

Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning

 ,
, 
Abstract
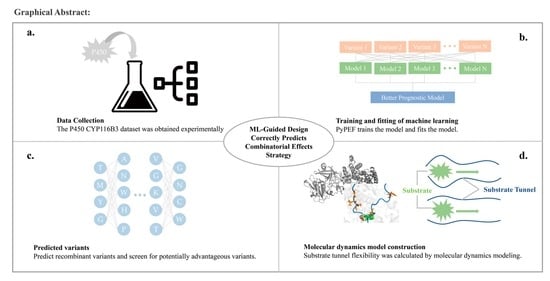
1. Introduction
2. Results and Discussion
2.1. Dataset
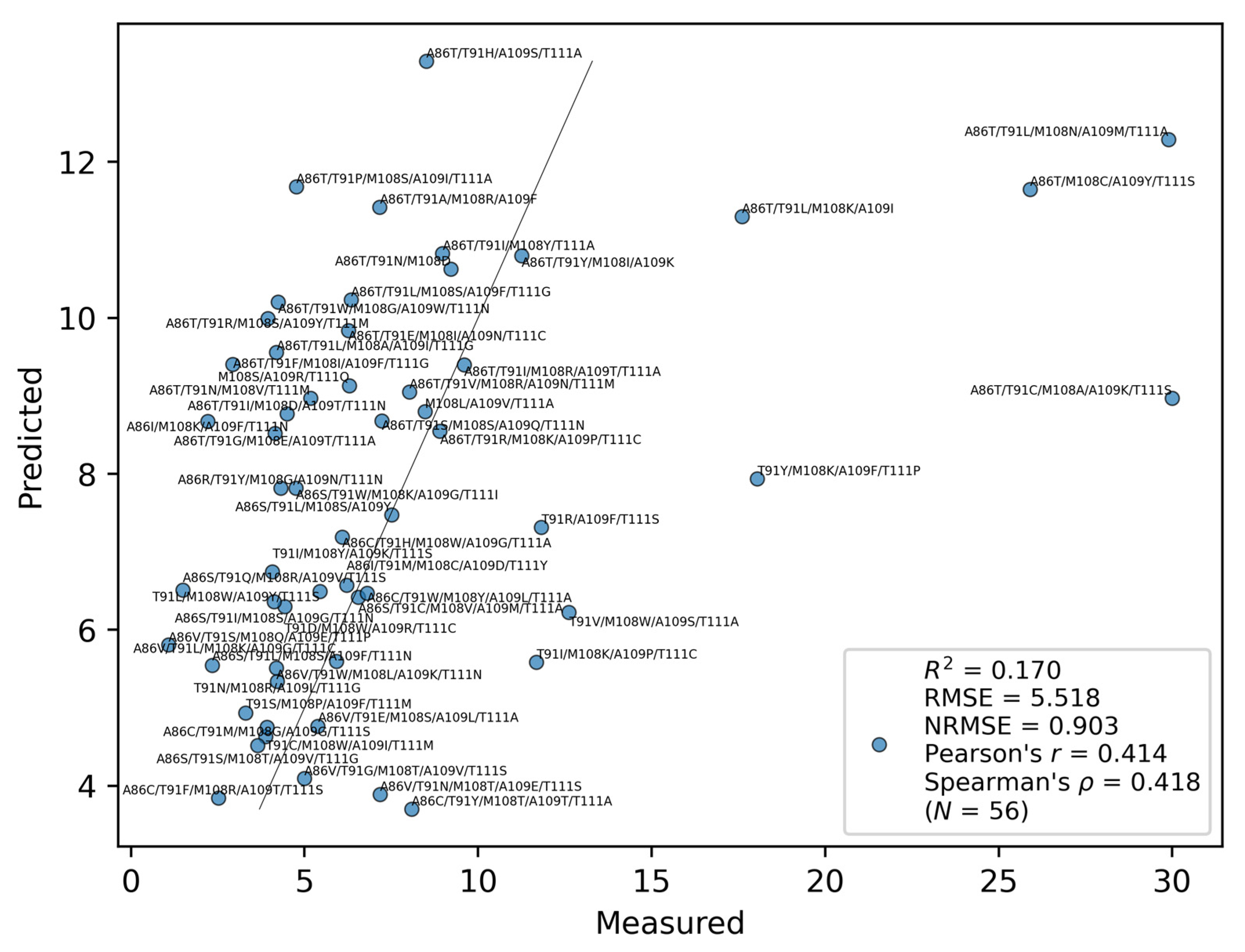
2.2. Training Result
- A small amount of data with poor correlation.
