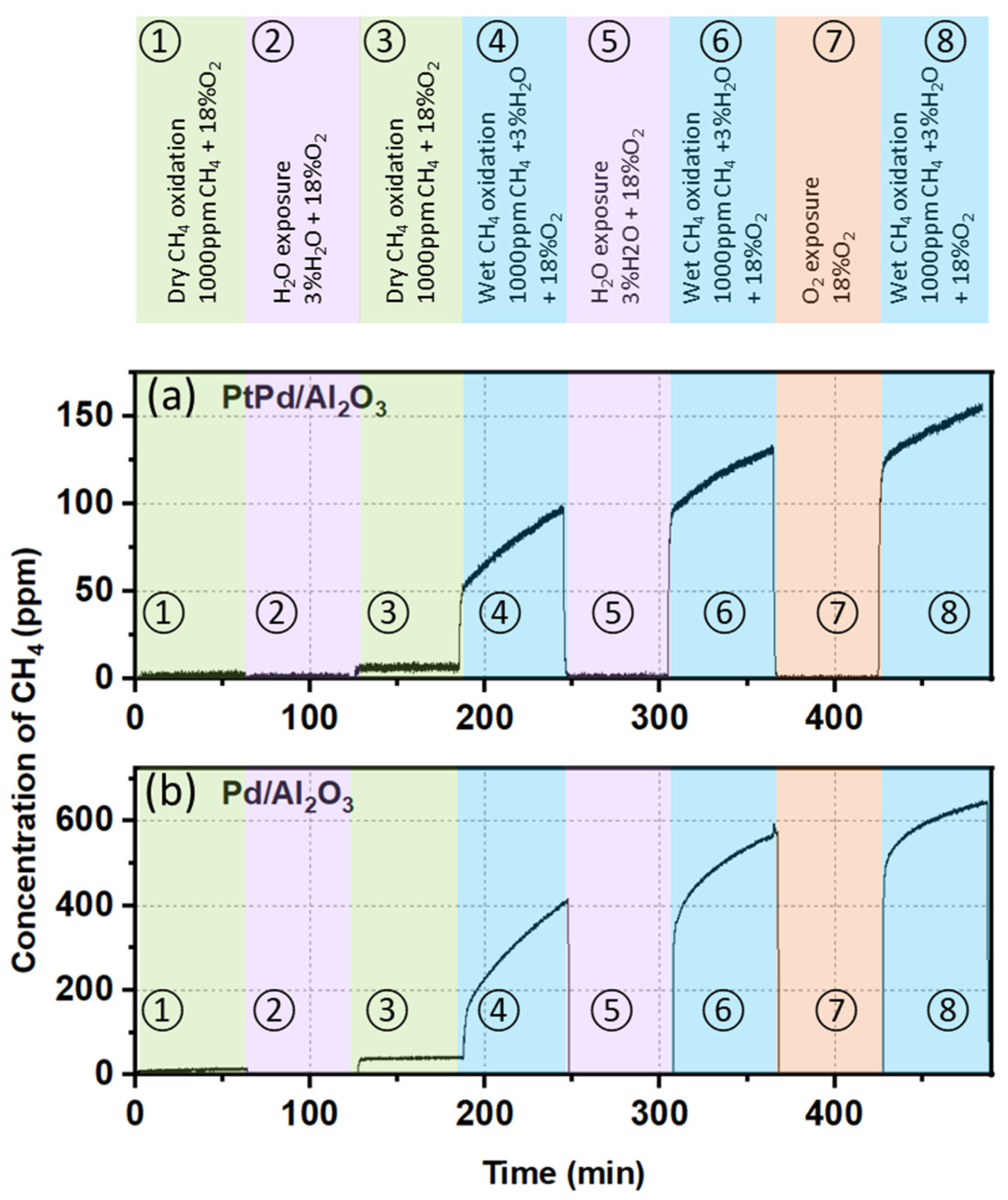
2.2. Mechanistic Analysis of the Reactivity and Stability of the PtPd/Al2O3 Catalyst
2.3. Kinetic Data Analysis
2.3.1. Kinetic Data Analysis by Power Law Expressions
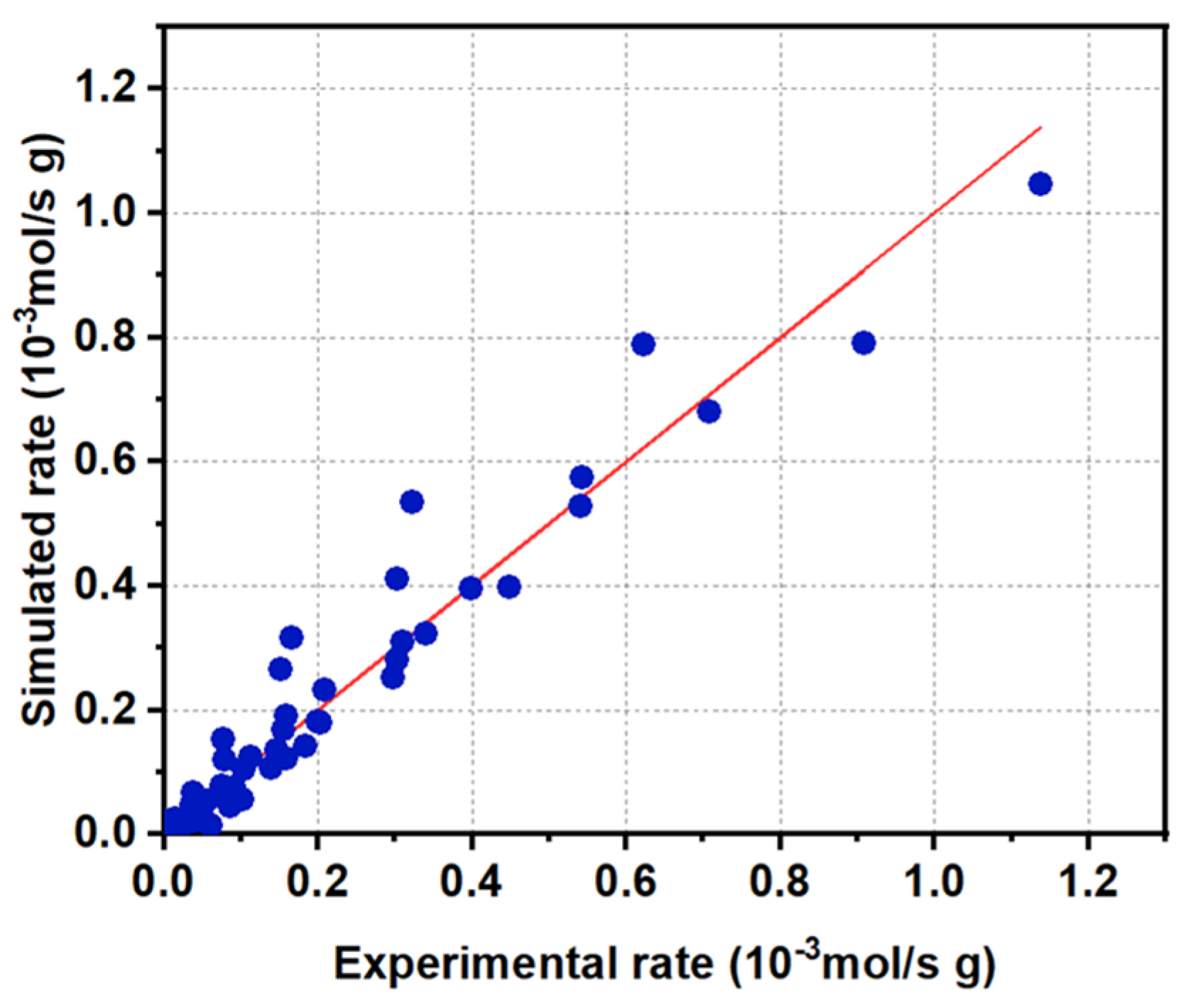
2.3.2. Kinetic Model Development
3. Materials and Methods
3.1. Catalyst Samples
3.2. Catalyst Stability Evaluation
3.3. Catalyst Characterization
3.4. Kinetic Data Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- EPA, U.S. Importance of Methane. Available online: https://www.epa.gov/gmi/importance-methane (accessed on 31 July 2024).
- EPA, U.S. Overview of Greenhouse Gases. Available online: https://www.epa.gov/ghgemissions/overview-greenhouse-gases#methane (accessed on 31 July 2024).
- EPA, U.S. Understanding Global Warming Potentials. Available online: https://www.epa.gov/ghgemissions/understanding-global-warming-potentials (accessed on 31 July 2024).
