Abstract
The increasing concentration of anthropogenic CO2 in the air is one of the main causes of global warming. The Paris Agreement at COP 21 aims to reach the global peak of greenhouse gas emissions in the second half of this century, with CO2 conversion towards valuable added compounds being one of the main strategies, especially in the field of heterogeneous catalysis. In the current search for new catalysts, the deposition of metallic nanoparticles (NPs) supported on metal oxides and metal carbide surfaces paves the way to new catalytic solutions. This review provides a comprehensive description and analysis of the relevant literature on the utilization of metal-supported NPs as catalysts for CO2 conversion to useful chemicals and propose that the next catalysts generation can be led by single-metal-atom deposition, since in general, small metal particles enhance the catalytic activity. Among the range of potential indicators of catalytic activity and selectivity, the relevance of NPs’ size, the strong metal–support interactions, and the formation of vacancies on the support are exhaustively discussed from experimental and computational perspective.
1. Introduction
Carbon dioxide (CO2) is the second most abundant greenhouse gas in the Earth’s atmosphere due to the vast and excessive emissions from human activities related to the burning of fossil fuels [1] with concomitant environmental problems [2]. It is estimated that around 80% of the world’s energy demand is supplied by fossil fuels. These anthropogenic emissions are considered to be the major contributors to climate change which may cause extreme events (high or low temperature, dryness, etc.) that are beyond human control. Some technologies have been introduced to reduce the CO2 emissions [3], although they have been increased since the 1960s to a current level of more than 400 ppm in 2020 [4], as illustrated in Figure 1. Moreover, predictions indicate that CO2 emissions will continue to increase until at least 2040 [5], with devastating consequences for the environment.
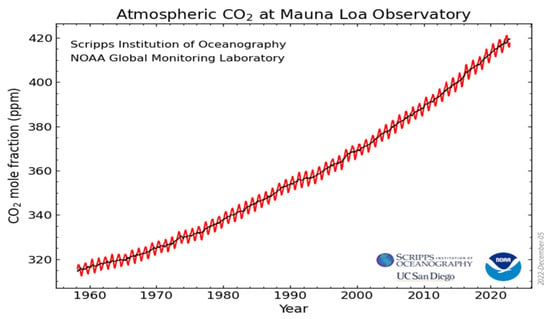
Figure 1.
The constant increase in CO2 concentrations in the atmosphere over the last sixty years. The red and black lines indicate the monthly mean values and the seasonal average, respectively. Adapted from https://www.esrl.noaa.gov/gmd/ccgg/trends/full.html (accessed date: 4 January 2022).
It has become urgent to mitigate the harmful effects of CO2 emissions, CO2 capture and storage (CCS), and especially its conversion towards valuable fuels and precursors. Many studies have been conducted with the aim of providing the effective capture and sequestration of CO2 [6,7]. The deployment of CCS schemes is a multifaceted problem that requires the shared vision and efforts of governments, policy makers, and economists, as well as scientists, engineers, and venture capitalists [8,9]. The Intergovernmental Panel on Climate Change estimates that CO2 emissions into the atmosphere could be reduced by 80–90% for a modern conventional power plant equipped with carbon capture and storage technology [10]. CO2 adsorption is considered one of the most promising technologies for CCS [11], where natural zeolites, metal organic frameworks (MOFs) [12], ionic liquids [13,14], and Fe3O4-graphene are the conventional adsorbents given their capacity to adsorb CO2 (222–2640 mg CO2/g sorbent) at a laboratory scale [15,16].
Nevertheless, it seems clear that efforts should be directed towards the potential use of CO2 as an economical feedstock [17,18,19], because this means that two targets would be hit with one shot [20,21,22,23]. The number of emitted tons of CO2 can be reduced while producing useful chemicals [24,25,26]. In addition, the net amount of CO2 mitigated by conversion with renewable energy is 20–40 times greater than sequestration over a 20-year period [27,28]. To substantially reduce CO2 emissions via catalytic conversion, only reactions that produce fuels or commodity chemicals can be considered viable and economically feasible solutions. The demand for fine chemicals is simply not great enough to effectively reduce emissions through a CO2 conversion process [29]. Due to this, Figure 2 summarizes different ways of CO2 conversion. CO2 reduction has become an interesting option since the CO produced could be used as feedstock in Fischer–Tropsch synthesis [30]. This is an industrial catalytic process used to produce synthetic hydrocarbons to be used as fuels, which has been known since the 1940s. Furthermore, CO is also a key ingredient of syngas, a mixture of CO, CO2, and H2 that is used in many industrial processes to produce tons of chemicals such as methane (CH4) [31,32] or methanol (MeOH) [33]. In this sense, the CO2 conversion to MeOH emerges as the most important method of CO2 recovery, since it is the most direct route for CO2 utilization. In addition, MeOH is a fuel for batteries and a precursor to many interesting chemicals [34], and it is very remarkable that the use of methanol as a transportation fuel presents economic advantages with respect to hydrogen-based fuel cells [35], which unfortunately have not been generalized yet in the automotive world because of the cost of Pt anodes [36]. Finally, the direct hydrogenation of CO2 to alkane species (CO2–Fischer–Tropsch) is possible in a reactor, since it is thermodynamically easier than the reverse water gas shift reaction (WGSR) because the overall process is exothermic [37].
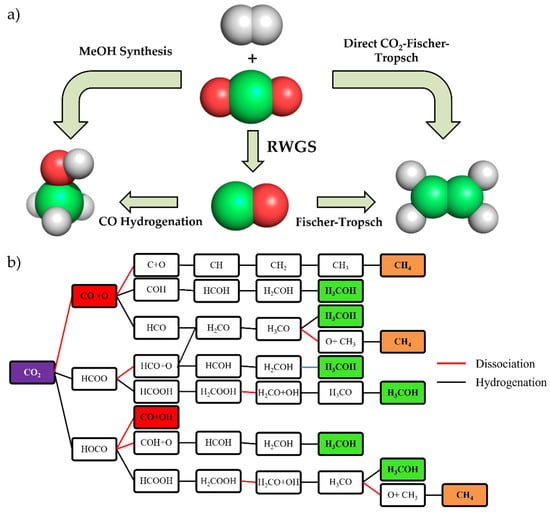
Figure 2.
(a) Simplified scheme about different CO2 hydrogenation pathways. (b) Reaction map of CO2 hydrogenation including reaction intermediates. Red, orange, and green panels show the final products.
Many examples of the use of nanoparticles (NPs) supported on metal oxides to collect CO2 are known. Different methods can be employed to synthesize NPs of different sizes and shapes, including top-down and bottom-up approaches [38]. As an example, on the use of NPs to convert CO2, the RWGS (CO2 + H2 → CO + H2O) reaction occurs on well-dispersed NPs supported on metal oxides to maximize the interface area between the particle and support [39]. Table 1 summarizes selected RWGS catalysts with conversion greater than 20%.

Table 1.
Summary of selected RWGS catalysts with a conversion superior to 20%, including reaction conditions.
On the other hand, the Cu/ZnO/Al2O3 composite [46,47] is the commercial catalyst for MeOH synthesis using a mixture of CO2, CO, and H2 as reactants. Nevertheless, this Cu-supported system requires complex activation steps, particle sintering promotes catalyst deactivation, high pressures are required to obtain good yields, and it is pyrophoric in nature [48]. The poor stability of Cu NPs is the bottleneck for industrial application [49]. A Cu-based catalyst is not only the most commonly used catalyst in methanol synthesis from CO2 hydrogenation but also the best candidate for the WGS and the RWGS reactions [50], which highlight the importance of Cu, since both CO2 and CO can be hydrogenated to MeOH [51]. The catalytic pathways controlling these processes catalyzed by Cu surfaces or Cu-supported metal particles have been extensively investigated in order to implement this sustainable chemistry on a large scale [52], i.e., to find a low-cost and highly active, selective, and stable catalyst for the conversion of CO2 into useful fuels [53,54,55,56]. Furthermore, hydrogenation to MeOH or CO is not the only way to recycle CO2. CO2 methanation highlights another alternative to produce useful hydrocarbons. In this sense, Ni is the most used catalyst [57,58]. Cu, Ni, Au, Pt, Pd, and Ru NPs supported on reducible metal oxides such as Al2O3, TiO2, SiO2, ZrO, and mainly CeO2 have been employed as dual-functional catalysts: the oxide supports provide oxygen vacancies to activate CO2, and metal active sites dissociate molecular hydrogen. Atomic hydrogen then spills over onto the support or onto interfacial sites/vacancies to hydrogenate the adsorbed CO2 [59,60]. The importance of oxygen vacancies on the support is essential for many reactions [61,62,63]. Oxide materials with ionic characteristics and wide band gaps (such as MgO and Al2O3 among others) are working just as supports of metal particles in many reactions, i.e., they do not participate in catalytic conversion [64]. However, metal oxides with lower ionic characteristics and small band gaps (for instance, CeO2) can participate in the catalytic reaction, not only working as simple spectators [65], but directly involving the metal and its support in the catalytic process [66]. In this sense, transition metal carbide materials appeared in the last two decades as a clear example of how metal↔support interactions can modify the catalytic activity of a system with respect to the metal and the support working independently.
In this paper, we briefly review a series of studies that explore the conversion of CO2 to CO, MeOH, and CH4 using metal NPs supported on metal oxides and transition metal carbides. Understanding the reaction mechanism is a rewarding goal that requires the combination of computational techniques based on multiscale modeling [67] together with sophisticated experiments able to determine the surface-active sites and metal–support interactions. This review therefore aims at determining the key descriptors about the performance of metal-supported NPs with the goal to find better (until now) NP–support combinations for carbon dioxide conversion, either to MeOH, CO, or CH4. We will emphasize (i) the relation of metal–support interactions and the catalytic activity of the system, (ii) the importance of NPs’ sizes and shapes, and (iii) the role of the support as an active phase or a mere spectator. In addition, we briefly describe the progress of metal single-atom-site catalysts, with special emphasis on supported metal atoms that maximize the reactivity of many catalytic applications.
2. Metal Nanoparticles Supported on Metal Oxides
2.1. Al2O3
Alumina is considered as a catalytically inert support although essential to enhance the catalytic activity of metal NPs and to store hydroxyl moieties, as we review in this section. Carbon dioxide hydrogenation on Cu/Al2O3 was studied by Lam and coworkers by combining experiments and theoretical calculations [68], concluding that this system works as a bifunctional catalyst with high activity for the hydrogenation of CO2 to MeOH, dimethyl ether, and CO, in contrast to what is observed with SiO2 or ZrO2 supports. Moreover, Lam et al. reported that binary Cu/Al2O3 systems work better than ternary ones (see Figure 3a). The main formation of CO as a key intermediate was previously determined by Chen et al. [40]. Song and coworkers recently revealed the role of the terminal hydroxyl groups on Cu/Al2O3 systems, being the active sites to generate HCOO species during the hydrogenation of CO2. The role of Cu NPs was to break H2 molecules and store the H atoms necessary for the hydrogenation process [69]. The proposed reaction mechanism by Song et al. is depicted in Figure 3b, illustrating how OH moieties adsorbed on the oxide support participate in the reaction. Promotion with Bi and K enhances the selectivity of Cu/Al2O3 catalysts to MeOH and CO, respectively, according to Bansode et al. [70]. Pastor-Pérez and coworkers prepared a series of bimetallic Cu-Fe catalysts supported on Alumina for a RWGS reaction, in which the addition of Cu and Cs particles as promoters increases the activity and stability toward CO [71].