- 2.
- Large amount of data with strong correlation.
- 3.
- A small amount of data with strong correlation.
- 4.
- A large amount of data with poor correlation.
2.3. Fitting of Databases

2.4. Prediction of P450 CYP116B3
2.5. MD Simulation Setup and Prepration
2.6. MD Simulation
2.7. Machine Learning Strategy
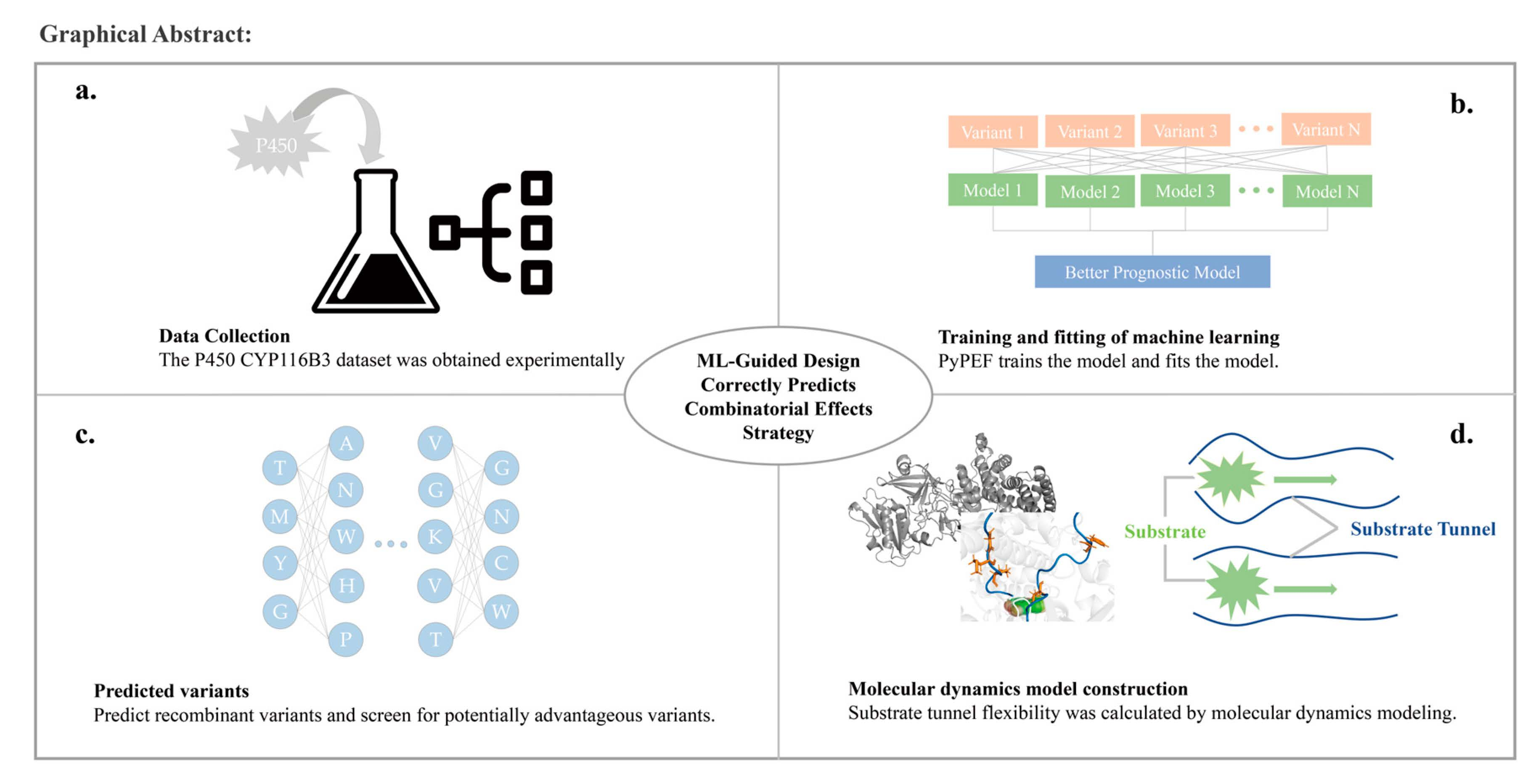
- Obtain an amount of data by experiment. Sort and group the data according to mutation position and mutation situation. Based on the effect of mutation on activity, select the relevant sequences suitable for recombination to create a mutation library. The library should be created according to the mutation code and relative parameters according to the PyPEF engineering framework, and the wild type data should be stored in “*.Fasta” format.
