
Scale-Up for the Conversion of Corn Stover-Derived Levulinic Acid into 2-Methyltetrahydrofuran

Abstract
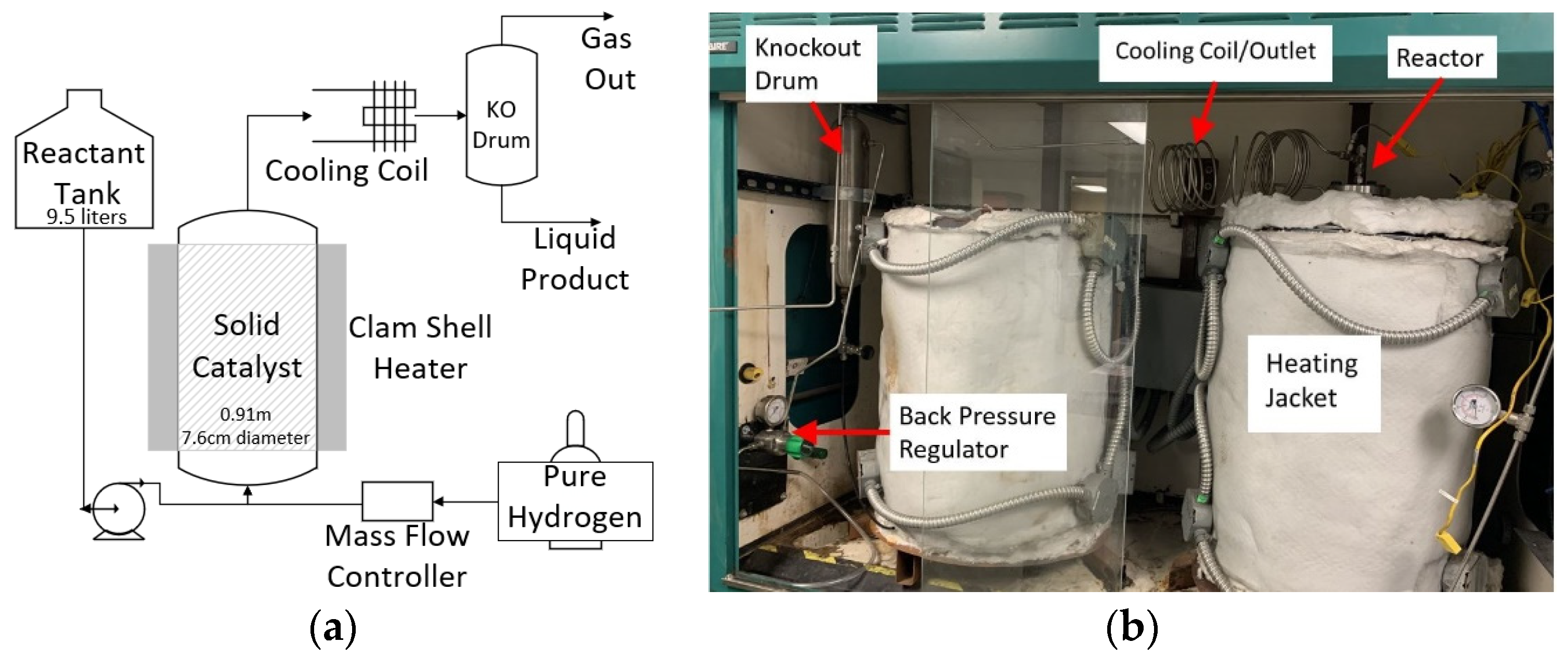
:1. Introduction
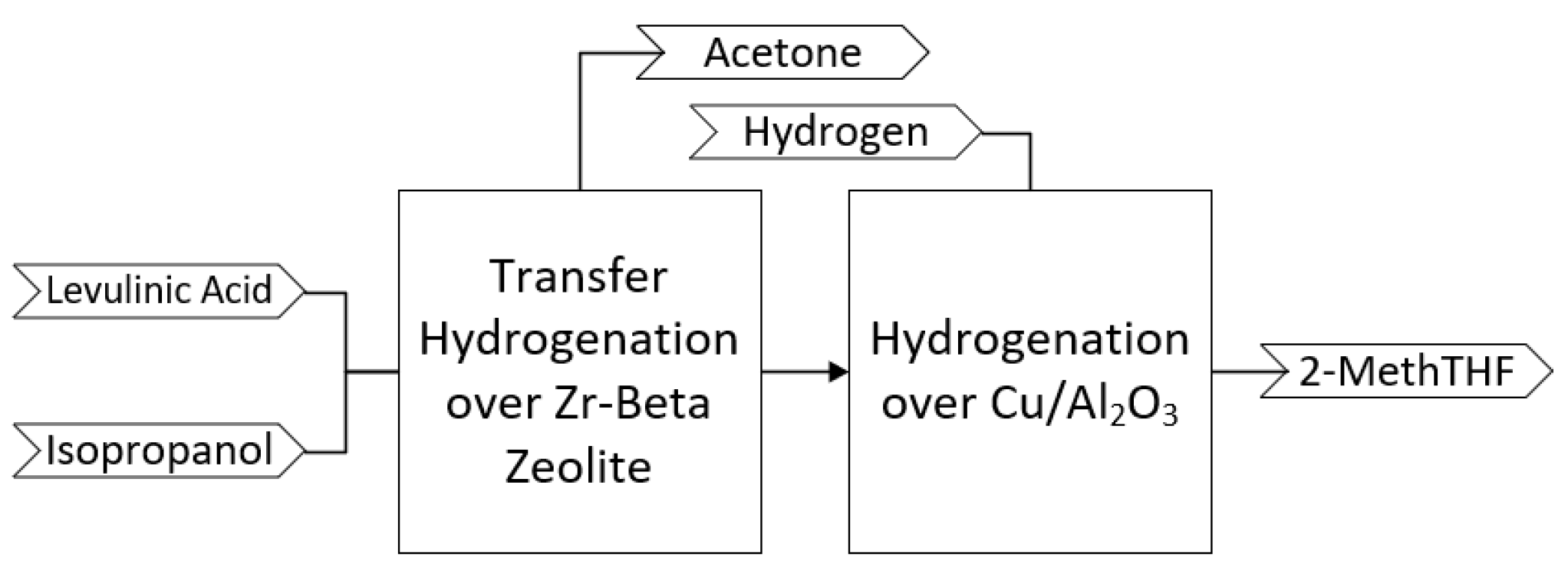
- Translate to the continuous bench-scale (0.613 kg/h production rate) a two-step reaction pathway to convert levulinic acid into 2-methyltetrahydrofuran (2-MeTHF).