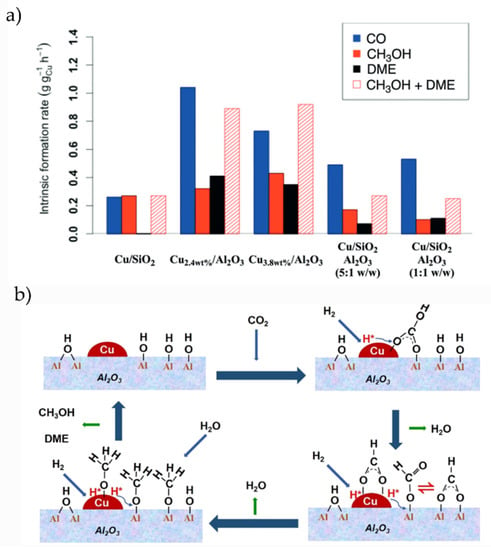
Figure 3.
(a) Intrinsic formation rates for CO, MeOH, and DME after the deposition of Cu NPs on different oxide supports. Extracted with permission from Ref. [66]. Copyright Wiley-VCH. (b) Proposed reaction mechanism for CO2 hydrogenation over Cu/Al2O3, extracted with permission from Ref. [68]. Copyright ACS.
Regarding CO2 methanation, several groups [72,73,74,75] investigated the use of small Ru particles supported on Al2O3. They concluded that Ru/Al2O3 is an excellent catalyst, obtaining 96% of methane yields without CO, a slightly higher yield than the obtained with Ni/Al2O3 [76]. It was revealed that the higher the number of Ni particles, the higher the CO production. The addition of vanadium improved the catalytic activity of Ni/Al2O3 towards CO2 methanation, slightly increasing the methane yield, and again, without detecting the CO generation [77]. In contrast, Italiano and coworkers detected the formation of a NiAl2O4 spinel, which lowers the quantity of active Ni species, decreasing the catalytic activity of the Ni/Al2O3 catalyst [78]. Quindimil et al. experimentally evaluated both Ni and Ru metal particles on Al2O3, reporting that Ni/Al2O3 presents high metal–support interactions, reducing the amount of metal active sites to catalyze CO2 conversion, whereas Ru is a more efficient metal particle for H2 dissociation [79]. Mihet and Lazar studied the influence of Pt, Pd, and Rh promotion on the Ni/Al2O3 system for the methanation of CO2, concluding that Rh shows lower catalytic activity than Ni/Al2O3, whereas Pd and Pt promotion increases the catalytic activity [80]. Regarding other noble metals, Schubert and coworkers proved that the high activity of Co/Al2O3 can be promoted by Pt, which enhances H2 dissociation [81]. Regarding the methanation process, one can summarize that Ru and Ni are the best candidates, although there are some discrepancies about which NP shows better performance.
2.2. ZrO2
Wambach and collaborators [82] exhaustively studied and compared the activity of several metal NPs supported on zirconia catalysts to investigate their performance in CO2 hydrogenation. They reported that Cu and Ag promoted MeOH formation, while Ni, Ru, and Rh tended to catalyze CO2 methanation. Note that these metals also promote the formation of methane using alumina as a support [77,78]. Less selectivity and low activity were found using Pd, Pt, and Au, in which the simultaneous formation of all the byproducts (MeOH, CO, and CH4) was reported. Regarding Cu NPs, their deposition on ZrO2 exhibits slightly higher activity for the conversion of CO2 into MeOH, displaying comparable yields to the commercial catalysts using a very low amount of Cu (1–2 wt %) [83]. Similar results were found by Nitta et al., who reported that Cu/ZrO2 is more effective for MeOH production than Cu/ZnO [84]. Nitta and coworkers also found that the addition of ZnO particles can help carbon dioxide conversion although it decreases the MeOH selectivity. In another study, Nitta et al. suggested that large Cu crystals favor MeOH production and selectivity, while the high dispersion of Zr species enhances carbon dioxide conversion [85]. As reported by Song using an alumina support [69], Meunier and coworkers demonstrated the key effect of hydroxyl groups on the oxide support, since they were characterized as the sites on which carbonate and formate moieties were hydrogenated to methoxy species [86].
Regarding Au NPs, studies by Bogdan et al. and Baiker et al. show that Au/ZrO2 is a very active catalyst for RWGS, with CO being the main product [87,88]. According to Wu and coworkers, the use of very small Au NPs exhibits good activity and selectivity towards MeOH [89]. The use of silver-supported clusters did not show better performance than Cu, even though some papers reported slightly high selectivity towards MeOH [90,91]. The addition of Ag NPs to Cu/ZrO2 did not show an improvement on the catalyst activity according to Tada and coworkers, although it increased the MeOH selectivity [92]. The use of Pd was discarded by Fujitani and coworkers since it is not able to hydrogenate CO2 [93]. Finally, it was found that a mixture of Pt and Co NPs supported on ZrO2 favors methane production [94].
2.3. SiO2
Wang and coworkers prepared a long-lived Cu/SiO2 catalyst synthetized using an ammonia evaporation method, showing excellent performance for CO2 hydrogenation and large catalyst stability. CO, MeOH, and only a low amount of CH4 were found at 260 °C, although the MeOH production drastically decreased at high temperatures [95]. Very recently, Shawabkeh and collaborators computationally studied the interaction of CO2 with a Cu/SiO2 catalyst, concluding that CO2 is physiosorbed and bent on the oxide surface, owing to moderate interaction with one of the oxygen surface atoms [96]. A Cu/SiO2 catalyst prepared via flame spray pyrolysis exhibited comparable catalytic performance to an active Cu/ZrO2 catalyst for MeOH synthesis from CO2, although the conversion was only 5.2%. This experimental investigation showed that Cu NPs avoid CO dissociation and allow its hydrogenation to MeOH [97]. The important role of Cu particles was also suggested by Wang and collaborators [98]. Lam et al. synthetized a Cu-based catalyst using surface organometallic chemistry starting from a material consisting of isolated Zn2+ surface sites dispersed on SiO2, and then generating CuZn alloys. This Cu-Zn/SiO2 material displayed high catalytic activity, methanol selectivity, and higher conversion compared to the benchmark Cu/ZnO/Al2O3 and most other catalysts [99]. Very recently, Fayisa and coworkers found that Pt could be an effective promoter to enhance the catalytic performance of Cu/SiO2 for the hydrogenation of CO2 to MeOH [100].
The combination of Cu and Ni particles on SiO2 was investigated for CO2 methanation, concluding that higher Ni content increases CO2 conversion and CH4 selectivity, and Cu-promoted samples favor the CO selectivity [101]. Therefore, it seems clear that Ni NPs favor methanation, whereas Cu NPs enhance hydrogenation into MeOH. The catalytic activity of Ni supported on SiO2 for CH4 production was reported by several authors [102,103,104], showing the high conversion of carbon dioxide and 100% selectivity to CH4 (Figure 4). The work by Wu and collaborators revealed the importance of Ni coverage and size, since at low Ni loading (0.5 wt%), the system showed comparatively higher catalytic activity for CO2 hydrogenation with large CO selectivity, whereas increasing the content to 10 wt%—9 nm particles—a selectivity switch was observed, favoring CH4 production [105]. The promotion of Ni/SiO2 with other metal particles such as Fe, Co, and Zn was studied by Dias and Perez-Lopez, showing that Fe and Co enhanced CO2 conversion and selectivity towards CH4, while Zn did not favor CO2 conversion and promoted the production of CO [106]. Huang et al. recently investigated the combination of Ni and Pt particles on SiO2, reporting that CH4 was the main product only when the Ni/Pt molar ratio was higher than 9, owing to the fact that Pt NPs can directly dissociate CO2 to CO [107]. The addition of Sn to Ni/SiO2 slightly improved the activity and the catalytic stability at 650 °C, reducing the carbon formation, although a very small amount of Sn had a negative effect on the activity and stability of the Ni/SiO2 catalyst [108]. Finally, Kattel and coworkers reported that Pt NPs are able to enhance the overall CO2 conversion due to the CO2 binding energy on SiO2 with oxygen vacancies [109].
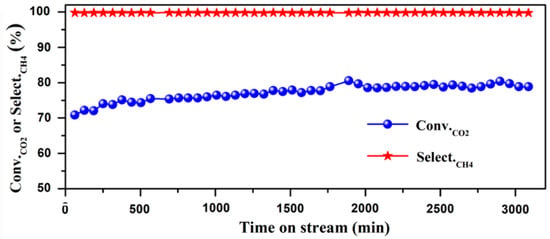
Figure 4.
Catalytic performance of CO2 methanation on Ni/SiO2 catalysts, reproduced with permission from Ref. [104]. Copyright Elsevier B. V.
2.4. TiO2
Titanium oxide is not an inert support. It exhibits great potential as an ideal and powerful photocatalyst for various significant reactions due to their chemical stability, nontoxicity, and high reactivity [110]. TiO2-supported metal catalysts have attracted interest due to the characteristics of TiO2 NPs, showing high activity for various reduction and oxidation processes [111].
As reported using other oxide supports, the deposition of Cu on TiO2 enhances the catalytic activity of these systems towards CO2 hydrogenation to CO, MeOH, and CH4 [112,113,114,115,116,117,118]. López-Caballero and collaborators used DFT simulations to prove that small Cu clusters catalyze the direct dissociation of CO2 due to C=O bond activation and the subsequent reduction in the energy barrier for bond breaking [119]. The same tendency was observed in all these investigations. The deposition of Cu NPs on TiO2 photocatalysts showed significantly higher photoactivity for CO2 reduction, and it was more efficient than bare TiO2. Nevertheless, the use of big NPs of Cu does not favor CO2 conversion. In order to achieve enhanced photoactivity, the concentration of Cu particles on TiO2 must be low [120]. It is important to mention that Liu et al. reported that the deposition of Cu on TiO2 nanotubes shows different results for the RWGS reaction with respect to the deposition on TiO2 NPs. This different behavior was attributed to the formation of more O vacancies on the anatase (001) surfaces of nanotubes [121].
Xie and collaborators evaluated the effect of noble metal NPs on the photocatalytic activity of TiO2 for CO2 hydrogenation. The rate of CH4 formation increased as follows: Ag < Rh < Au < Pd < Pt [122]. Jin et al. showed that group XI of transition metals enhances CO selectivity, while group X promotes the methane production [123]. Li and coworkers studied Pd/TiO2 catalysts, showing the importance of Pd-TiO2 interaction and the importance of controlling the size of Pd particles [124]. The performance with respect to CO2 hydrogenation was better than bare TiO2. In addition, Pd particles not only can capture migrating electrons to separate electron−hole pairs but also promote the activation and bending of CO2. Regarding Ni NPs, Zhou and coworkers reported the excellent performance of Ni particles on TiO2 for the methanation of CO2, highlighting the important role of Ni (111) exposed faces and CO as a key intermediate [125]. Li et al. found that Ni/TiO2 NPs enhance the formation of CO2δ− species in the CO2 methanation process, showing 76% conversion. Furthermore, they used DFT simulations to prove that electrons are transferred from Ti to Ni [126]. The study of Vrijburg and collaborators showed that the addition of Mn to Ni/TiO2 catalysts leads to significantly enhanced CO2 methanation activity [127].