- The library needs to be learned several times by machine learning, and after a certain number of learning cycles, the coding method is counted and the prediction model is selected.
- The original data are used to fit the prediction model and determine the accuracy of the model.
- The appropriate prediction model is used to reorganize the mutation.
- Model building and validation of variants by AI. The number of models is determined according to the model accuracy to ensure that the best variants are obtained.
3. Materials and Methods
3.1. Identification and Prediction Methods PyPEF
3.2. Data Format
3.3. Learning Set and Validation Set
3.4. Regression Mode Selection
3.5. Selection of Model
3.6. Prediction
3.7. MD Simulations
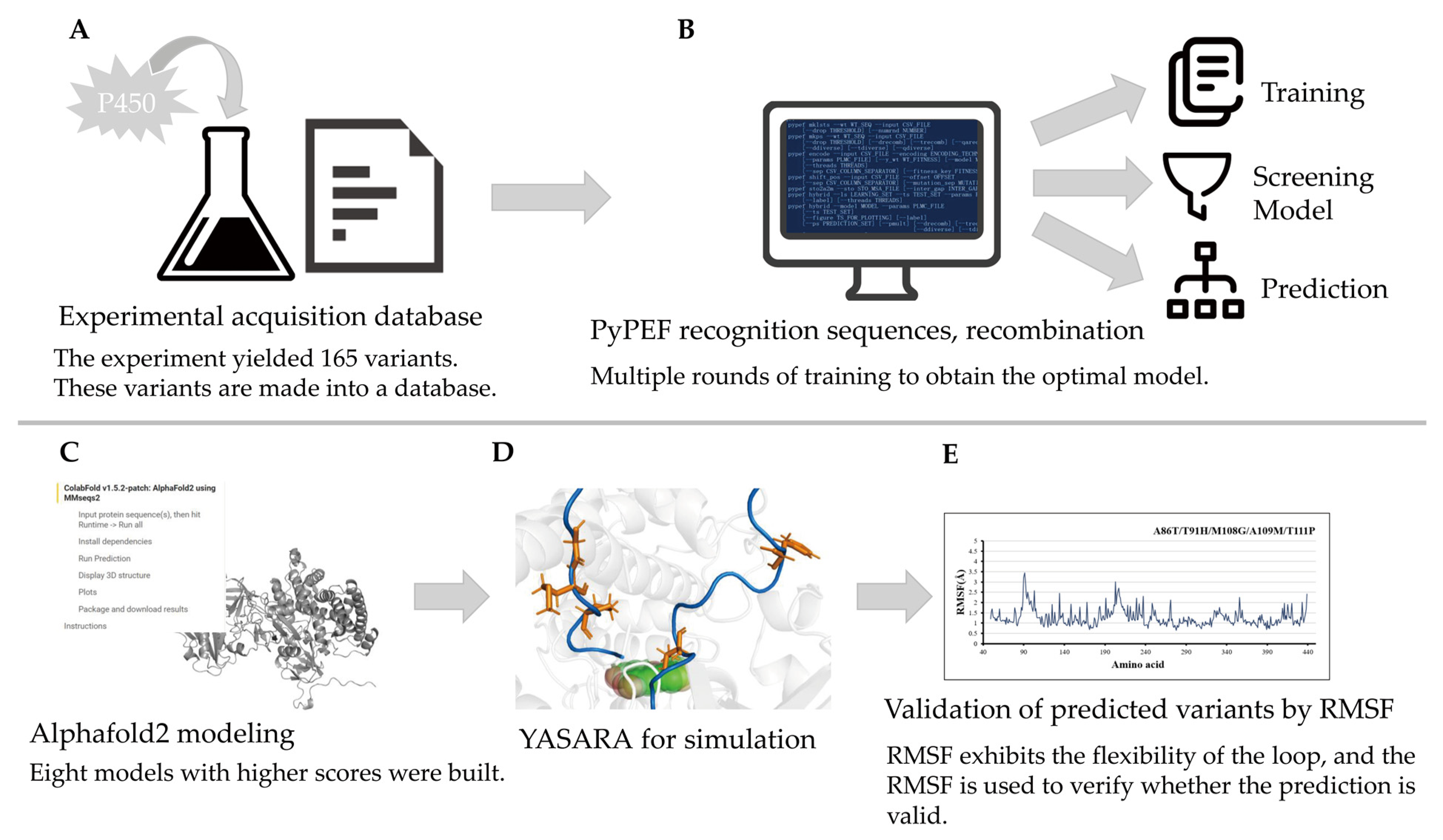
- The molecular dynamics model was established by AlphaFold2. Although there are no related proteins in Alphafold2’s library, online structure prediction can be used. This method requires only the use of sequences. After the establishment of models, choose the optimal model according to the prompt of AlphaFold2.