- Determine the optimum reaction conditions for each reaction to maximize the production of 2-MeTHF through a careful study of the effects of residence time, reaction temperature, reaction pressure, and percent excess solvent used on conversion and yield in each reaction.
2. Results and Discussion
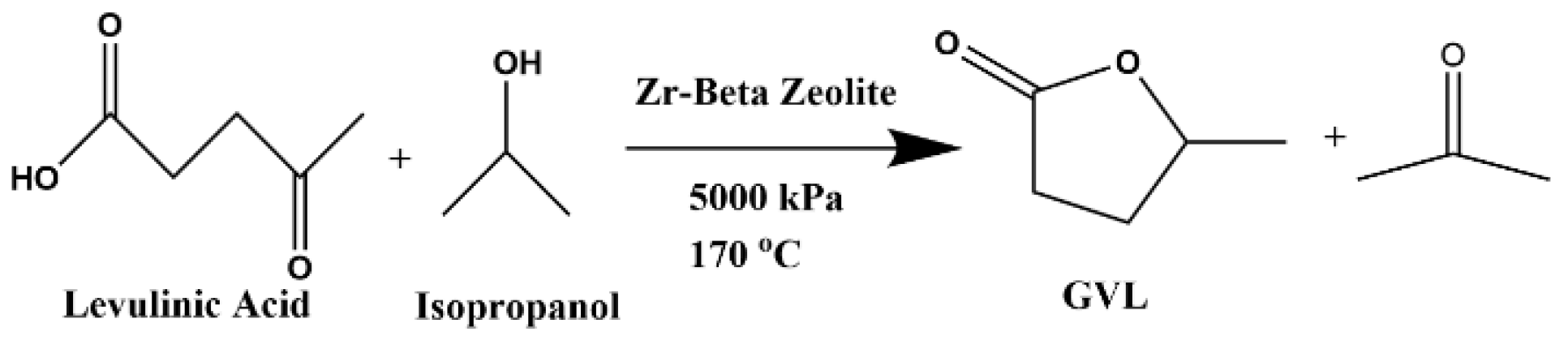
2.1. Preliminary Batch Lab-Scale Experiments—First Reaction
2.2. Bench-Scale Continuous Flow Conversion of Levulinic Acid to GVL
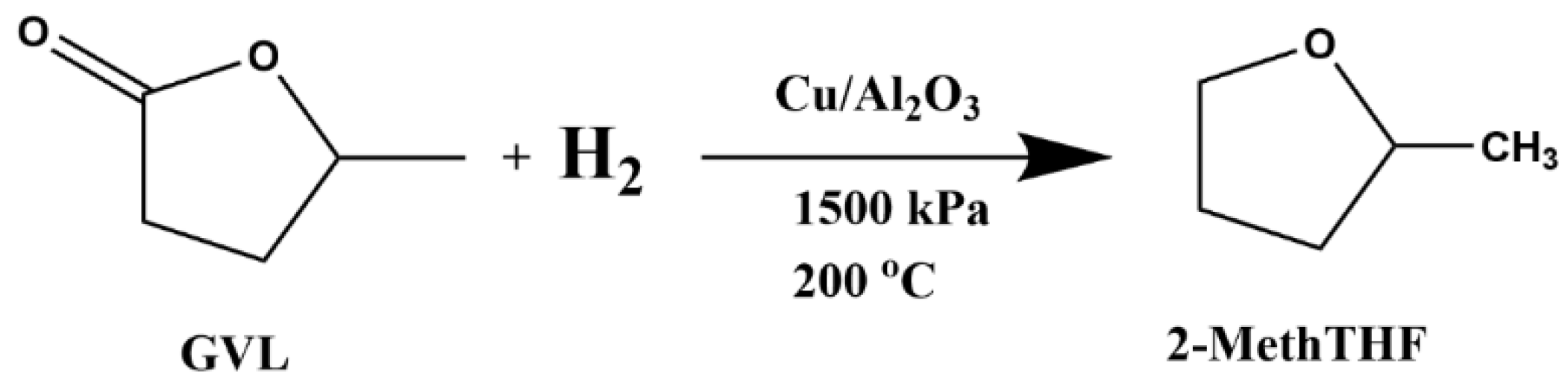
2.3. Batch Lab-Scale Conversion of GVL to 2-MeTHF
2.4. Bench-Scale Continuous Flow Conversion of GVL to 2-MeTHF

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Temp. (°C) | Pressure (MPa) | Residence Time (hours) | Excess H2 (mol%) | 2-MeTHF Yield (mol%) | Standard Error (mol%) | 2-MeTHF Selectivity (mol%) | Standard Error (mol%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 200 | 1.5 | 2.5 | 30 | 89 | 0.3 | 95 | 0.2 |
| 2 | 250 | 1.5 | 2.5 | 20 | 90 | 0.8 | 92 | 0.9 |
| 3 | 150 | 1.5 | 2 | 10 | 83 | 0.4 | 83 | 0.7 |
| 4 | 200 | 2 | 1.5 | 20 | 71 | 0.9 | 94 | 0.1 |
| 5 | 150 | 1 | 2 | 20 | 85 | 1.1 | 83 | 2.4 |
| 6 | 200 | 1 | 1.5 | 20 | 66 | 0.5 | 94 | 0.4 |
| 7 | 200 | 1 | 2.5 | 20 | 89 | 0.2 | 96 | 0.8 |
| 8 | 250 | 1.5 | 2 | 10 | 86 | 0.7 | 89 | 0.2 |
| 9 | 200 | 1.5 | 1.5 | 30 | 68 | 0.3 | 98 | 0.1 |
| 10 | 250 | 1.5 | 1.5 | 20 | 66 | 0.5 | 95 | 0.3 |