Kohno and collaborators reported that Rh/TiO2 produced CO and a very small amount of CH4, thus enhancing the RWGS reaction [128]. Zhou and coworkers concluded that metal–support interactions using anatase or rutile phases of TiO2 can critically control metal–support interactions in Ru/TiO2 systems and their catalytic performances for CO2 hydrogenation. They reported that annealing Ru/rutile-TiO2 in air can enhance CO2 conversion to methane. However, the use of anatase decreases CO2 conversion and promotes CO formation due to the strong metal–support interaction [129]. Zhang and collaborators demonstrated that the pretreatment temperature of Ir/TiO2 catalysts can tune the selectivity of CO2 hydrogenation, enhancing CH4 production or even generating only CO. Encapsulated Ir NPs favor CO, while exposed Ir particles promote methane production [130]. The deposition of Pt has been extensively studied by several authors. Liu et al. found that Pt deposition on TiO2 ultrathin nanosheets exhibited excellent photocatalytic efficiency for the conversion of CO2 into CH4 and CO. Strong metal–support interactions due to the formation of O vacancies improved the capability to bind CO2 [131]. Zhang and coworkers suggested that the methanation process is the preferred reaction for Pt/TiO2 catalysts, with the ability to activate CO2 without H2 assistance. They suggested that the methanation reaction proceeds via the activation of carbon dioxide, the subsequent CO2δ− hydrogenation into HCOO− moiety, its dissociation to CO, and finally, the hydrogenation to CHO, with CH4 being the final product [132]. Kattel and coworkers also studied Pt deposition on TiO2, concluding that the effect of Pt NP is better for TiO2 supports than for SiO2. Carbon monoxide is the main product in both Pt-supported oxide supports, although Pt/TiO2 is slightly more selective to CH4 than Pt/SiO2 [109]. Qiu-Ye and coworkers also found that the deposition of Pt improved the efficiency of CO2 conversion to CH4 [133]. The recent work of Permporn and collaborators also confirmed the excellent performance of Pt. They loaded Pd, Pt, Cu, and Ni on TiO2, concluding that Pt/TiO2 is the best system for CO2 activation and conversion, since Pt provides the largest work function when formed in heterojunction with TiO2 [134]. In conclusion, Pt/TiO2 does not catalyze MeOH production; otherwise, it favors CO and CH4 generation.
With respect to bimetallic combinations, Neatu et al. revealed that the deposition of Au and Cu particles on a TiO2 photocatalyst is an extremely efficient material for the solar-light reduction of CO2 to CH4 [135]. Reñones et al. showed that bimetallic Au and Ag NPs supported on TiO2 are good catalysts for the photocatalytic conversion of CO2 using water as the reducing agent, reporting that the supported bimetallic particles switch selectivity, enhancing methane production [136]. Singhal and Kumar reported that the deposition of two metal particles on TiO2, for instance, AgPd significantly improves methane formation [137].
2.5. ZnO
The commercial catalyst for CO2 hydrogenation towards MeOH is composed of Cu together with ZnO NPs supported on alumina [138]. Therefore, Cu-ZnO interaction has been exhaustively investigated to elucidate the key interactions for this excellent performance. One of the most relevant works was carried out by Kattel and collaborators. Model systems Cu(111), ZnCu(111), ZnO/Cu(111), Cu/ZnO(000ī), and ZnO/Cu/ZnO(000ī) were experimentally synthesized for systematic comparison, including theoretical models of ZnCu and ZnO/Cu(111) [139]. This work reported that ZnO/Cu(111) presents the highest catalytic activity, as well as ZnCu(111), since the Zn is oxidized to ZnO at reaction conditions due to the direct dissociation of carbon dioxide and Zn oxidation. This system showed higher activity than the model of the commercial catalyst, Cu/ZnO(000ī), suggesting that the inverse system (ZnO NPs on Cu surfaces) is more effective than the traditional Cu NPs on ZnO supports. The morphology of a ZnO support was evaluated by Lei et al., concluding that filament-ZnO support is more active than ZnO nanorods after the deposition of Cu NPs [140]. Mahapatra et al. discussed the size of ZnO NPs, concluding that large ZnO NPs are not as chemically active as small ZnO NPs [141]. The work of Le Valant and collaborators revealed that CuZn is not an active site. The key is the presence of a ZnOx shell, in agreement with the work of Kattel [139]. The co-precipitated Cu–ZnO catalyst produces a large amount of CO according to Le Valant et al. [142]. Marcos and collaborators reported that Cu/ZnO/ZrO2 is a promising system for MeOH production [143]. It is likely that CO2 is activated by the generated oxygen vacancies of ZnO, and the Cu phase at the interface assists the molecular rearrangement. The use of a ZnO plate shows higher MeOH selectivity than a ZnO rod, although the latter shows slightly high catalytic activity [144]. Phongprueksathat et al. presented a new method to synthetize a Cu/ZnO catalyst in the absence of alumina, showing excellent catalytic activity [145]. Very recently, Guzmán and coworkers reported that Cu NPs and ZnO exhibited a synergistic effect in hydrogenating CO2 with respect to pure Cu-based catalysts, in which MeOH and CO were the only products, displaying that the selectivity to MeOH changes from 100% at 200 °C to 23% at 300 °C [146].
Going beyond Cu NPs, in 1993, Sakurai and coworkers reported that Au/ZnO showed the highest selectivity to date and yields of CO2 hydrogenation to MeOH in comparison with other metal oxide supports [147]. Chen et al. studied Au deposition on ZnO, concluding that increasing Au loading is directly related with an increase in the Au particle size, leading to a decrease in catalyst activity but enhanced selectivity to MeOH [148]. Hartadi and coworkers reported that the activity of the Au/ZnO catalyst for CO2 hydrogenation is significantly higher than that for CO hydrogenation, and they concluded that both reaction pathways can be produced simultaneously [149]. Later, Abdel-Mageed and collaborators found a rapid formation of O-vacancies in the ZnO surface region in the initial stages of the reaction with the subsequent formation of negatively charged Au sites. This fact implies an increase in CO adsorption and MeOH production [150]. To complement these conclusions, Behm and coworkers demonstrated that increasing the ZnO particle size and maintaining the Au NP size and loading at constant values, the activity and selectivity increased [151]. The variation in MeOH production can be rationalized by optimizing the concentration of oxygen vacancies. On the other hand, the use of ZnO particles enhances the Ni/CeO2 catalyst; it was reported that the addition of ZnO into a Ni/CeO2 catalyst markedly alters the catalyst’s properties and hampers the methanation of CO2, albeit it favors the production of CO via the RWGS reaction. Liao and coworkers recently investigated Ni and Ru deposition on different ZnO support morphologies, concluding that ZnO nanoplates show strong metal–support interactions since Ru NPs exhibits slightly lower interaction with the support and enhance CH4 selectivity [152]. The deposition of Ru on ZnO enhances CO2 methanation with respect to the bare support, where RWGS is the preferred reaction mechanism according to Dreyer et al. [153]. Nevertheless, the Ru/CeO2 system presents great activity. Regarding Pd NPs, the use of light irradiation on Pd/ZnO catalyst showed a higher MeOH yield [154].
In summary, we have clearly highlighted the important role of ZnO for MeOH production. ZnO-supported NPs work remarkably better than ZnO surfaces. The main evidence of this is the structure and performance of the commercial catalyst for CO2 hydrogenation to MeOH, where Cu and ZnO NPs are deposited on alumina, which works as an innocent support.
2.6. CeO2
Ceria has been used as a catalyst support for a long time due to its unique structural properties resulting from the stability of Ce4+ and Ce3+ species, which promotes the formation of oxygen vacancies [155]. Therefore, ceria does not act as an innocent support; otherwise, these O vacancies are the key in most reaction processes [156].
One of the pioneering works in the use of the Cu/CeO2 catalyst was carried out by Graciani and coworkers, combining experiments and DFT simulations. The computed energy barriers for the HCO→H2CO, H2CO→H3CO, and H3CO→H3COH hydrogenation steps were 5.3, 3.5, and 5.0 kcal/mol, respectively, which were very easy to overcome at 450 K. In contrast to the commercial Cu/ZnO/Al2O3 catalyst, the reaction pathway using Cu/CeO2 was presented in general exothermic steps [157]. In the work of Wang et al., Cu NPs were introduced to facilitate the O vacancy concentration on the CeO2 support due to the formation of Ce3+ species, promoting the photocatalytic activity of the Cu/CeO2 system, ∼26 times larger than the bare CeO2 support [158]. Lin and coworkers investigated the Cu deposition on ceria nanorods, revealing that the (110) termination of ceria showed better performance because it enhanced the oxygen vacancy formation. This surface termination also promoted more effective CO2 activation and the formation of bidentate carbonate and formate moieties, characterized by DFT simulations [159]. Figuereido et al. reported that a higher concentration of Cu NPs on the surface under reaction conditions was more reactive [160]. The addition of a second metal NP to Cu/CeO2 did not show the same behavior; the work of Yan and collaborators evaluated the addition of W to Cu/CeO2 for CO2 hydrogenation. W increased and stabilized the concentration of Ce3+, enhancing the methanol production and its selectivity [161]. In contrast, the addition of In into Cu/CeO2 decreased the catalyst activity dispersion of Cu NPs and the formation of oxygen vacancies on CeO2 due to the obstruction of In [162]. The work of Yang et al. revealed that the bimetallic Cu-Fe NPs supported on mixed CeO2-Al2O3 showed the higher conversion of CO2 than the monometallic NPs, boosting H2 dissociation and CO2 activation simultaneously [163].
Wang and collaborators published a series of papers investigating the Au-CeO2 system. In 2013, they found that CO2 interacts with pre-reduced Au/CeO2, reporting the presence of oxygen, thus suggesting the direct dissociation of the RWGS reaction at temperatures above 200 °C [164]. In another study, it was concluded that the RWGS reaction was dominated by the hydrogenation of carbon dioxide at lower temperatures (120 °C) [165]. The deposition of Au exhibits high activity, 10 times higher towards the RWGS reaction under light irradiation compared to the photothermal reaction, exhibiting a selectivity of around 100% to CO according to Lu et al. [166]. Rezvani et al. studied CO2 hydrogenation to MeOH on Au/CeO2, showing slightly lower activity than a Au/ZnO catalyst under similar conditions. The key of the Au/CeO2 system is the support reducibility, i.e., the O vacancy generation and the subsequent Au NP dispersion [167]. On the other hand, although this paper reviews the most relevant binary systems based on metal NPs supported in one support, it is important to mention the role of ceria in ternary systems, as in the case of Au/CeO2/TiO2. The work of Yang and collaborators reported that the addition of 0.1 monolayers of ceria to TiO2 support stabilized the formation of small Au nanoparticles, promoting the catalyst activity in both CO and MeOH production and improving their selectivity towards MeOH with respect to Cu NPs, as illustrated in Figure 5 [168].