- Use YASARA to minimize the energy of these variants in YAMBER3 force field. The YAMBER3 force field is an improvement made by the YASARA team based on the AMBER force field.
- Use supercomputing platform to predict the motion of the model in 20 ns. After the simulation of 20 ns, the protein configuration tends to be stable, so the experimental simulation simulates the motion state of P450 in 20 ns. The method and procedure of md have been described in the previous article, and the method is the same as the previous study (Flexibility Regulation of Loops Surrounding the Tunnel Entrance in Cytochrome P450 Enhanced Substrate Access Substantially) [14].
- Confirm the binding degree of the mutation site by RMSF [14].
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Wang, B.; Wang, Z.; Chen, T.; Zhao, X. Advances in the Research of Protein Directed Evolution. Prog. Biochem. Biophys. 2015, 42, 123–131. [Google Scholar]
- Misiura, M.; Shroff, R.; Thyer, R.; Kolomeisky, A.B. DLPacker: Deep learning for prediction of amino acid side chain conformations in proteins. Proteins 2022, 90, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qin, W.; Qian, X. Application of deep learning method in biological mass spectrometry and proteomics. Prog. Biochem. Biophys. 2018, 45, 1214–1223. [Google Scholar]
- Siedhoff, N.E.; Schwaneberg, U.; Davari, M.D. Machine learning-assisted enzyme engineering. Methods Enzym. 2020, 643, 281–315. [Google Scholar]
- Singhal, A.; Kumari, N.; Ghosh, P.; Singh, Y.; Garg, S.; Shah, M.P.; Jha, P.K.; Chauhan, D. Optimizing cellulase production from Aspergillus flavus using response surface methodology and machine learning models. Environ. Technol. Innov. 2022, 27, 102805. [Google Scholar] [CrossRef]
- Wu, Z.; Kan, S.B.J.; Lewis, R.D.; Wittmann, B.J.; Arnold, F.H. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. USA 2019, 116, 8852–8858. [Google Scholar] [CrossRef]
- Cui, H.; Cao, H.; Cai, H.; Jaeger, K.; Davari, M.D.; Schwaneberg, U. Computer-Assisted Recombination (CompassR) Teaches us How to Recombine Beneficial Substitutions from Directed Evolution Campaigns. Chemistry 2020, 26, 643–649. [Google Scholar] [CrossRef]
- Cui, H.; Jaeger, K.E.; Davari, M.D.; Schwaneberg, U. CompassR Yields Highly Organic-Solvent-Tolerant Enzymes through Recombination of Compatible Substitutions. Chemistry 2021, 27, 2789–2797. [Google Scholar] [CrossRef]
- Herrmann, K.R.; Brethauer, C.; Siedhoff, N.E.; Hofmann, I.; Eyll, J.; Davari, M.D.; Schwaneberg, U.; Ruff, A.J. Evolution of E. coli Phytase Toward Improved Hydrolysis of Inositol Tetraphosphate. Front. Chem. Eng. 2022, 4, 838056. [Google Scholar] [CrossRef]
- Siedhoff, N.E.; Illig, A.M.; Schwaneberg, U.; Davari, M.D. PyPEF-An Integrated Framework for Data-Driven Protein Engineering. J. Chem. Inf. Model. 2021, 61, 3463–3476. [Google Scholar] [CrossRef]