| 11 | 200 | 2 | 2.5 | 20 | 88 | 1 | 96 | 0.2 |
| 12 | 150 | 1.5 | 2.5 | 20 | 87 | 0.4 | 84 | 0.4 |
| 13 | 200 | 2 | 2 | 10 | 83 | 0.3 | 93 | 0.5 |
| 14 | 200 | 1 | 2 | 10 | 86 | 0.8 | 96 | 0.2 |
| 15 | 250 | 2 | 2 | 20 | 84 | 0.4 | 92 | 0.8 |
| 16 | 200 | 1.5 | 2.5 | 10 | 91 | 0.3 | 97 | 0.9 |
| 17 | 150 | 1.5 | 2 | 30 | 87 | 0.8 | 85 | 0.4 |
| 18 | 250 | 1.5 | 2 | 30 | 81 | 0.3 | 90 | 0.1 |
| 19 | 200 | 1.5 | 2 | 10 | 93 | 0.5 | 98 | 0.7 |
| 20 | 200 | 1.5 | 2 | 20 | 86 | 0.4 | 96 | 0.5 |
| 21 | 250 | 1 | 2 | 20 | 79 | 0.2 | 91 | 0.4 |
| 22 | 200 | 1 | 2 | 30 | 82 | 0.9 | 97 | 0.9 |
| 23 | 150 | 2 | 2 | 20 | 83 | 2.2 | 83 | 10 |
| 24 | 200 | 1.5 | 2 | 20 | 81 | 0.1 | 94 | 0.2 |
| 25 | 200 | 1.5 | 1.5 | 10 | 72 | 3.2 | 95 | 0.4 |
| 26 | 200 | 2 | 2 | 30 | 85 | 0.6 | 94 | 0.2 |
| 27 | 150 | 1.5 | 1.5 | 20 | 71 | 0.7 | 81 | 0.1 |
| 28 | 200 | 1.5 | 3 | 10 | 93 | 0.7 | 97 | 0.3 |
| 29 | 200 | 1.5 | 3 | 10 | 92 | 0.5 | 98 | 0.5 |
3. Methods
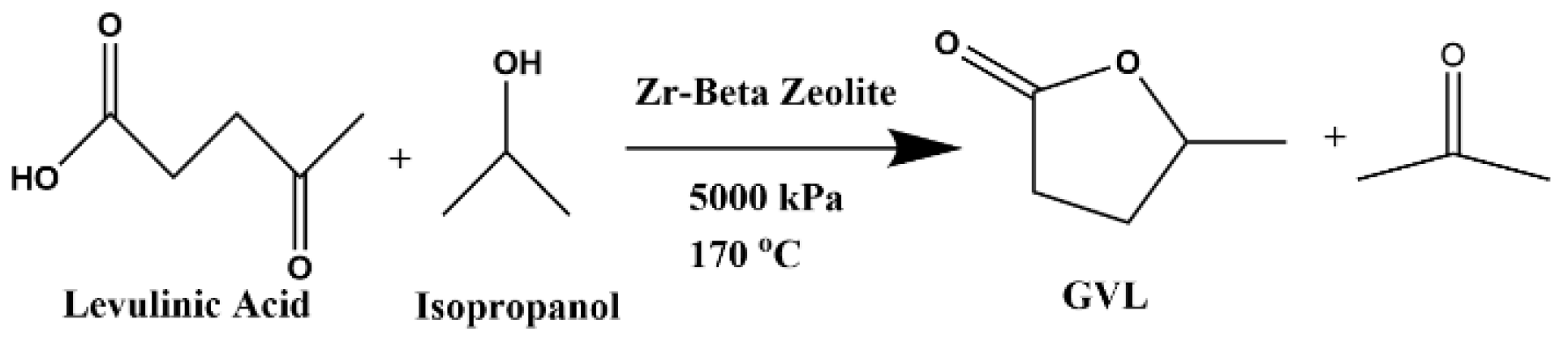
3.1. Reactants and Catalyst Materials
3.2. Zr-Beta Preparation
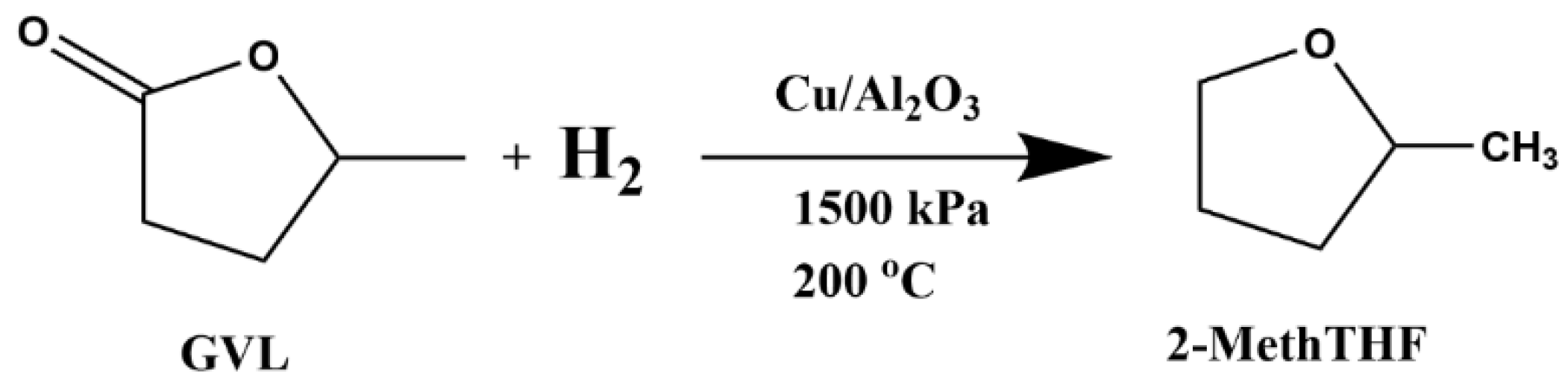
3.3. CuO/Al2O3 Catalyst Preparation
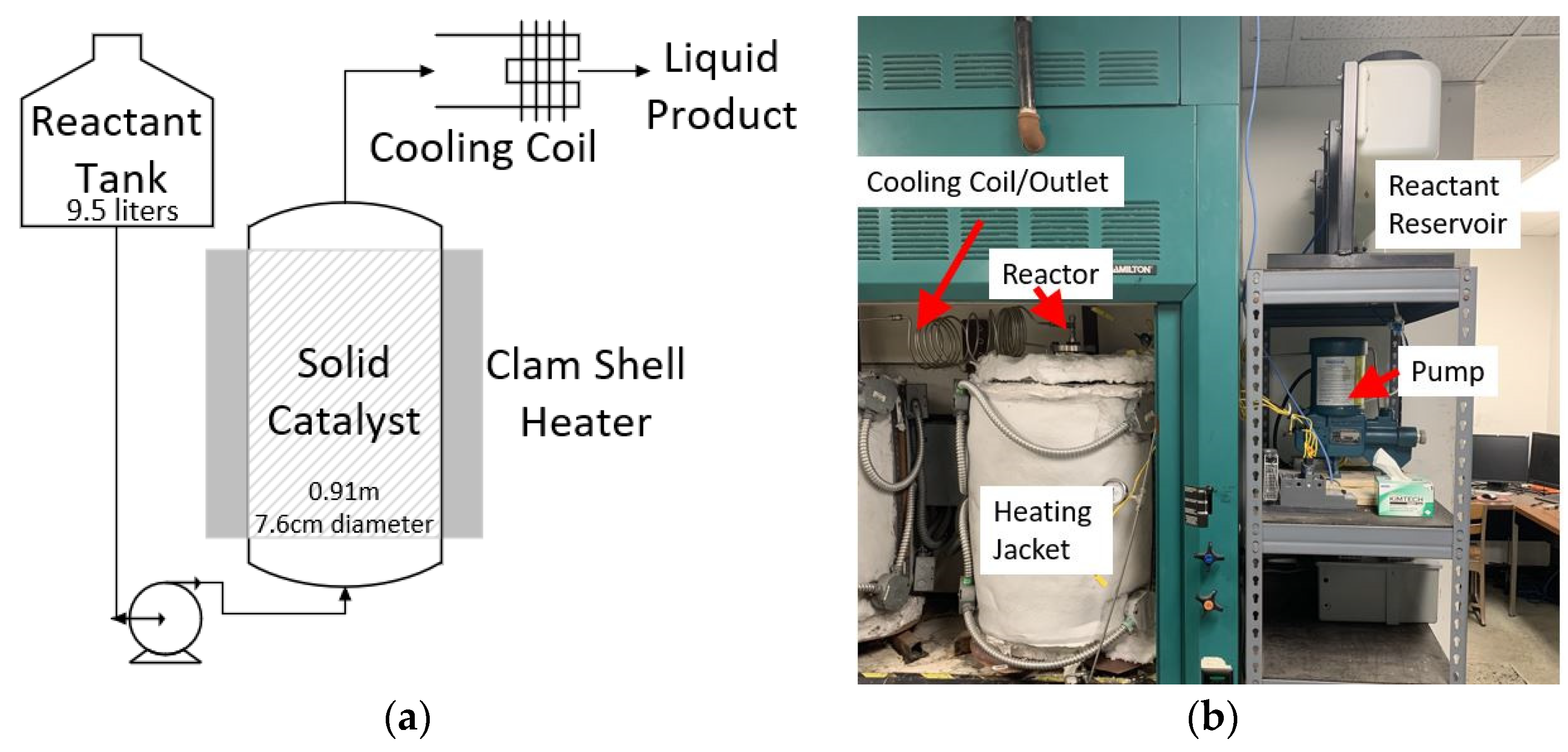
3.4. Batch Reactor
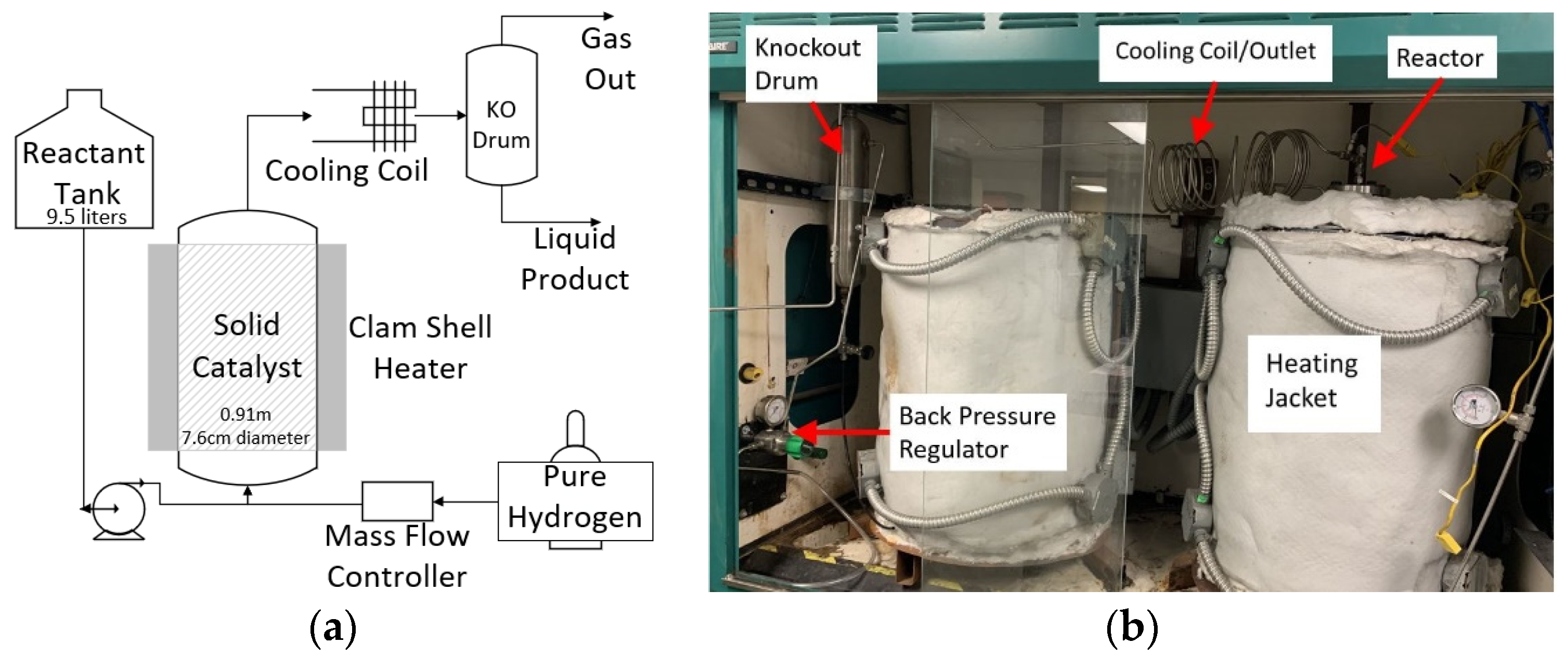
3.5. Reaction 1 Continuous Flow Bench-Scale Reactor Setup
3.6. Reaction 2 Continuous Flow Bench-Scale Reactor Setup
3.7. First-Step Batch Reaction
3.8. Second-Step Batch Reaction