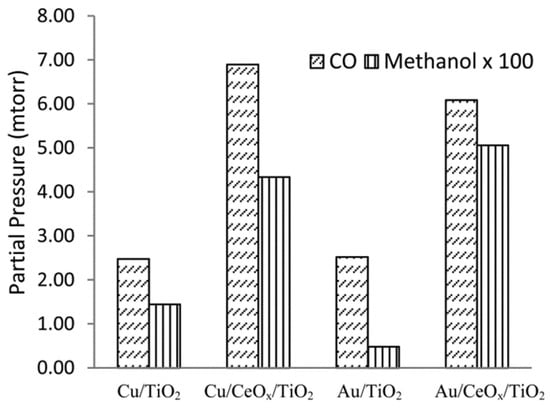
Figure 5.
TPR at 573K of CO2 hydrogenation over Cu/TiO2 and Cu/CeOx/TiO2, Au/TiO2, and Au/CeOx/TiO2 to produce CO and MeOH reproduced with permission from Ref. [168]. Copyright ACS.
The deposition of Ni particles on ceria has been extensively studied to carry out CO2 methanation. Several experimental studies report that ceria is the best support for Ni nanoparticles since this system exhibits higher catalytic activity than other oxide supports [169,170,171]. Due to the oxygen vacancy generation and the dispersion of Ni particles, Ni/CeO2 was reported as the best system for CO2 methanation, with almost 100% selectivity to methane. Rui et al. synthetized Ni/CeO2 catalysts via the decomposition of a nickel precursor using gas discharge plasma, reporting very high activity due to the rich interfacial Ni sites, showing 99% selectivity to methane [172]. Bian and coworkers carried out a kinetic study, again revealing that the oxygen vacancies were thought to be active sites for the formate route [173]. The work of Jomjaree and collaborators studied the effect of ceria morphology, showing that Ni supported on ceria nanopolyhedrons displayed the highest activity for CO2 methanation. They expected ceria nanorods to exhibit the highest catalytic activity due to their high surface area, large amounts of oxygen vacancies, and very strong interaction with Ni. Nevertheless, this strong interaction has a negative impact on CO2 conversion at low temperatures [174]. Vavroutis et al. reported that, surprisingly, larger Ni particles (20 nm) exhibit the highest catalytic activity supported on ceria nanorods compared to Ni particles from 10 to 25 nm, obtaining CH4 selectivity of 99% and a yield of 92%, the highest ever reported below 300 °C [175]. The study of Lin and collaborators also evaluated the importance of Ni particles on ceria surfaces. The Ni particle of 8 nm exhibited superior methanation selectivity over the 4 and 2 nm NPs. In addition, the methanation activity in terms of TOF was 10 times and 70 times higher than the 4 and 2 nm NPs, respectively. The 8 nm Ni NPs supported on CeO2 surfaces were revealed to enhance the formate hydrogenation [176]. Winter et al. studied the addition of Fe in a 3:1 molar ratio of Ni to Fe, reporting similar activity to the Ni catalyst with slightly improved CO selectivity. The catalysts that contained the same amount of Ni and Fe improved the selectivity to CO, but the catalytic activity was lower [177]. Sun and collaborators [178] investigated yttrium doping to Ni/CeO2. CO2 methanation showed that the 2 wt% Y-doping of the Ni/CeO2 catalyst exhibited the best CO2 conversion and high selectivity towards CH4, since this amount of Y enhances the formation of oxygen vacancies in ceria. Doping with La has been recently investigated by Alvarez-Galvan and coworkers, reporting that the optimum value of La concentration is 10%, which corresponds to a maximum oxygen vacancy concentration and the highest CO yield [179]. Moreover, again, they reported the importance of O vacancy formation on ceria support. The system reached an average conversion of 52% and 100% CO selectivity. On the other hand, it has been observed that Ni NPs supported on mixed CeO2 and Al2O3 are a very stable and active catalyst with high CO2 conversion for the RWGS reaction [180].
Regarding other transition metals, Xie and collaborators [181] studied the role of Co nanoparticles on different ceria morphologies, finding that ceria nanorods show great catalytic activity, as it was found for Ni particles [174]. This work shows that Co0 and oxygen vacancies improve the catalytic performance, with 91% selectivity in CH4. Nguyen et al. suggested that CO2 methanation proceeds by means of the CO2 associative pathway, in which carbonate, bicarbonate, and formate intermediates are detected. However, direct dissociation was also observed on Co NPs [182]. López-Rodríguez and coworkers reported that 2.5 wt. Ru% is the optimal loading for Ru/CeO2, since it exhibits good CO2 adsorption and dissociation capacity with the efficient hydrogenation of intermediates on the ceria surface [183]. López-Rodríguez et al. completed this investigation by studying the role of Ru and ceria for CO2 methanation including DFT simulations [184]. They revealed that both metal and supports can activate and dissociate H2 almost without an energy barrier, leading to high H and OH coverages on the catalyst. This fact is suggested to be positive for ceria but negative for Ru. Different Ru sizes were tested by Guo et al., who reported that Ru NPs show the best performance for CO2 methanation [185]. The combination of Ru with Fe NPs promoted the RWGS reaction and the formation of CO. Again, the importance of oxygen vacancies on ceria was highlighted [186]. Iron NPs were found to be active for carbon dioxide conversion on mixed CeO2-Al2O3 supports with high selectivity for RWGS at low temperatures [187]. Moreover, the addition of Ni and Cu particles enhances the catalytic activity and the catalyst robustness. As it was found for all the supports reviewed in this work, Ni doping promotes CO2 methanation and Cu enhances the formation of CO. The use of Pd dimers supported on CeO2 displayed a selectivity of 99% towards ethanol [188]. Jiang et al. revealed that the presence of Pd NPs promotes the formation of oxygen vacancies on the ceria support. Moreover, they performed DFT calculations showing that MeOH formation is likely from the formate (HCOO*) pathway via C−O bond cleavage in H2COOH*, with the reduction of HCOO* to HCOOH* as the rate-limiting step [189]. The deposition of small Pt particles on ceria provides evidence of strong metal−support interactions in Pt/CeO2(111) and Pt/CeO2 powders, leading to systems that bind CO2 well at room temperature [190]. Nevertheless, in 2004, Goguet and coworkers displayed that large amounts of CO on Pt/CeO2 lead to carbon deposition with subsequent deactivation [191].
Finally, it is important to highlight that for single-metal-atom catalysts, the minimum size of metal NPs has been employed for CO2 conversion using ceria support. Zheng and coworkers reported that doping with Ti NPs enhances the catalytic activity of a single Rh atom supported on CeO2 for ethanol production [192].
3. Transition Metal Carbides
Transition metal carbides (TMCs) are formed when carbon atoms, produced, for example, by the decomposition of hydrocarbon molecules, are incorporated into metal interstitial sites. TMCs have unique physical and chemical properties which combine the characteristic features of three different classes of materials: they show the extreme hardness and brittleness of covalent solids, high melting temperatures and simple crystal structures, typical in ionic crystals, and electronic and magnetic properties similar to transition metals [193,194]. TMCs have become a family of materials with an increasing role in heterogeneous catalysis in recent decades because of their chemical properties and low cost. The first and key landmark in this topic was the work of Levy and Boudart, who suggested that tungsten carbides displayed Pt-like behavior in several catalytic reactions such as hydrogenation, dehydrogenation, isomerization, and desulfurization reactions [195]. Since the pioneering work of Levy and Boudart in the 1970s, the number of reactions catalyzed by TMCs has greatly increased [196,197]. Apart from their catalytic activity per se, one of the most promising properties of some TMCs is their capability to modify the electronic structure of small supported metal particles and dramatically increase their catalytic activity through strong metal↔support interactions [198].
3.1. Metal Nanoparticles on TiC(001)
The first attempt to deposit metal NPs on TMCs involved the deposition of Au on TiC, carried out by Roldan-Cuenya and coworkers [199,200]. Theoretical calculations evidenced strong Au↔TiC interactions, in which TiC activated the deposited Au particles [198]. This fact promoted the deposition of different metal particles such as Cu or Ni. It was experimentally and computationally tested that small NPs show better results towards CO2 hydrogenation than big NPs [201,202], and usually, these systems have been modeled with square and planar clusters containing four metal atoms. Focusing on CO2 conversion, it is important to mention that CO2 does not show strong interaction with metals, although the deposition of small metal particles on TiC exhibits the excellent conversion of CO2 to CO and MeOH. Vidal and coworkers demonstrated that TiC produces a slightly lower amount of MeOH and CO as products of CO2 hydrogenation with respect the Cu/ZnO(0001) system, a model of the commercial catalysts, but larger than the Cu(111) surface. However, Cu/ZnO(0001) cannot reach the catalytic performance of Au/TiC and Cu/TiC systems, with the MeOH and CO production being 5–12 times higher than the model of the commercial catalyst, as illustrated in Figure 6a [203]. CO2 activation plays a very important role in the catalyst’s performance. DFT simulations showed a binding energy of −0.62 eV on a bare TiC(001) surface, where the molecule is bent and both C=O bonds are equally elongated (1.29 Å). The interaction became stronger when the CO2 molecule was adsorbed on top of the metal-supported particle, Au4/TiC(001) and Cu4/TiC(001), being −0.68 and −1.12 eV, respectively. According to these studies, the deposition of small particles of Au and Cu metal particles on TiC(001) increased the catalytic activity of CO2 hydrogenation compared to a bare Cu(111) surface and Cu/ZnO(0001). Thus, TiC enhanced the reactivity of the supported metals by 1–2 orders of magnitude. The major product was CO, which was produced via the RWGS reaction. In addition, a substantial amount of methanol was also produced, especially using Cu NPs. Nevertheless, the deposition of Ni NPs changed the selectivity of the reaction. The main product for CO2 hydrogenation using the Ni/TiC system was CH4, which was not observed using Cu- or Au-supported particles [204]. The amount of produced CO was even higher with respect to Cu and Au supported on TiC. A recent computational study revealed that CO2 interaction is stronger in Ni/TiC(001) systems in comparison to bare Ni(111) and TiC(001), with lower dissociation energy barriers. According to experimental studies, small and 2D Ni particles deposited on TiC(001) exhibited larger binding energies and lower dissociation barriers for CO2 compared to 3D particles [205,206].
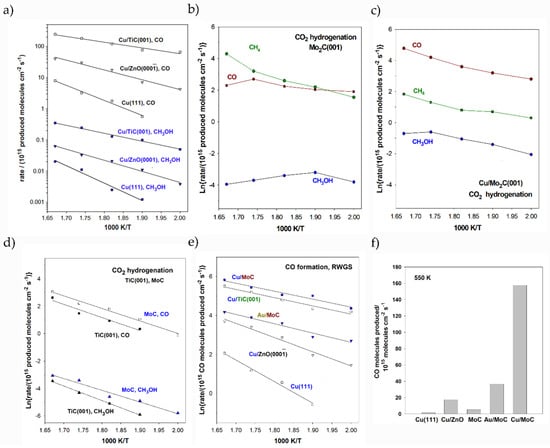
Figure 6.
Arrhenius plots for CO, MeOH, and CH4 production on different bare and metal-supported carbides catalysts at different temperatures. Panel (a) shows the performance of Cu/TiC in comparison to Cu surfaces and the model of commercial catalysts for CO2 hydrogenation. Panel (b,c) illustrate the products for CO2 hydrogenation using bare Mo2C and Cu/Mo2C respectively. Panel (d) compares the performance of TiC and MoC for CO2 hydrogenation evaluating the formation of CO and MeOH. Panels (e,f) compares the CO production among different catalysts. All the reported values correspond to the optimized NP sizes and loadings. Figures adapted with permission from Refs [203,204,207,208]. Copyright ACS, Elsevier B. V., and Royal Chemistry Society.