- Setiawan, A.; Kennedy, E.M.; Stockenhuber, M. Development of Combustion Technology for Methane Emitted from Coal-Mine Ventilation Air Systems. Energy Technol. 2017, 5, 521–538. [Google Scholar] [CrossRef]
- Setiawan, A.; Friggieri, J.; Kennedy, E.M.; Dlugogorski, B.Z.; Stockenhuber, M. Catalytic combustion of ventilation air methane (VAM)—long term catalyst stability in the presence of water vapour and mine dust. Catal. Sci. Technol. 2014, 4, 1793–1802. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, L.; Li, D.; Tian, S.; Zhu, X.; Wang, H.; He, C.; Li, K. Progress and key challenges in catalytic combustion of lean methane. J. Energy Chem. 2022, 75, 173–215. [Google Scholar] [CrossRef]
- Hinde, P.; Mitchell, I.; Riddell, M. COMET—A new Ventilation Air Methane (VAM) abatement technology. Johns. Matthey Technol. Rev. 2016, 60, 211–221. [Google Scholar] [CrossRef]
- Yin, J.; Su, S.; Yu, X.; Bae, J.-S.; Jin, Y.; Villella, A.; Jara, M.; Ashby, M.; Cunnington, M.; Loney, M. Site Trials and Demonstration of a Novel Pilot Ventilation Air Methane Mitigator. Energy Fuels 2020, 34, 9885–9893. [Google Scholar] [CrossRef]
- Karakurt, I.; Aydin, G.; Aydiner, K. Mine ventilation air methane as a sustainable energy source. Renew. Sustain. Energy Rev. 2011, 15, 1042–1049. [Google Scholar] [CrossRef]
- Wang, W.; Ren, J.; Li, X.; Li, H.; Li, D.; Li, H.; Song, Y. Enrichment experiment of ventilation air methane (0.5%) by the mechanical tower. Sci. Rep. 2020, 10, 7276. [Google Scholar] [CrossRef]
- IEA. Global Methane Tracker 2023. Available online: https://www.iea.org/reports/global-methane-tracker-2023 (accessed on 31 July 2024).
- Raj, A. Methane emission control: A review of mobile and stationary source emissions abatement technologies for natural gas engines. Johns. Matthey Technol. Rev. 2016, 60, 228–235. [Google Scholar] [CrossRef]
- Monai, M.; Montini, T.; Gorte, R.J.; Fornasiero, P. Catalytic Oxidation of Methane: Pd and Beyond. Eur. J. Inorg. Chem. 2018, 2018, 2884–2893. [Google Scholar] [CrossRef]
- He, L.; Fan, Y.; Bellettre, J.; Yue, J.; Luo, L. A review on catalytic methane combustion at low temperatures: Catalysts, mechanisms, reaction conditions and reactor designs. Renew. Sustain. Energy Rev. 2020, 119, 109589. [Google Scholar] [CrossRef]
- Gélin, P.; Primet, M. Complete oxidation of methane at low temperature over noble metal based catalysts: A review. Appl. Catal. B Environ. 2002, 39, 1–37. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic Combustion of Methane over Palladium-based Catalysts. Catal. Rev. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Oh, J.; Boucly, A.; van Bokhoven, J.A.; Artiglia, L.; Cargnello, M. Palladium Catalysts for Methane Oxidation: Old Materials, New Challenges. Acc. Chem. Res. 2024, 57, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Coney, C.; Stere, C.; Millington, P.; Raj, A.; Wilkinson, S.; Caracotsios, M.; McCullough, G.; Hardacre, C.; Morgan, K.; Thompsett, D.; et al. Spatially-resolved investigation of the water inhibition of methane oxidation over palladium. Catal. Sci. Technol. 2020, 10, 1858–1874. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Lugo-Jose, Y.; Fedeyko, J.; Toops, T.; Fidler, J. Deactivation Mechansims of Pd/Pd/γ-Al2O3 Catalysts for Lean Methane Oxidation. In Proceedings of the 28th North American Catalysis Society Meeting, Providence, RI, USA, 18–23 June 2023. [Google Scholar]
- Chen, H.-Y.; Lugo-Jose, Y.; Fedeyko, J.M.; Toops, T.J.; Fidler, J. Irreversible Catalyst Deactivation Mechanisms of PdO/γ-Al2O3 Catalysts for Lean Methane Oxidation. ACS Catal. 2024, 14, 15751–15763. [Google Scholar] [CrossRef]
- DOE, U.S. Catalytic Oxidation of Ventilation Air Methane. Available online: https://arpa-e.energy.gov/technologies/projects/catalytic-oxidation-ventilation-air-methane (accessed on 31 July 2024).