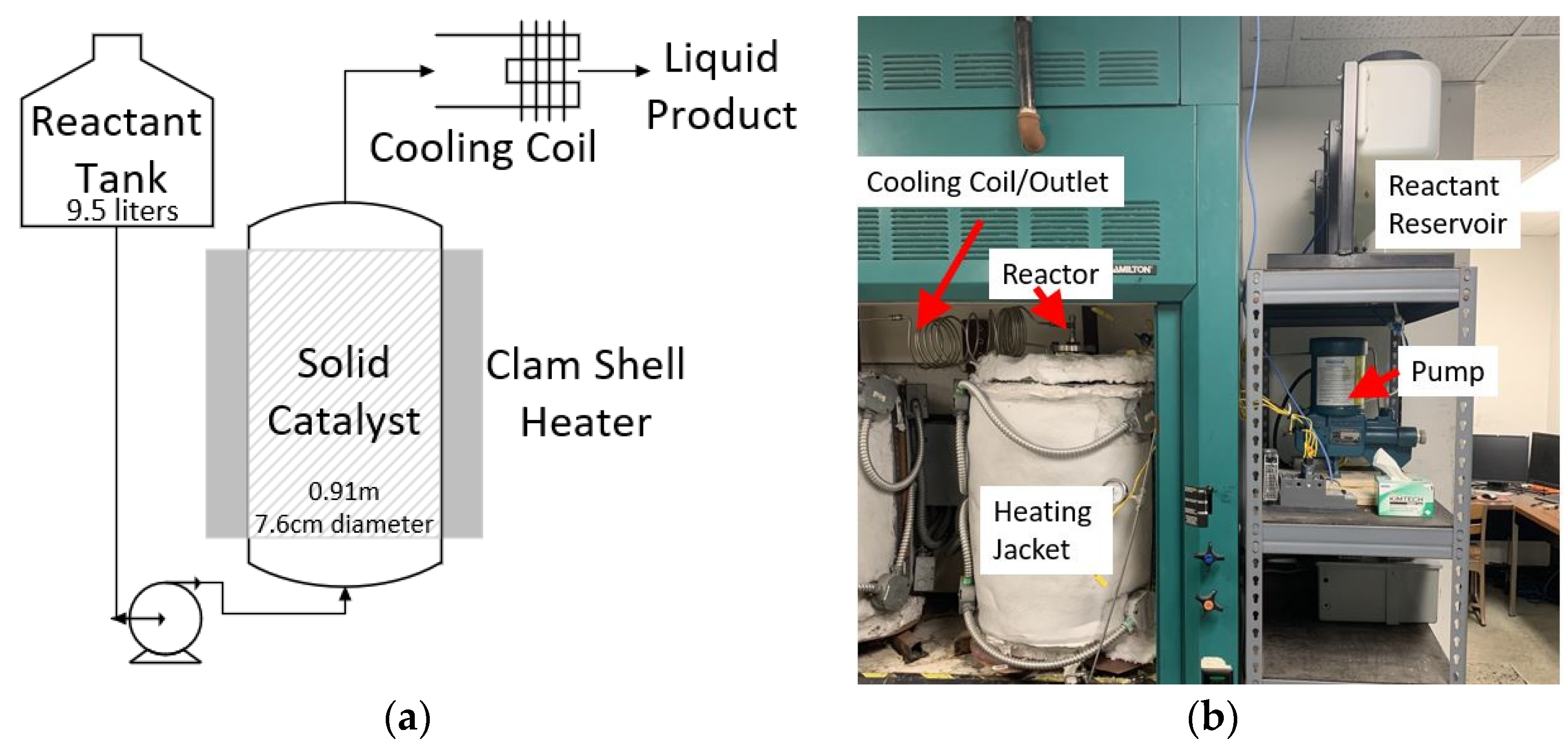
3.9. First-Step Bench-Scale Continuous Reaction Experiments
3.10. Second-Step Bench-Scale Continuous Reaction Experiments
3.11. Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohler, A.; Seames, W.; Foerster, I.; Kadrmas, C. Catalytic Formation of Lactic and Levulinic Acids from Biomass Derived Monosaccarides through Sn-Beta Formed by Impregnation. Catalysts 2020, 10, 1219. [Google Scholar] [CrossRef]
- Toif, M.E.; Hidayat, M.; Rochmadi; Budiman, A. Glucose to Levulinic acid, a versatile building block chemical. In Proceedings of the International Conference on Science and Applied Science (ICSAS2020), Surakarta, Indonesia, 7 July 2020. [CrossRef]
- Mika, L.T.; Horváth, I.T. Homogeneous transition metal catalyzed conversion of levulinic acid to gamma-valerolactone. Catal. Biomass Convers. 2021, 77, 1–25. [Google Scholar] [CrossRef]
- Gogate, A.; Kemmer, M.; Myskewitz, E.; Stegner, L. Scoping Study for a Project to Produce Polyether Polyols from Biobased 1,4-Pentanediol; University of North Dakota Course CHE412 Report; University of North Dakota: Grand Forks, ND, USA, 2021. [Google Scholar]
- Sun, D.; Saito, T.; Otsuka, S.; Ozawa, T.; Yamada, Y.; Sato, S. Selective hydrogenation of γ-valerolactone to 2-methyltetrahydrofuran over Cu/Al2O3 catalyst. Appl. Catal. A Gen. 2020, 590, 117309. [Google Scholar] [CrossRef]