3.2. Metal Nanoparticles on Molybdenum Carbides
Despite the great performance of titanium carbide material, it is a cumbersome support due to the difficulty of obtaining catalytically active supported NPs on working conditions. In this sense, molybdenum carbides, in particular cubic (δ) MoC and orthorhombic (β) Mo2C, have emerged as excellent alternatives because they are more active and do not require special conditions for their synthesis. Experimental and theoretical investigations were performed to explore the suitability of these Mo carbide phases. The performance of β-Mo2C for CO2 conversion was impressive, since it is able to activate and dissociate CO2 and even CO without the hydrogen assistance [209,210]. Owing to the fact that the β-Mo2C(001) single-crystal surface contains two possible terminations, C or Mo, DFT calculations were essential to elucidate the contribution of each different termination to CO2 hydrogenation, revealing that a Mo-terminated surface is very active and responsible for large amounts of catalytic activity, while a C-terminated surface is able to activate carbon dioxide [207]. Figure 7 represents the CO2 adsorption modes on C- and Mo-terminated surfaces, clearly showing that C-terminated surfaces enlarge one of the C=O bonds. Theoretical calculations showed that the energy barrier for the first C=O scission was only 0.21 eV on the Mo-terminated surface and 0.50 eV for the C-terminated surface, in which the second one was more energy-demanding: 0.86 eV for Mo-terminated and higher than 1.5 eV for C-terminated surfaces [207].
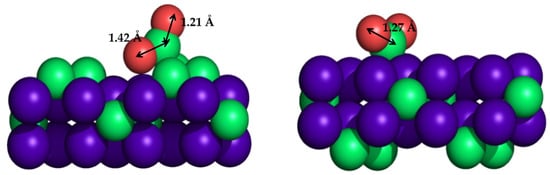
Figure 7.
Sketches of CO2 adsorption mode on orthorhombic (β)-Mo2C(001) surface, showing the C-terminated (left) and Mo-terminated (right) slab models.
The major products for CO2 hydrogenation were CH4 and CO, while the amount of methanol was very low according to the experiments (see Figure 6b). The deposition of Cu NPs on β-Mo2C drastically modified the catalysts’ selectivity. The CO2 conversion increased under the presence of H2, increasing the production of MeOH as well with respect to bare surfaces (Figure 6c) and decreasing the amount of produced CH4. Posada-Pérez and coworkers reported that the larger the Cu NP, the lower the CH4 production [207]. Nevertheless, big Cu particles and Cu monolayers clearly decreased the catalytic activity of β-Mo2C. Theoretical models of Cu-supported clusters were reported by Illas and coworkers [211], showing that Cun clusters in contact with β-Mo2C(001) adopted a planar configuration, independently of the surface termination. The Cu4 NPs were anchored to a Mo-terminated surface to model the Cu/β-Mo2C system. In detail, Posada-Pérez et al. determined that Cu NPs on Cu/β-Mo2C catalysts have a dual function [207]. On the one hand, Cu NPs occupy active surface sites for direct CO2 and CO dissociation, blocking the fast conversion to CH4. On the other hand, DFT simulations conclude that Cu NPs open a new pathway to generate MeOH. Cu-supported particles cannot directly dissociate CO2, but they favor its hydrogenation to COOH and HCOO intermediates, generating a new route to produce MeOH at the Cu-carbide interface. The experimental work of Heracleous and coworkers proved that the addition of 20 wt% Cu slightly decreases the catalysts’ reactivity, although it enhances the MeOH selectivity with respect to methane [212]. Furthermore, Zhang and coworkers computationally proved that Cu-supported particles facilitate CO hydrogenation to an HCO moiety compared to bare β-Mo2C [213], in agreement with the studies of Posada-Pérez [207,208]. The study of Jing and coworkers [214] also demonstrated that the addition of Cu exhibits better catalytic activity for the hydrogenation of CO2, and Zhang et al. demonstrated that Cs and Cu supported on β-Mo2C have a positive impact on the selectivity and activity [215]. As it was found with metal NPs supported on TiC, Cu/β-Mo2C produced larger amounts of MeOH than the model of the commercial catalysts. However, the commercialization of Cu/β-Mo2C is not viable due to its large deactivation. The large reactivity of the carbide support, allowing the CO2 and CO direct dissociations, promotes O deposition, which decreases the catalytic activity of the system [216,217]. On the other hand, the use of supported Ni particles on Mo2C was explored for CO2 methanation. In this particular case, the Ni/Mo2C system was supported on Al2O3. Again, the large influence of metal↔carbide interaction on the catalytic activity was demonstrated, since the conversions of CO2 for the reduced Ni-Mo/Al2O3 catalyst and Ni-Mo2C/Al2O3 catalysts were 5.3% and 13.8%, respectively, with a corresponding selectivity in CH4 of 10.0% and 98.1% [218]. Therefore, the Ni↔Mo2C interaction is more efficient for CO2 methanation than Ni↔Mo interaction.
In order to improve the stability and selectivity of Mo carbides towards the hydrogenation of CO2, the reaction was investigated by decreasing the metal:carbon ratio on the carbide. The surface of MoC experimentally synthetized was best described as polycrystalline [219]. Owing to the complex phase diagram of MoC [220], a well-defined (001) termination of the δ-MoC (001) surface is difficult to synthetize. The ideal δ-MoC(001) surface was modeled computationally [209], exhibiting promising behavior: it can activate the CO2 molecule as it was observed using β-Mo2C but with a moderate adsorption energy and a large dissociation energy barrier, which avoids the formation of atomic oxygen and self-poisoning [208,211,221]. As it was observed for TiC and Cu/TiC systems, δ-MoC and Cu/δ-MoC catalysts only produce CO and MeOH, while CH4 is not detected (Figure 6d). This is because the direct dissociation towards CO is energetically demanding, avoiding the formation of C* + O*, which is directly related with CH4 production and Mo-carbide deactivation [207,220]. Furthermore, δ-MoC shows slightly more major production of CO and MeOH than TiC(001). The deposition of small Cu NPs increases the CO and MeOH production in both β-Mo2C and δ-MoC(001) surfaces (Figure 6e) and increases the selectivity towards both products on β-Mo2C, reducing the methane generation. Therefore, the metal:carbon ratio on the carbide plays a relevant role in the function of Cu NPs: on β-Mo2C, Cu NPs block surface sites for O generation and CH4 production and promote CO2 hydrogenation [222]. In contrast, the deposition of Cu on δ-MoC(001) enhances direct CO2 dissociation to CO on top of Cu clusters [208]. Figure 6f compares all the carbides and models of commercial catalysts for CO2 hydrogenation to CO, highlighting the catalytic activity of Cu/δ-MoC.
On the other hand, other transition metals were deposited on molybdenum carbides, although these systems demonstrated high catalytic activity for the water gas shift reaction instead of the reverse water gas shift. For instance, Nagai and coworkers showed that Co supported on Mo2C is more active than Mo2C, partially avoiding the catalysts’ deactivation [223]. Similar results were reported by Rodriguez and coworkers, using Pt supported on δ-MoC(001) [224], showing high activity, stability, and selectivity at low temperature, and better performance than the typical industrial Cu/ZnO catalyst.
In summary, molybdenum carbides in particular and TMCs in general exhibit excellent catalytic activity towards the hydrogenation of CO2. Nevertheless, the deposition of metal NPs becomes essential to increase the selectivity towards CO and MeOH, especially when the metal/carbon ratio on the carbide is 2. This is related with the large O-carbide interaction, which enhances CO2 and CO dissociation. Thus, anchoring metals with lower oxygen binding energy can avoid these dissociation processes, suppressing methane generation [225]. This was observed for other reactions, such as MeOH decomposition studied by Kelly et al. using Ni-, Rh-, and Au-modified WC surfaces [226]. Ni-, Rh-, and Pt-modified WC surfaces preferably catalyze the C-H scission instead of C-O, disfavoring methane production.
4. Perspective: Single-Metal-Atom Catalysts
One of the general remarks that can be highlighted from this review is the fact that, in general, small metal NPs enhance the catalytic activity of the metal–support system, while the catalytic activity decreases, increasing the NP size. As it has been reported in this work, the support morphology and NP size can drastically modify the reaction selectivity. Focusing on the industrial applications of these heterogeneous catalysts, its facile separation from the products is a very strong point that helps to reduce the operating costs, although it is complicated to control the selectivity. In this sense, homogeneous catalysts avoid this problem since, in general, they are very selective, although the use of precious metals and catalysts’ recovery practically discard their commercialization. Single-atom catalysts (SACs) hold great promise to bridge homogeneous and heterogeneous catalysts since, on the one hand, SACs maximize the metal utilization due to the low coordination of the single metal atom [227], and they have tunability of the catalytic site on the support [228,229,230,231,232,233]. On the other hand, they can reach the selectivity obtained with homogeneous catalysts. Furthermore, SACs contribute to lowering the cost, because the amount of metal decreases. Since the seminal work of Zhang and coworkers, who reported in 2011 that the Pt1/FeOx SAC was three times more active than its nano-Pt counterpart for CO oxidation [234], SACs have become a new frontier in heterogeneous catalysis.
For example, Au single atoms likely occupy surface ceria vacancy sites, thus achieving higher metal loading [235]. Ceria can stabilize transition metals and favor metal dispersion due to the high number of vacancies, becoming an excellent support for many reactions such as the WGS reaction, CO oxidation, CO2 reduction, or CH4 oxidation. The deposition of single metal atoms on a TiO2 support was exhaustively investigated by Chen and coworkers, reporting that the direct dissociation of CO2 to CO using a Cu single atom presents an energy barrier lower than 0.2 eV, i.e., the dissociation is almost spontaneous [236]. Using a Pt single atom, the energy barrier is higher (0.43 eV). Very recently, two-dimensional transition carbides (MXenes) have emerged as very active catalysts for several reactions, since the catalytic activity increases with respect to 3D materials [237]. The deposition of single metal atoms shows excellent catalytic activity; for instance, the Pt1/Ti3−xC2Ty catalyst, in the presence of a diverse range of amines and silanes, could readily fixate CO2 and yield value-added amides with very high conversion and selectivity [238]. Bare MXenes are very reactive, with high metal:carbon ratios, which implies low stability. The deposition of single metal atoms may enhance the support stabilization and improve the catalytic activity, especially focusing on carbon dioxide conversion. These studies, together with the excellent results reviewed in this work about highly active small metal NPs supported on non-innocent metal oxide and metal carbide supports, encourage the investigation of the deposition of single-metal-atom catalysts for CO2 conversion.