- Illig, A.M.; Siedhoff, N.E.; Schwaneberg, U.; Davari, M.D. A hybrid model combining evolutionary probability and machine learning leverages data-driven protein engineering. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, L.; Schmid, R.D.; Urlacher, V.B. Cloning, expression, and characterization of a self-sufficient cytochrome P450 monooxygenase from Rhodococcus ruber DSM 44319. Appl. Microbiol. Biotechnol. 2006, 72, 876–882. [Google Scholar] [CrossRef]
- Li, Z.; Meng, S.; Nie, K.; Schwaneberg, U.; Davari, M.D.; Xu, H.; Ji, Y.; Liu, L. Flexibility Regulation of Loops Surrounding the Tunnel Entrance in Cytochrome P450 Enhanced Substrate Access Substantially. ACS Catal. 2022, 12, 12800–12808. [Google Scholar] [CrossRef]
- Tao, S.; Gao, Y.; Li, K.; Lu, Q.; Qiu, C.; Wang, X.; Chen, K.; Ouyang, P. Engineering substrate recognition sites of cytochrome P450 monooxygenase CYP116B3 from Rhodococcus ruber for enhanced regiospecific naphthalene hydroxylation. Mol. Catal. 2020, 493, 111089. [Google Scholar] [CrossRef]
- Liu, L.; Schmid, R.D.; Urlacher, V.B. Engineering cytochrome P450 monooxygenase CYP 116B3 for high dealkylation activity. Biotechnol. Lett. 2010, 32, 841–845. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kreß, N.; Halder, J.M.; Rapp, L.R.; Hauer, B. Unlocked potential of dynamic elements in protein structures: Channels and loops. Curr. Opin. Chem. Biol. 2018, 47, 109–116. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Thapa, N.; Chaudhari, M.; McManus, S.; Roy, K.; Newman, R.H.; Saigo, H.; Kc, D.B. Correction: DeepSuccinylSite: A deep learning based approach for protein succinylation site prediction. BMC Bioinform. 2022, 23, 349. [Google Scholar] [CrossRef]
- Hie, B.L.; Yang, K.K. Adaptive machine learning for protein engineering. Curr. Opin. Struct. Biol. 2022, 72, 145–152. [Google Scholar] [CrossRef]
- Oh, S.-H. Protein Disorder Prediction Using Multilayer Perceptrons. Int. J. Contents 2013, 9, 11–15. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef] [PubMed]
- He, J. Research and Application of Machine Learning Algorithm Based on Gaussian Process Model. Ph.D. Thesis, Dalian University of Technology, Dalian, China, 2012; pp. 130–131. [Google Scholar]
- Wittmund, M.; Cadet, F.; Davari, M.D. Learning Epistasis and Residue Coevolution Patterns: Current Trends and Future Perspectives for Advancing Enzyme Engineering. ACS Catal. 2022, 12, 14243–14263. [Google Scholar] [CrossRef]
- Carkli Yavuz, B.; Yurtay, N.; Ozkan, O. Prediction of Protein Secondary Structure with Clonal Selection Algorithm and Multilayer Perceptron. IEEE Access 2018, 6, 45256–45261. [Google Scholar] [CrossRef]
- Xiong, W.; Liu, B.; Shen, Y.; Jing, K.; Savage, T.R. Protein engineering design from directed evolution to de novo synthesis. Biochem. Eng. J. 2021, 174, 108096. [Google Scholar] [CrossRef]
- Diaz, D.J.; Kulikova, A.V.; Ellington, A.D.; Wilke, C.O. Using machine learning to predict the effects and consequences of mutations in proteins. Curr. Opin. Struct. Biol. 2023, 78, 102518. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Times | Index | Times |
|---|---|---|---|
| (a) | |||
| TANS770102 | 6 | RICJ880102 | 1 |