- Deng, L.; Li, J.; Lai, D.-M.; Fu, Y.; Guo, Q.-X. Catalytic conversion of biomass-derived carbohydrates into γ-Valerolactone without using an external H2supply. Angew. Chem. Int. Ed. 2009, 48, 6529–6532. [Google Scholar] [CrossRef] [PubMed]
- Al-Shaal, M.G.; Dzierbinski, A.; Palkovits, R. Solvent-free γ-valerolactone hydrogenation to 2-methyltetrahydrofuran catalysed by RU/c: A reaction network analysis. Green Chem. 2014, 16, 1358–1364. [Google Scholar] [CrossRef]
- López-Aguado, C.; Paniagua, M.; Melero, J.A.; Iglesias, J.; Juárez, P.; López Granados, M.; Morales, G. Stable continuous production of γ-valerolactone from biomass-derived levulinic acid over zr–al-beta zeolite catalyst. Catalysts 2020, 10, 678. [Google Scholar] [CrossRef]
- Kerkel, F.; Markiewicz, M.; Stolte, S.; Müller, E.; Kunz, W. The Green Platform Molecule Gamma-valerolactone—Ecotoxicity, biodegradability, solvent properties, and potential applications. Green Chem. 2021, 23, 2962–2976. [Google Scholar] [CrossRef]
- Guarinos, J.M.; Cirujano, F.G.; Rapeyko, A.; Llabrés i Xamena, F.X. Conversion of levulinic acid to γ-valerolactone over ZR-containing metal-organic frameworks: Evidencing the role of Lewis and Brønsted Acid Sites. Mol. Catal. 2021, 515, 111925. [Google Scholar] [CrossRef]
- Yuan, Q.; van de Bovenkamp, H.H.; Zhang, Z.; Piskun, A.S.; Sami, S.; Havenith RW, A.; Heeres, H.J.; Deuss, P.J. Mechanistic investigations into the catalytic levulinic acid hydrogenation, insight in H/D exchange pathways, and a synthetic route to D8-γ-valerolactone. ACS Catal. 2021, 11, 10467–10477. [Google Scholar] [CrossRef]