The use of highly dispersed SACs is a promising way to increase the catalytic activity of a support. Nevertheless, as it was reported with metal NPs, SACs are very sensitive to the structural and electronic properties of the support. It is commonly assumed that SACs can activate the adsorbed molecules working analogously to homogeneous catalysts, but one should bear in mind that the structure of an active support site and possible defects may play a major role [239]. NPs’ deposition on or close to surface vacancies enhance the catalytic activity of the system, together with the capability of the support to anchor reaction intermediates, such as hydroxyl groups, which have been demonstrated to play a relevant role in CO2 activation. In our opinion, this fact may be more decisive in the performance of SACs. Therefore, to further comprehend and control the catalytic activity and selectivity of SACs, it is mandatory to explore the local coordination environment of the metal on the support, avoiding possible SAC absorption on the material, with the subsequent suppression of the catalytic activity of SACs [240]. The use of SACs may bring new opportunities in CO2 conversion in particular and green chemistry reactions in general, owing to the precise building of catalytically active sites in dimensions of the sub-nanometer scale.
5. Conclusions
In the present review, we discussed selected examples of metallic NPs supported on metal oxides and metal carbide surfaces for the conversion of CO2, covering the most relevant produced fuels and chemicals. A combined analysis of the state of the art of density functional simulations and sophisticated experiments was used to find suitable metal NP and support combinations for the efficient conversion of CO2. The discussion is focused on the evaluation of suitable catalysts to identify opportunities by exploiting these harmful greenhouse gases as economical feedstock, following the main principles of green chemistry.
In this work, we have reported the catalytic activity and selectivity of several metallic NPs supported on different innocent and a priori non-innocent supports, highlighting important descriptors that govern the activity, selectivity and stability of these catalysts, such as metal–support interactions, NP sizes, and support morphology. Some general conclusions can be extracted independently of the metal oxide and metal carbide structure and morphology. In general, Cu stands out among all metal NPs in its enhancement of the RWGS reaction and the subsequent hydrogenation of CO towards MeOH. The use of alumina as a unique support is preferred with respect the ternary catalysts. It was proved that alumina can host hydroxyl moieties, which play a relevant role in the facilitation of CO2 activation and formate production. Furthermore, it is important to highlight the role of ZnO in MeOH production, where experiments and simulations proved that it is more active as a supported NP than as support. In addition, CeO2 is also presented as an excellent support for Cu NPs to enhance the conversion of carbon dioxide. The facile transformation of Ce4+ to Ce3+ species promotes the formation of oxygen vacancies, an essential fact to become a suitable support for catalytic purposes. Regarding the use of transition metal carbides as supports, the formation of CH4 using Cu supported in metal carbide particles was computationally and experimentally observed, which in general was not observed using metal oxides. Nevertheless, it was proved that the large catalytic activity of the support enhances the double bond cleavage of CO2 molecules, favoring the formation of methane, i.e., Cu NPs do not participate in the methanation process. This is because the metal:carbide ratio plays a relevant role in the metal Cu activity. For Mo2C, with a metal:carbon ratio of 2, Cu NPs block active surface sites for direct CO2 and CO dissociations, which lead to the formation of oxycarbide surfaces with the subsequent catalysts’ deactivation. Cu NPs open a new catalytic route for CO2 and CO hydrogenations since they bind weakly–moderately to Cu NPs. For carbides with metal:carbon ratios of 1, such as TiC and MoC, Cu NPs can enhance CO2 hydrogenation and also promote its direct dissociation to CO. For these systems, the methane production was not observed.
Regarding other metal particles, the deposition of Au NPs follows the same trend as Cu, although its performance is lower than Cu NPs. This is not bad for the prospects of catalyst commercialization, since Cu is cheaper than Au. Some works revealed that Au is more effective for the WGS reaction. The use of Ni NPs clearly promotes CO2 methanation, independently of metal oxide and metal carbide support. Other metal particles such as Pt, Pd, and Ru have been reported in this review to show excellent catalytic activity for CO2 methanation, although the take-home message should be simplified, highlighting Cu NPs for CO and MeOH production and the use of Ni NPs for CO2 methanation. No general conclusions can be extracted for the combination of bimetallic particles, since they do not always improve the catalytic performance of the system, either for MeOH production or CO2 methanation. Each combination must be evaluated individually.
Another general conclusion that can be gathered from these selected works is the strong dependence of the catalytic activity of the system and the capability of metal oxide supports to generate oxygen vacancies. These defects have a double role: they favor NP dispersion and enhance CO2 activation. On the other hand, it is well-known and assumed by the research community that strong metal–support interactions govern the catalytic activity of the system, since the stronger the interaction is, the higher the catalytic performance is. Nevertheless, in this review, we reported a few works that suggest that the support morphology with the highest interaction with the metal NPs does not always show the best activity (for instance, CeO2 nanorods and Ni and Ru supported on Al2O3).
The last important descriptor to analyze is the metal NP sizes, concluding that, in general, small metal particles enhance the catalytic activity of CO2 conversion. Therefore, it seems clear that the efforts should be routed towards research regarding a single-metal-atom catalyst, due to its large activity, selectivity, and the reduction in the amount of metal, which clearly benefits its possible commercialization. However, as it has been presented in this review, the number of variables that affect the performance of metal-supported particles (or single metal atoms) is large, such as the formation of oxygen vacancies, the morphology of the support, and the metal–support interactions, among others. Therefore, to investigate the suitability of single-atom catalysts, it is necessary to further explore the interaction of these single atoms with surface defects and different active sites, using computational and experimental techniques, with the goal to correlate the catalytic performance with the coordination site and the coordination environment around the support.
To summarize, the use of CO2 as feedstock is a clear benefit that helps to reduce the impacts of global warming compared to those of current technologies only based on CO2 capture. One of the keys to accelerate this reduction is to find low-cost, stable, selective, and highly active catalysts for the potential conversion of CO2 to fuels and chemicals. Despite the fact that the descriptors that govern this process cannot be overgeneralized, some take-home messages can be derived from our revision. Cu and Ni NPs supported on metal oxides and metal carbides are promising catalysts for the future commercialization of more efficient catalysts, although the full puzzle that combines low kinetic barriers and the thermodynamic stability of catalysts at reaction conditions is not complete. In addition, the inclusion of single metal atoms as a new puzzle piece will promote the investigation of metal–support interactions. We hope that the most relevant descriptors analyzed in this work can help researchers to make progress in CO2 utilization.
Author Contributions
Writing, review and editing, S.P.-P., M.S. and A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministerio de Ciencia e Innovación (projects PID2021-127423NB-I00 to A.P. and PID2020-113711GB-I00 to M.S.) and the Generalitat de Catalunya (project 2021 SGR0623 and ICREA Academia prize 2019 to A.P.). S.P.-P. appreciates the economic support of the Marie Curie fellowship (H2020-MSCA-IF-2020-101020330).
Acknowledgments
A.P. is a Serra Húnter Fellow and thanks the ICREA Academia prize 2019. S.P.-P. thanks the Marie Curie fellowship.
Conflicts of Interest
There are no conflict of interest to declare.
Abbreviations
| CCS | CO2 capture and storage |
| MOF | Metal organic framework |
| NP | Nanoparticle |
| RWGS | Reverse water gas shift reaction |
| SAC | Single-atom catalyst |
| TMC | Transition metal carbides |
| WGSR | Water gas shift reaction |
References
- Lim, L. How to make the most of carbon dioxide. Nature 2015, 526, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Intergovernmental Panel on Climate Change. Climate Change 2013—The Physical Science Basis, 1st ed.; Cambridge University Press: Cambridge, UK, 2014.
- Aresta, A. (Ed.) Carbon Dioxide as Chemical Feedstock; Wiley-VCH: New York, NY, USA, 2010. [Google Scholar]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- U.E.I. Administration. International Energy Outlook 2013. 2013. Available online: http://www.eia.gov/forecasts/ieo/pdf/0484%282013%29.pdf (accessed on 24 December 2022).
- Vummaleti, S.V.C.; Nolan, S.P.; Cavallo, L.; Talarico, G.; Poater, A. How easy is CO2 fixation by M–C bond containing complexes (M = Cu, Ni, Co, Rh, Ir)? Org. Chem. Front. 2016, 3, 19–23. [Google Scholar] [CrossRef]
- Vummaleti, S.V.C.; Nolan, S.P.; Cavallo, L.; Talarico, G.; Poater, A. Mechanism of CO2 fixation by Ir–X Bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 4653–4657. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Review of solutions to global warming, air pollution, and energy security. Energy Environ. Sci. 2009, 2, 148–173. [Google Scholar] [CrossRef]
- D'Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed]