- Ribeiro, F.H.; Chow, M.; Dallabetta, R.A. Kinetics of the Complete Oxidation of Methane over Supported Palladium Catalysts. J. Catal. 1994, 146, 537–544. [Google Scholar] [CrossRef]
- Fujimoto, K.-i.; Ribeiro, F.H.; Avalos-Borja, M.; Iglesia, E. Structure and Reactivity of PdOx/ZrO2Catalysts for Methane Oxidation at Low Temperatures. J. Catal. 1998, 179, 431–442. [Google Scholar] [CrossRef]
- Zhu, G.; Han, J.; Zemlyanov, D.Y.; Ribeiro, F.H. Temperature Dependence of the Kinetics for the Complete Oxidation of Methane on Palladium and Palladium Oxide. J. Phys. Chem. B 2005, 109, 2331–2337. [Google Scholar] [CrossRef]
- Au-Yeung, J.; Chen, K.; Bell, A.T.; Iglesia, E. Isotopic Studies of Methane Oxidation Pathways on PdO Catalysts. J. Catal. 1999, 188, 132–139. [Google Scholar] [CrossRef]
- Groppi, G.; Ibashi, W.; Tronconi, E.; Forzatti, P. Structured reactors for kinetic measurements in catalytic combustion. Chem. Eng. J. 2001, 82, 57–71. [Google Scholar] [CrossRef]
- Abbasi, R.; Wu, L.; Wanke, S.E.; Hayes, R.E. Kinetics of methane combustion over Pt and Pt–Pd catalysts. Chem. Eng. Res. Des. 2012, 90, 1930–1942. [Google Scholar] [CrossRef]
- Habibi, A.H.; Semagina, N.; Hayes, R.E. Kinetics of Low-Temperature Methane Oxidation over SiO2-Encapsulated Bimetallic Pd–Pt Nanoparticles. Ind. Eng. Chem. Res. 2018, 57, 8160–8171. [Google Scholar] [CrossRef]
- Mihai, O.; Smedler, G.; Nylén, U.; Olofsson, M.; Olsson, L. The effect of water on methane oxidation over Pd/Al2O3 under lean, stoichiometric and rich conditions. Catal. Sci. Technol. 2017, 7, 3084–3096. [Google Scholar] [CrossRef]
- Kikuchi, R.; Maeda, S.; Sasaki, K.; Wennerström, S.; Eguchi, K. Low-temperature methane oxidation over oxide-supported Pd catalysts: Inhibitory effect of water vapor. Appl. Catal. A Gen. 2002, 232, 23–28. [Google Scholar] [CrossRef]
- Alyani, M.; Smith, K.J. Kinetic Analysis of the Inhibition of CH4 Oxidation by H2O on PdO/Al2O3 and CeO2/PdO/Al2O3 Catalysts. Ind. Eng. Chem. Res. 2016, 55, 8309–8318. [Google Scholar] [CrossRef]
- Todorova, S.; Naydenov, A.; Kolev, H.; Ivanov, G.; Ganguly, A.; Mondal, S.; Saha, S.; Ganguli, A.K. Reaction kinetics and mechanism of complete methane oxidation on Pd/Mn2O3 catalyst. React. Kinet. Mech. Catal. 2018, 123, 585–605. [Google Scholar] [CrossRef]
- Hurtado, P.; Ordóñez, S.; Sastre, H.; Díez, F.V. Development of a kinetic model for the oxidation of methane over Pd/Al2O3 at dry and wet conditions. Appl. Catal. B Environ. 2004, 51, 229–238. [Google Scholar] [CrossRef]
- Stotz, H.; Maier, L.; Boubnov, A.; Gremminger, A.T.; Grunwaldt, J.D.; Deutschmann, O. Surface reaction kinetics of methane oxidation over PdO. J. Catal. 2019, 370, 152–175. [Google Scholar] [CrossRef]
- Keller, K.; Lott, P.; Stotz, H.; Maier, L.; Deutschmann, O. Microkinetic Modeling of the Oxidation of Methane Over PdO Catalysts—Towards a Better Understanding of the Water Inhibition Effect. Catalysts 2020, 10, 922. [Google Scholar] [CrossRef]