- Damayanti, A.P.; Dewi, H.P. Selective hydrogenation of levulinic acid to γ-valerolactone using bimetallic Pd-Fe catalyst supported on titanium oxide. IOP Conf. Ser. Mater. Sci. Eng. 2020, 980, 012013. [Google Scholar] [CrossRef]
- González, G.; Area, M.C. An overview of the obtaining of biomass-derived gamma-valerolactone from levulinic acid or esters without H2 supply. BioResources 2021, 16, 8417–8444. [Google Scholar] [CrossRef]
- Dutta, S.; Yu, I.K.M.; Tsang, D.C.W.; Ng, Y.H.; Ok, Y.S.; Sherwood, J.; Clark, J.H. Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: A critical review. Chem. Eng. J. 2019, 372, 992–1006. [Google Scholar] [CrossRef]
- Hengne, A.M.; Kadu, B.S.; Biradar, N.S.; Chikate, R.C.; Rode, C.V. Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported ni catalysts. RSC Adv. 2016, 6, 59753–59761. [Google Scholar] [CrossRef]
- Omoruyi, U.; Page, S.; Hallett, J.; Miller, P.W. Homogeneous catalyzed reactions of levulinic acid: To γ-valerolactone and beyond. ChemSusChem 2016, 9, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Chia, M.; Dumesic, J.A. Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ-valerolactone over metal oxide catalysts. Chem. Commun. 