| QIAN880126 | 3 | RACS820102 | 1 |
| QIAN880102 | 2 | KARS160111 | 1 |
| CHAM830107 | 2 | ISOY800108 | 1 |
| PALJ810107 | 2 | RICJ880110 | 1 |
| WILM950102 | 2 | CHAM830106 | 1 |
| GEOR030105 | 2 | KRIW790102 | 1 |
| WERD780101 | 2 | FAUJ880104 | 1 |
| HUTJ700101 | 2 | GEOR030106 | 1 |
| QIAN880125 | 1 | GEOR030101 | 1 |
| RICJ880101 | 1 | ||
| (b) | |||
| TANS770102 | 8 | OOBM850105 | 1 |
| PRAM820103 | 7 | VASM830102 | 1 |
| AURR980120 | 6 | AURR980102 | 1 |
| KARS160120 | 5 | QIAN880129 | 1 |
| RICJ880108 | 4 | MONM990101 | 1 |
| GEOR030105 | 2 | SNEP660104 | 1 |
| CHOC760104 | 2 | GEOR030102 | 1 |
| QIAN880113 | 2 | PRAM820101 | 1 |
| QIAN880125 | 2 | ISOY800108 | 1 |
| QIAN880123 | 2 | RACS820112 | 1 |
| RICJ880102 | 1 | PALJ810107 | 1 |
| FINA770101 | 1 | QIAN880101 | 1 |
| CHAM830108 | 1 | QIAN880138 | 1 |
| HUTJ700101 | 1 | RICJ880101 | 1 |
| CIDH920101 | 1 | RACS820101 | 1 |
| Index | Spearman’s | R2 | RMSE | NRMSE | Pearson’s r |
|---|---|---|---|---|---|
| TANS770102 | 0.42 | 0.17 | 5.52 | 0.90 | 0.41 |
| PRAM820103 | 0.40 | 0.23 | 5.33 | 0.87 | 0.450 |
| GEOR030105 | 0.38 | 0.17 | 5.51 | 0.90 | 0.42 |
| RICJ880108 | 0.36 | 0.15 | 5.58 | 0.91 | 0.39 |
| CHOC760104 | 0.32 | 0.15 | 5.55 | 0.91 | 0.40 |
| Model | Precision |
|---|---|
| TANS770102 | 0.1933 |
| PRAM82013 | 0.1790 |
| RICJ880108 | 0.1526 |
| Variant | Activity |
|---|---|
| A86T/T91H/M108G/A109M/T111P | 15.37 |
| A86T/T91H/M108V/A109M/T111P | 15.32 |
| A86T/T91H/M108N/A109M/T111P | 15.29 |
| A86W/T91H/M108G/A109M/T111P | 15.28 |
| A86T/T91P/M108G/A109M/T111P | 15.26 |
| A86T/T91P/M108V/A109M/T111P | 15.21 |
| A86T/T91H/M108S/A109M/T111P | 15.20 |
| A86W/T91H/M108V/A109M/T111P | 15.19 |
| A86T/T91P/M108N/A109M/T111P | 15.18 |
| A86W/T91H/M108N/A109M/T111P | 15.17 |
| A86W/T91P/M108G/A109M/T111P | 15.14 |
| A86W/T91P/M108V/A109M/T111P | 15.12 |
| A86T/T91P/M108S/A109M/T111P | 15.09 |
| A86W/T91H/M108S/A109M/T111P | 15.08 |
| A86W/T91P/M108N/A109M/T111P | 15.06 |
| A86T/T91W/M108G/A109M/T111P | 15.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Li, Z.; Cao, C.; Zhang, X.; Meng, S.; Davari, M.D.; Xu, H.; Ji, Y.; Schwaneberg, U.; Liu, L. Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning. Catalysts 2023, 13, 1228. https://doi.org/10.3390/catal13081228
Liu Y, Li Z, Cao C, Zhang X, Meng S, Davari MD, Xu H, Ji Y, Schwaneberg U, Liu L. Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning. Catalysts. 2023; 13(8):1228. https://doi.org/10.3390/catal13081228
Chicago/Turabian StyleLiu, Yiheng, Zhongyu Li, Chenqi Cao, Xianzhi Zhang, Shuaiqi Meng, Mehdi D. Davari, Haijun Xu, Yu Ji, Ulrich Schwaneberg, and Luo Liu. 2023. "Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning" Catalysts 13, no. 8: 1228. https://doi.org/10.3390/catal13081228
APA StyleLiu, Y., Li, Z., Cao, C., Zhang, X., Meng, S., Davari, M. D., Xu, H., Ji, Y., Schwaneberg, U., & Liu, L. (2023). Engineering of Substrate Tunnel of P450 CYP116B3 though Machine Learning. Catalysts, 13(8), 1228. https://doi.org/10.3390/catal13081228