- Jørgensen, M.; Grönbeck, H. First-Principles Microkinetic Modeling of Methane Oxidation over Pd(100) and Pd(111). ACS Catal. 2016, 6, 6730–6738. [Google Scholar] [CrossRef]
- Keller, K.; Lott, P.; Tischer, S.; Casapu, M.; Grunwaldt, J.-D.; Deutschmann, O. Methane Oxidation over PdO: Towards a Better Understanding of the Influence of the Support Material. ChemCatChem 2023, 15, e202300366. [Google Scholar] [CrossRef]
- Weaver, J.F.; Zhang, F.; Pan, L.; Li, T.; Asthagiri, A. Vacancy-Mediated Processes in the Oxidation of CO on PdO(101). Acc. Chem. Res. 2015, 48, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Pan, L.; Li, T.; Diulus, J.T.; Asthagiri, A.; Weaver, J.F. CO Oxidation on PdO(101) during Temperature-Programmed Reaction Spectroscopy: Role of Oxygen Vacancies. J. Phys. Chem. C 2014, 118, 28647–28661. [Google Scholar] [CrossRef]
- Martin, N.M.; Van den Bossche, M.; Grönbeck, H.; Hakanoglu, C.; Zhang, F.; Li, T.; Gustafson, J.; Weaver, J.F.; Lundgren, E. CO Adsorption on Clean and Oxidized Pd(111). J. Phys. Chem. C 2014, 118, 1118–1128. [Google Scholar] [CrossRef]
- Velin, P.; Ek, M.; Skoglundh, M.; Schaefer, A.; Raj, A.; Thompsett, D.; Smedler, G.; Carlsson, P.-A. Water Inhibition in Methane Oxidation over Alumina Supported Palladium Catalysts. J. Phys. Chem. C 2019, 123, 25724–25737. [Google Scholar] [CrossRef]
- Tessier, D.; Rakai, A.; Bozon-Verduraz, F. Spectroscopic study of the interaction of carbon monoxide with cationic and metallic palladium in palladium–alumina catalysts. J. Chem. Soc. Faraday Trans. 1992, 88, 741–749. [Google Scholar] [CrossRef]
- Lear, T.; Marshall, R.; Antonio Lopez-Sanchez, J.; Jackson, S.D.; Klapötke, T.M.; Bäumer, M.; Rupprechter, G.; Freund, H.-J.; Lennon, D. The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J. Chem. Phys. 2005, 123, 174706. [Google Scholar] [CrossRef]
- Gelin, P.; Siedle, A.R.; Yates, J.T., Jr. Stoichiometric adsorbate species interconversion processes in the chemisorbed layer. An infrared study of the carbon monoxide/palladium system. J. Phys. Chem. 1984, 88, 2978–2985. [Google Scholar] [CrossRef]
- Jang, E.J.; Lee, J.; Oh, D.G.; Kwak, J.H. CH4 Oxidation Activity in Pd and Pt–Pd Bimetallic Catalysts: Correlation with Surface PdOx Quantified from the DRIFTS Study. ACS Catal. 2021, 11, 5894–5905. [Google Scholar] [CrossRef]
- Xu, J.; Ouyang, L.; Mao, W.; Yang, X.-J.; Xu, X.-C.; Su, J.-J.; Zhuang, T.-Z.; Li, H.; Han, Y.-F. Operando and Kinetic Study of Low-Temperature, Lean-Burn Methane Combustion over a Pd/γ-Al2O3 Catalyst. ACS Catal. 2012, 2, 261–269. [Google Scholar] [CrossRef]
- Oh, D.G.; Aleksandrov, H.A.; Kim, H.; Koleva, I.Z.; Khivantsev, K.; Vayssilov, G.N.; Kwak, J.H. Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation. Molecules 2023, 28, 1957. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, F.; Bisson, L.; Armaroli, T.; Verdon, C.; Lemaitre, L.; Thomazeau, C. Characterization of well faceted palladium nanoparticles supported on alumina by transmission electron microscopy and FT-IR spectroscopy of CO adsorption. Appl. Catal. A: Gen. 2009, 352, 50–60. [Google Scholar] [CrossRef]