2011, 47, 12233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, Z.; Arai, M.; Zhang, C.; Liu, K.; Shi, R.; Wu, P.; Wang, Z.; Lin, W.; Cheng, H.; et al. Transformation of γ-valerolactone into 1,4-pentanediol and 2-methyltetrahydrofuran over zn-promoted Cu/Al2O3 Catalysts. Catal. Sci. Technol. 2020, 10, 4412–4423. [Google Scholar] [CrossRef]
- Obregón, I.; Gandarias, I.; Ocio, A.; García-García, I.; Alvarez de Eulate, N.; Arias, P.L. Structure-activity relationships of Ni-Cu/Al 2 o 3 catalysts for γ-Valerolactone conversion to 2-methyltetrahydrofuran. Appl. Catal. B Environ. 2017, 210, 328–341. [Google Scholar] [CrossRef]
- Pothu, R.; Challa, P.; Rajesh, R.; Boddula, R.; Balaga, R.; Balla, P.; Perugopu, V.; Radwan, A.B.; Abdullah, A.M.; Al-Qahtani, N. Vapour-phase selective hydrogenation of γ-Valerolactone to 2-methyltetrahydrofuran biofuel over silica-supported copper catalysts. Nanomaterials 2022, 12, 3414. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-L.; Bi, Q.-Y.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Tunable copper-catalyzed chemoselective hydrogenolysis of biomass-derived γ-valerolactone into 1,4-pentanediol or 2-methyltetrahydrofuran. Green Chem. 2012, 14, 935. [Google Scholar] [CrossRef]
- 2-Methyl-Tetrahydrofuran-Market Size, Share, Analysis & Forecast. Verified Market Research. 2023. Available online: https://www.verifiedmarketresearch.com/product/2-methyl-tetrahydrofuran-market/ (accessed on 29 March 2023).