- IPCC. IPCC Special Report on Carbon Dioxide Capture and Storage; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Evaluation of Carbon Dioxide Absorption Characteristics Lithium Ortho-Silicate in Chemical Heat Storage. Int. J. Greenh. Gas Control 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Poater, J.; Gimferrer, M.; Poater, A. Covalent and Ionic Capacity of MOFs To Sorb Small Gas Molecules. Inorg. Chem. 2018, 57, 6981–6990. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Posada-Pérez, S.; Brotons-Rufes, A.; Pajski, J.J.; Vajiha; Kumar, G.; Mateen, A.; Poater, A.; Solà, M.; Chawla, M.; et al. Selective absorption of H2S and CO2 by azole based protic ionic liquids: A combined Density Functional Theory and Molecular Dynamics study. J. Mol. Liq. 2022, 367, 120558. [Google Scholar] [CrossRef]
- Shaikh, A.R.; Ashraf, M.; AlMayef, T.; Chawla, M.; Poater, A.; Cavallo, L. Amino acid ionic liquids as potential candidates for CO2 capture: Combined density functional theory and molecular dynamics simulations. Chem. Phys. Lett. 2020, 745, 137239. [Google Scholar] [CrossRef]
- Cavenati, S.; Grande, C.A.; Rodrigues, A.E. Adsorption Equilibrium of Methane, Carbon Dioxide, and Nitrogen on Zeolite 13X at High Pressures. J. Chem. Eng. Data 2004, 49, 1095–1101. [Google Scholar] [CrossRef]
- Millward, A.R.; Yaghi, O.M. Metal−Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Pomelli, C.S.; Tomasi, J.; Solà, M. Theoretical Study on the Thermodynamics of the Elimination of Formic Acid in the Last Step of the Hydrogenation of CO2 Catalyzed by Rhodium Complexes in the Gas Phase and Supercritical CO2. Organometallics 1998, 17, 3164–3168. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Aomchad, V.; Del Globo, S.; Poater, A.; D’Elia, V. Exploring the potential of Group III salen complexes for the conversion of CO2 under ambient conditions. Catal. Today 2021, 375, 324–334. [Google Scholar] [CrossRef]
- Natongchai, W.; Posada-Pérez, S.; Phungpanya, C.; Luque Urrutia, J.A.; Solà, M.; D'Elia, V.; Poater, A. Enhancing the catalytic performance of group I, II metal halides in the cycloaddition of CO2 to epoxides under atmospheric conditions by cooperation with homogeneous and heterogeneous highly nucleophilic aminopyridines: Experimental and theoretical study. J. Org. Chem. 2022, 87, 2873–2886. [Google Scholar] [CrossRef] [PubMed]
- Arayachukiat, S.; Yingcharoen, P.; Vummaleti, S.V.C.; Cavallo, L.; Poater, A.; D’Elia, V. Cycloaddition of CO2 to challenging N-tosyl aziridines using a halogen-free niobium complex: Catalytic activity and mechanistic insights. Mol. Catal. 2017, 443, 280–285. [Google Scholar] [CrossRef]
- Al Maksoud, W.; Saidi, A.; Samantaray, M.K.; Abou-Hamad, E.; Poater, A.; Ould-Chikh, S.; Guo, X.; Guan, E.; Ma, T.; Gates, B.C.; et al. Docking of tetra-methyl zirconium to the surface of silica: A well-defined pre-catalyst for conversion of CO2 to cyclic carbonate. Chem. Commun. 2020, 56, 3528–3531. [Google Scholar] [CrossRef]
- Natongchai, W.; Luque-Urrutia, J.A.; Phungpanya, C.; Solà, M.; D’Elia, V.; Poater, A.; Zipse, H. Cycloaddition of CO2 to epoxides by highly nucleophilic 4-aminopyridines: Establishing a relationship between carbon basicity and catalytic performance by experimental and DFT investigations. Org. Chem. Front. 2021, 8, 613–627. [Google Scholar] [CrossRef]
- Sodpiban, O.; Del Gobbo, S.; Barman, S.; Aomchad, V.; Kidkhunthod, P.; Ould-Chikh, S.; Poater, A.; D’Elia, V.; Basset, J.-M. Synthesis of Well-defined Yttrium-based Lewis Acids by Capture of a Reaction Intermediate and Catalytic Application for cycloaddition of CO2 to Epoxides Under Atmospheric Pressure. Catal. Sci. Technol. 2019, 9, 6152–6165. [Google Scholar] [CrossRef]
- Coufourier, S.; Gaignard-Gaillard, Q.; Lohier, J.-F.; Poater, A.; Gaillard, S.; Renaud, J.-L. Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes. ACS Catal. 2020, 10, 2108–2116. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef]
- Perathoner, S.; Centi, G. CO2 recycling: A key strategy to introduce green energy in the chemical production chain. ChemSusChem 2014, 7, 1274–1282. [Google Scholar] [CrossRef]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.; Millar, G.J. Carbon Dioxide Reforming of Methane to Produce Synthesis Gas over Metal-Supported Catalysts: State of the Art. Energy Fuels 1996, 10, 896–904. [Google Scholar] [CrossRef]
- Caballero, A.; Pérez, P.J. Methane as raw material in synthetic chemistry: The final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef]
- Liu, X.M.; Lu, G.Q.; Yan, Z.F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Xiaoding, X.; Moulijn, J.A. Mitigation of CO2 by Chemical Conversion: Plausible Chemical Reactions and Promising Products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas: The Methanol Economy; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. (Eds.) Green Carbon Dioxide: Advances in CO2 Utilization; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Baig, N.; Kammakakam, I.; Falath, W. Nanomaterials: A review of synthesis, methods, properties, recent progress, and challenges. Mater. Adv. 2021, 2, 1821–1871. [Google Scholar] [CrossRef]
- Lu, B.W.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D. Study of bimetallic Cu–Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrogen Energy 1999, 24, 351–354. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Takassi, M.A. A novel γ-alumina supported Fe-Mo bimetallic catalyst for reverse water gas shift reaction. Chin. J. Chem. Eng. 2013, 21, 1007–1014. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Ostadi, M. Development of Ni-Mo/Al2O3 Catalyst for Reverse Water Gas Shift (RWGS) Reaction. J. Nanosci. Nanotechnol. 2014, 14, 6841–6847. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support's reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Xu, W.; Ramírez, P.J.; Stacchiola, D.; Brito, J.L.; Rodriguez, J.A. The Carburization of Transition Metal Molybdates (MxMoO4, M = Cu, Ni or Co) and the Generation of Highly Active Metal/Carbide Catalysts for CO2 Hydrogenation. Catal. Lett. 2015, 145, 1365–1373. [Google Scholar] [CrossRef]
- Laudenschleger, D.; Ruland, H.; Muhler, M. Identifying the nature of the active sites in methanol synthesis over Cu/ZnO/Al2O3 catalysts. Nat. Commun. 2020, 11, 3898. [Google Scholar] [CrossRef]
- Beck, A.; Zabilskiy, M.; Newton, M.A.; Safonova, O.; Willinger, M.G.; van Bokhoven, J.A. Following the structure of copper-zinc-alumina across the pressure gap in carbon dioxide hydrogenation. Nat. Catal. 2021, 4, 488–497. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Chen, C.S.; Lin, J.H.; You, J.H.; Chen, C.R. Properties of Cu(thd)2 as a precursor to prepare Cu/SiO2 catalyst using the atomic layer epitaxy technique. J. Am. Chem. Soc. 2006, 128, 15950–15951. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive review of Cu-based CO2 hydrogenation to CH3OH: Insights from experimental work and theoretical analysis. Int. J. Hydrogen Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Liu, Y.M.; Liu, J.T.; Liu, S.Z.; Li, J.; Gao, Z.H.; Zuo, Z.J.; Huang, W. Reaction mechanisms of methanol synthesis from CO/CO2 hydrogenation on Cu2O(111): Comparison with Cu(111). J. CO2 Util. 2017, 20, 59–65. [Google Scholar] [CrossRef]
- Samantaray, M.K.; D'Elia, V.; Pump, E.; Falivene, L.; Harb, M.; Ould Chikh, S.; Cavallo, L.; Basset, J.-M. The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis. Chem. Rev. 2020, 120, 734–813. [Google Scholar] [CrossRef]
- Yang, Y.; Mims, C.A.; Mei, D.H.; Peden, C.H.F.; Campbell, C.T. Mechanistic studies of methanol synthesis over Cu from CO/CO2/H2/H2O mixtures: The source of C in methanol and the role of water. J. Catal. 2013, 298, 10–17. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, K.H.; Lee, S.Y.; Kim, Y.G. A comparative study of methanol synthesis from CO2/H2 and CO/H2 over a Cu/ZnO/Al2O3 catalyst. J. Catal. 1993, 144, 414–424. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Kasatkin, I.; Kurr, P.; Kniep, B.; Trunschke, A.; Schlögl, R. Role of lattice strain and defects in copper particles on the activity of Cu/ZnO/Al2O3 catalysts for methanol synthesis. Angew. Chem. Int. Ed. 2007, 46, 7324–7327. [Google Scholar] [CrossRef]
- Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. 2017, 59, 95–164. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Conner, W.C.; Falconer, J.L. Spillover in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 759–788. [Google Scholar] [CrossRef]
- Shen, H.; Li, H.; Yang, Z.; Li, C. Magic of hydrogen spillover: Understanding and application. Green Energy Environ. 2022, 7, 1161–1198. [Google Scholar] [CrossRef]
- Massaro, A.; Pecoraro, A.; Hernandez, S.; Talarico, G.; Munoz-Garcia, A.B.; Pavone, M. Oxygen evolution reaction at the Mo/W-doped bismuth vanadate surface: Assessing the dopant role by DFT calculations. Mol. Catal. 2022, 517, 112036. [Google Scholar] [CrossRef]
- Zheng, Y.; Fu, K.; Yu, Z.; Su, Y.; Han, R.; Liu, Q. Oxygen vacancies in a catalyst for VOCs oxidation: Synthesis, characterization, and catalytic effects. J. Mater. Chem. A 2022, 10, 14171–14186. [Google Scholar] [CrossRef]
- Baiano, C.; Schiavo, E.; Gerbaldi, C.; Bella, F.; Meligrana, G.; Talarico, G.; Maddalena, P.; Pavone, M.; Muñoz-García, A.B. Role of surface defects in CO2 adsorption and activation on CuFeO2 delafossite oxide. Mol. Catal. 2020, 496, 111181. [Google Scholar] [CrossRef]
- Flórez, E.; Feria, L.; Viñes, F.; Rodriguez, J.A.; Illas, F. Effect of the Support on the Electronic Structure of Au Nanoparticles Supported on Transition Metal Carbides: Choice of the Best Substrate for Au Activation. Phys. Chem. Chem. Phys. 2012, 14, 427–438. [Google Scholar] [CrossRef]
- Anderson, J.A.; Fernández-García, M. (Eds.) Supported Metals in Catalysis; Catalytic Science Series; Imperial College Press: London, UK, 2005; Volume 5. [Google Scholar]
- Rodriguez, J.A.; Stacchiola, D. Catalysis and the nature of mixed-metal oxides at the nanometer level: Special properties of MOx/TiO2(110) {M= V, W, Ce} surfaces. Phys. Chem. Chem. Phys. 2010, 12, 9557–9565. [Google Scholar] [CrossRef]
- Samanta, B.; Morales-García, A.; Illas, F.; Goga, N.; Anta, J.A.; Calero, S.; Bieberle-Hütter, A.; Libisch, F.; Muñoz-García, A.B.; Pavone, M.; et al. Challenges of modeling nanostructured materials for photocatalytic water splitting. Chem. Soc. Rev. 2022, 51, 3794–3818. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 Hydrogenation on Cu/Al2O3: Role of the Metal/Support Interface in Driving Activity and Selectivity of a Bifunctional Catalyst. Angew. Chem. Int. Ed. 2019, 58, 13989–13996. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yang, C.; Li, X.; Wang, Z.; Pei, C.; Zhao, Z.J.; Gong, J. On the Role of Hydroxyl Groups on Cu/Al2O3 in CO2 Hydrogenation. ACS Catal. 2022, 12, 14162–14172. [Google Scholar] [CrossRef]