- Lentz, C.; Jand, S.P.; Melke, J.; Roth, C.; Kaghazchi, P. DRIFTS study of CO adsorption on Pt nanoparticles supported by DFT calculations. J. Mol. Catal. A Chem. 2017, 426, 1–9. [Google Scholar] [CrossRef]
- Tankov, I.; Cassinelli, W.H.; Bueno, J.M.C.; Arishtirova, K.; Damyanova, S. DRIFTS study of CO adsorption on praseodymium modified Pt/Al2O3. Appl. Surf. Sci. 2012, 259, 831–839. [Google Scholar] [CrossRef]
- Raskó, J. CO-induced surface structural changes of Pt on oxide-supported Pt catalysts studied by DRIFTS. J. Catal. 2003, 217, 478–486. [Google Scholar] [CrossRef]
- Anderson, J.R.; Boudart, M. Catalysis Science and Technology, Vol 8; Spinger: New York, NY, USA, 1987. [Google Scholar]
- Tang, M.; Shiraiwa, M.; Pöschl, U.; Cox, R.; Kalberer, M. Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations. Atmos. Chem. Phys. 2015, 15, 5585–5598. [Google Scholar] [CrossRef]
- Vannice, M.A. Kinetic of Catalytic Reactions; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber | Assignment | References |
|---|---|---|
| 2135 cm−1 | CO atop bonded on PdCUS sites | [20,38,39,40] |
| 2100 cm−1 | CO atop bonded on isolated Pd0 on flat planes | [41,42,43,44,45,46,47,48] |
| 2076 cm−1 | CO atop bonded Pd0 at the corners or edges | [41,42,43,44,45,46,47,48] |
| 2060 cm−1 | CO atop bonded on Pt0 sites | [49,50,51] |
| 1976 cm−1 | CO bridge bonded on Pd0 sites | [41,42,43,44,45,46,47,48] |
| 1934 cm−1 | CO hollow bonded on Pd0 sites | [41,42,43,44,45,46,47,48] |
| Feed Gases or Reaction Temperature | Matrix | # of Variables |
|---|---|---|
| CH4 (%) * | 0.05, 0.1, 0.2, 0.4 | 4 |
| H2O (%) * | 0, 1, 3, 5 | 4 |
| O2 (%) *,** | 18 | 1 |
| Temperature (℃) | 450, 500, 550, 600, 650, 700 | 6 |
| Total gas flow rate (mL/min, STP) | 1250 | 1 |
| A0 (s−1 ∙ g−1 catal.) | E0 (kJ/mol) | A1 (s−1 ∙ g−1 catal.) | E1 (kJ/mol) | A2 (s−1 ∙ g−1 catal.) | E2 (kJ/mol) |
|---|---|---|---|---|---|
| 2.2 × 105 | 96.7 | 2.15 | −31.0 | 1.85 × 104 | 24.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Chen, H.-Y.; Lugo-Jose, Y.; Fedeyko, J.M.; Toops, T.J.; Fidler, J. Mechanistic and Kinetic Analysis of Complete Methane Oxidation on a Practical PtPd/Al2O3 Catalyst. Catalysts 2024, 14, 847. https://doi.org/10.3390/catal14120847
Wang M, Chen H-Y, Lugo-Jose Y, Fedeyko JM, Toops TJ, Fidler J. Mechanistic and Kinetic Analysis of Complete Methane Oxidation on a Practical PtPd/Al2O3 Catalyst. Catalysts. 2024; 14(12):847. https://doi.org/10.3390/catal14120847
Chicago/Turabian StyleWang, Min, Hai-Ying Chen, Yuliana Lugo-Jose, Joseph M. Fedeyko, Todd J. Toops, and Jacqueline Fidler. 2024. "Mechanistic and Kinetic Analysis of Complete Methane Oxidation on a Practical PtPd/Al2O3 Catalyst" Catalysts 14, no. 12: 847. https://doi.org/10.3390/catal14120847
APA StyleWang, M., Chen, H.-Y., Lugo-Jose, Y., Fedeyko, J. M., Toops, T. J., & Fidler, J. (2024). Mechanistic and Kinetic Analysis of Complete Methane Oxidation on a Practical PtPd/Al2O3 Catalyst. Catalysts, 14(12), 847. https://doi.org/10.3390/catal14120847