| Temp. (°C) | Pressure (MPa) | Reaction Time (hours) | Excess C3H8O mol% | GVL Yield (mol%) | Selectivity (mol%) |
|---|---|---|---|---|---|
| 150 | 5 | 3 | 20 | 68 | 94 |
| 150 | 5 | 3 | 20 | 67 | 93 |
| 150 | 5 | 3 | 20 | 70 | 96 |
| Run | Temp (°C) | Pressure (MPa) | Excess C3H8O (mol%) | Residence Time (hours) | GVL Yield (mol%) | Standard Error (mol%) | GVL Selectivity (mol%) | Standard Error (mol%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 170 | 5.35 | 20 | 3 | 77 | 0.2 | 94 | 0.1 |
| 2 | 160 | 5 | 30 | 2 | 54 | 0.5 | 91 | 0.6 |
| 3 | 150 | 5.35 | 40 | 3 | 71 | 0.1 | 95 | 0.2 |
| 4 | 150 | 5.35 | 30 | 4 | 83 | 1.5 | 96 | 0.9 |
| 5 | 160 | 5.7 | 30 | 2 | 53 | 0.4 | 90 | 0.1 |
| 6 | 150 | 5.7 | 30 | 3 | 69 | 0.9 | 94 | 0.7 |
| 7 | 160 | 5.35 | 30 | 3 | 73 | 0.3 | 93 | 0.5 |
| 8 | 170 | 5.35 | 40 | 3 | 76 | 0.3 | 95 | 0.9 |
| 9 | 160 | 5.35 | 30 | 3 | 71 | 0.8 | 94 | 0.1 |
| 10 | 170 | 5 | 20 | 4 | 87 | 0.4 | 98 | 0.2 |
| 11 | 170 | 5.7 | 30 | 3 | 74 | 0.2 | 95 | 0.8 |
| 12 | 160 | 5.7 | 30 | 4 | 83 | 0.9 | 98 | 0.4 |
| 13 | 160 | 5.7 | 40 | 3 | 70 | 0.7 | 94 | 0.2 |
| 14 | 160 | 5 | 40 | 3 | 69 | 0.5 | 93 | 0.6 |
| 15 | 150 | 5.35 | 30 | 2 | 52 | 0.6 | 89 | 0.3 |
| 16 | 160 | 5 | 30 | 4 | 84 | 0.1 | 96 | 0.3 |
| 17 | 160 | 5.7 | 20 | 3 | 68 | 2.1 | 92 | 0.9 |
| 18 | 160 | 5.35 | 40 | 4 | 83 | 0.3 | 98 | 0.5 |
| 19 | 170 | 5.35 | 30 | 2 | 57 | 0.4 | 87 | 0.1 |
| 20 | 170 | 5.35 | 30 | 4 | 86 | 0.6 | 97 | 0.7 |
| 21 | 150 | 5 | 30 | 3 | 70 | 0.2 | 94 | 0.3 |
| 22 | 160 | 5 | 20 | 3 | 71 | 18 | 94 | 1.1 |
| 23 | 150 | 5.35 | 20 | 3 | 71 | 0.4 | 93 | 0.3 |
| 24 | 160 | 5.35 | 40 | 2 | 55 | 0.7 | 90 | 0.1 |
| 25 | 160 | 5.35 | 20 | 2 | 58 | 0.5 | 88 | 0.7 |
| 26 | 160 | 5.35 | 20 | 4 | 85 | 0.4 | 96 | 0.4 |
| 27 | 160 | 5.35 | 30 | 3 | 72 | 0.7 | 95 | 0.6 |
| 28 | 180 | 5 | 20 | 4 | 86 | 0.4 | 98 | 0.3 |
| 29 | 170 | 5 | 20 | 5 | 88 | 0.2 | 97 | 0.1 |
| Temp (°C) | Pressure (MPa) | Reaction Time (hours) | Excess H2 (mol%) | 2-MeTHF Yield (mol%) | Selectivity (mol%) |
|---|---|---|---|---|---|
| 200 | 1.5 | 1.5 | 20 | 75 | 98 |
| 200 | 1.5 | 2 | 20 | 84 | 96 |
| 250 | 1.5 | 1.5 | 20 | 77 | 97 |
| Reaction | Temperature (°C) | Pressure (MPa) | Residence Time (hours) | Excess H Donor (mol%) |
|---|---|---|---|---|
| Levulinic to GVL | 150, 160, 170 | 5, 5.7 | 2, 3, 4, 5 | 20, 30, 40 |
| GVL to 2-MeTHF | 150, 200, 250 | 1, 1.5, 2 | 1.5, 1, 2, 2.5 | 10, 20, 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peske, E.A.; Foerster, I.M.; Seames, W.S. Scale-Up for the Conversion of Corn Stover-Derived Levulinic Acid into 2-Methyltetrahydrofuran. Catalysts 2023, 13, 972. https://doi.org/10.3390/catal13060972
Peske EA, Foerster IM, Seames WS. Scale-Up for the Conversion of Corn Stover-Derived Levulinic Acid into 2-Methyltetrahydrofuran. Catalysts. 2023; 13(6):972. https://doi.org/10.3390/catal13060972
Chicago/Turabian StylePeske, Eli A., Ian M. Foerster, and Wayne S. Seames. 2023. "Scale-Up for the Conversion of Corn Stover-Derived Levulinic Acid into 2-Methyltetrahydrofuran" Catalysts 13, no. 6: 972. https://doi.org/10.3390/catal13060972
APA StylePeske, E. A., Foerster, I. M., & Seames, W. S. (2023). Scale-Up for the Conversion of Corn Stover-Derived Levulinic Acid into 2-Methyltetrahydrofuran. Catalysts, 13(6), 972. https://doi.org/10.3390/catal13060972