- Bansode, A.; Tidona, B.; von Rohr, P.R.; Urakawa, A. Impact of K and Ba promoters on CO2 hydrogenation over Cu/Al2O3 catalysts at high pressure. Catal. Sci. Technol. 2013, 3, 767–778. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-García, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The effect of CO on CO2 methanation over Ru/Al2O3 catalysts: A combined steady-state reactivity and transient DRIFT spectroscopy study. Appl. Catal. B Environ. 2019, 256, 117791. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szany, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, C.; Chu, W.; Zhou, Y.; Köhler, K. CO2 Methanation over Supported Ru/Al2O3 Catalysts: Mechanistic Studies by In situ Infrared Spectroscopy. ChemistrySelect 2016, 1, 3197–3203. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Magistri, L.; Busca, G. A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure. Int. J. Hydrogen Energy 2014, 39, 11557–11565. [Google Scholar] [CrossRef]
- Garbarino, G.; Kowalik, P.; Riani, P.; Antoniak-Jurak, K.; Pieta, P.; Lewalska-Graczyk, A.; Lisowski, W.; Nowakowski, R.; Busca, G.; Pieta, I.S. Improvement of Ni/Al2O3 Catalysts for Low-Temperature CO2 Methanation by Vanadium and Calcium Oxide Addition. Ind. Eng. Chem. Res. 2021, 60, 6554–6564. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Ferraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B Environ. 2020, 264, 118494. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; González-Marcos, J.A.; Bueno-López, A.; González-Velasco, J.R. Effect of metal loading on the CO2 methanation: A comparison between alumina supported Ni and Ru catalysts. Catal. Today 2020, 356, 419–432. [Google Scholar] [CrossRef]
- Mihet, M.; Lazar, M.D. Methanation of CO2 on Ni/γ-Al2O3: Influence of Pt, Pd or Rh promotion. Catal. Today 2018, 306, 294–299. [Google Scholar] [CrossRef]
- Schubert, M.; Pokhrel, S.; Thomé, A.; Zielasek, V.; Gesing, T.M.; Roessner, F.; Madler, L.; Baumer, M. Highly active Co–Al2O3-based catalysts for CO2 methanation with very low platinum promotion prepared by double flame spray pyrolysis. Catal. Sci. Technol. 2016, 6, 7449–7460. [Google Scholar] [CrossRef]
- Wambach, J.; Baiker, A.; Wokaun, A. CO2 hydrogenation over metal/zirconia catalysts. Phys. Chem. Chem. Phys. 1999, 1, 5071–5080. [Google Scholar] [CrossRef]
- Denise, B.; Cherifi, O.; Bettahar, M.M.; Sneeden, R.P.A. Supported Copper Catalysts Prepared from Copper(II) Formate: Hydrogenation of Carbon Dioxide Containing Feedstocks. Appl. Catal. 1989, 48, 365–372. [Google Scholar] [CrossRef]
- Nitta, Y.; Suwata, O.; Ikeda, Y.; Okamoto, Y.; Imanaka, T. Copper-zirconia catalysts for methanol synthesis from carbon dioxide: Effect of ZnO addition to Cu-ZrO2 catalysts. Catal. Lett. 1994, 26, 345–354. [Google Scholar] [CrossRef]
- Nitta, Y.; Fujimatsu, T.; Okamoto, Y.; Imanaka, T. Effect of starting salt on catalytic behaviour of Cu-ZrO2 catalysts in methanol synthesis from carbon dioxide. Catal. Lett. 1993, 17, 157–165. [Google Scholar] [CrossRef]
- Meunier, F.C.; Dansette, I.; Eng, K.; Schuurman, Y. Differentiating the Reactivity of ZrO2-Bound Formates Formed on Cu/ZrO2 during CO2 Hydrogenation. Catalysts 2022, 12, 793. [Google Scholar] [CrossRef]
- Bogdan, V.I.; Koklin, A.E.; Nikolaev, S.A.; Kustov, L.M. Carbon dioxide hydrogenation on Au nanoparticles supported on TiO2, ZrO2 and sulphated ZrO2 under supercritical conditions. Top. Catal. 2016, 59, 1104–1109. [Google Scholar] [CrossRef]
- Baiker, A.; Kilo, M.; Maciejewski, M.; Menzi, S.; Wokaun, A.; In Guczi, L.; Solymosi, F. Hydrogenation of CO2 over copper, silver and gold/zirconia catalysts: Comparative study of catalyst properties and reaction pathways. Stud. Surf. Sci. Catal. 1993, 75, 1257–1272. [Google Scholar]
- Wu, C.Y.; Zhang, P.; Zhang, Z.F.; Zhang, L.J.; Yang, G.Y.; Han, B.X. Efficient hydrogenation of CO2 to methanol over supported subnanometer gold catalysts at low temperature. ChemCatChem 2017, 9, 3691–3696. [Google Scholar] [CrossRef]
- Grabowski, R.; Sloczynski, J.; Sliwa, M.; Mucha, D.; Socha, R.P.; Lachowska, M.; Skrzypek, J. Influence of polymorphic ZrO2 phases and the silver electronic state on the activity of Ag/ZrO2 catalysts in the hydrogenation of CO2 to methanol. ACS Catal. 2011, 1, 266–278. [Google Scholar] [CrossRef]
- Fröhlich, C.; Köppel, R.A.; Baiker, A.; Kilo, M.; Wokaun, A. Hydrogenation of carbon dioxide over silver promoted copper/ zirconia catalysts. Appl. Catal. A 1993, 106, 275–293. [Google Scholar] [CrossRef]
- Tada, S.; Watanabe, F.; Kiyota, K.; Shimoda, N.; Hayashi, R.; Takahashi, M.; Nariyuki, A.; Igarashi, A.; Satokawa, S. Ag addition to CuO-ZrO2 catalysts promotes methanol synthesis via CO2 hydrogenation. J. Catal. 2017, 351, 107–118. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I. Methanol synthesis from CO and CO2 hydrogenations over supported palladium catalysts. Bull. Chem. Soc. Jpn. 2002, 75, 1393–1398. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.T.; Yang, X.F.; Yan, B.H.; Huang, Y.Q.; Wan, W.M.; Liu, P.; Chen, J.G.G. CO2 Hydrogenation over oxide supported PtCo catalysts: The role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Xu, Z.-N.; Peng, S.-Y.; Zhang, M.-J.; Lu, G.; Cehn, Q.-S.; Chen, Y.; Guo, G.-C. High-Performance and Long-Lived Cu/SiO2 Nanocatalyst for CO2 Hydrogenation. ACS Catal. 2015, 5, 4255–4259. [Google Scholar] [CrossRef]
- Shawabkeh, R.A.; Faqir, N.M.; Rawajfeh, K.M.; Hussein, I.A.; Hamza, A. Adsorption of CO2 on Cu/SiO2 nano-catalyst: Experimental and theoretical study. Appl. Surf. Sci. 2022, 586, 152726. [Google Scholar] [CrossRef]
- Yu, J.; Yang, M.; Zhang, J.; Ge, Q.; Zimina, A.; Pruessmann, T.; Zheng, L.; Grunwaldt, J.D.; Sun, J. Stabilizing Cu+ in Cu/SiO2 Catalysts with a Shattuckite-Like Structure Boosts CO2 Hydrogenation into Methanol. ACS Catal. 2020, 10, 14694–14706. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Xu, Z.-N.; Zhang, M.-J.; Chen, Q.-S.; Chen, Y.; Guo, G.-C. Insight into composition evolution in the synthesis of high-performance Cu/SiO2 catalysts for CO2 hydrogenation. RSC Adv. 2016, 6, 25185–25190. [Google Scholar] [CrossRef]
- Lam, E.; Noh, G.; Larmier, K.; Safonova, O.V.; Copéret, C. CO2 Hydrogenation on Cu-Catalysts Generated from ZnII Single-Sites: Enhanced CH3OH Selectivity Compared to Cu/ZnO/Al2O3. J. Catal. 2021, 394, 266–272. [Google Scholar] [CrossRef]
- Fayisa, B.A.; Xi, Y.; Yang, Y.; Gao, Y.; Li, A.; Wang, M.-Y.; Lv, J.; Huang, S.; Wang, Y.; Ma, X. Pt-modulated Cu/SiO2 catalysts for efficient hydrogenation of CO2-derived ethylene carbonate to methanol and ethylene glycol. Chin. J. Chem. Eng. 2022, 41, 366–373. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. Carbon dioxide methanation over Ni-Cu/SiO2 catalysts. Energy Convers. Manag. 2020, 203, 112214. [Google Scholar] [CrossRef]
- Le, T.A.; Kang, J.K.; Park, E.D. CO and CO2 Methanation Over Ni/SiC and Ni/SiO2 Catalyst. Top. Catal. 2018, 61, 1537–1544. [Google Scholar] [CrossRef]
- Ye, R.-P.; Gong, W.; Sun, Z.; Sheng, Q.; Shi, X.; Wang, T.; Yao, Y.; Razink, J.J.; Lin, L.; Zhou, Z.; et al. Enhanced stability of Ni/SiO2 catalyst for CO2 methanation: Derived from nickel phyllosilicate with strong metal-support interactions. Energy 2019, 188, 116059. [Google Scholar] [CrossRef]
- Ye, R.-P.; Liao, L.; Ramirez Reina, T.; Liu, J.; Chevella, D.; Jin, Y.; Fan, M.; Liu, J. Engineering Ni/SiO2 catalysts for enhanced CO2 methanation. Fuel 2021, 285, 119151. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Dias, Y.R.; Perez-Lopez, O.W. CO2 conversion to methane using Ni/SiO2 catalysts promoted by Fe, Co and Zn. J. Environ. Chem. Eng. 2021, 9, 104629. [Google Scholar] [CrossRef]
- Huang, Z.; Yuan, Y.; Song, M.; Hao, Z.; Xiao, J.; Cai, D.; Ibrahim, A.-R.; Zhan, G. CO2 hydrogenation over mesoporous Ni-Pt/SiO2 nanorod catalysts: Determining CH4/CO selectivity by surface ratio of Ni/Pt. Chem. Eng. Sci. 2022, 247, 117106–117119. [Google Scholar] [CrossRef]
- Pantaleo, G.; La Parola, V.; Testa, M.L.; Venezia, A.M. CO2 Reforming of CH4 over SiO2-Supported Ni Catalyst: Effect of Sn as Support and Metal Promoter. Ind. Eng. Chem. Res. 2021, 60, 18684–18694. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Julkapli, N.M.; Hamid, S.B.A. Titanium Dioxide as a Catalyst Support in Heterogeneous Catalysis. Sci. World J. 2014, 727496. [Google Scholar] [CrossRef] [PubMed]
- In, S.I.; Vaughn, D.D.; Schaak, R.E. Hybrid CuO-TiO2−xNx Hollow Nanocubes for Photocatalytic Conversion of CO2 into Methane under Solar Irradiation. Angew. Chem. Int. Ed. 2012, 51, 3915–3918. [Google Scholar] [CrossRef]
- Yang, C.C.; Yu, Y.H.; van der Linden, B.; Wu, J.C.; Mul, G. Artificial Photosynthesis over Crystalline TiO2-Based Catalysts: Fact or Fiction? J. Am. Chem. Soc. 2010, 132, 8398–8406. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Liu, M.-H.; Ura, K.; Noguchi, T.G.; Yoshizawa, A.; Kato, K.; Sugiyama, T.; Yamauchi, M. Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane. Catalysts 2022, 12, 478. [Google Scholar] [CrossRef]
- Tseng, I.-H.; Wu, J.C.S.; Chou, H.-Y. Effects of sol–gel procedures on the photocatalysis of Cu/TiO2 in CO2 photoreduction. J. Catal. 2004, 221, 432–440. [Google Scholar] [CrossRef]
- Slamet, N.; Nasution, H.; Purnama, E.; Kosela, S.; Gunlazuardi, J. Photocatalytic Reduction of CO2 on Copper-Doped Titania Catalysts Prepared by Improved-Impregnation Method. Catal. Commun. 2005, 6, 313–319. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, W.; Miao, W.; Yuan, Z.; Yang, G.; Kong, F.; Yan, T.; Chen, J.; Huang, B.; An, C.; et al. Living Atomically Dispersed Cu Ultrathin TiO2 Nanosheet CO2 Reduction Photocatalyst. Adv. Sci. 2019, 6, 1900289. [Google Scholar] [CrossRef]
- Gonell, F.; Puga, A.V.; Julián-López, B.; García, H.; Corma, A. Copper-doped titania photocatalysts for simultaneous reduction of CO2 and production of H2 from aqueous sulfide. Appl. Catal. B 2016, 180, 263–270. [Google Scholar] [CrossRef]
- López-Caballero, P.; Hauser, A.W.; de Lara-Castells, M.P. Exploring the Catalytic Properties of Unsupported and TiO2-Supported Cu5 Clusters: CO2 Decomposition to CO and CO2 Photoactivation. J. Phys. Chem. C 2019, 123, 23064–23074. [Google Scholar] [CrossRef]
- Lan, Y.; Xie, Y.; Chen, J.; Hu, Z.; Cui, D. Selective photocatalytic CO2 reduction on copper–titanium dioxide: A study of the relationship between CO production and H2 suppression. Chem. Commun. 2019, 55, 8068–8071. [Google Scholar] [CrossRef] [PubMed]
- Chao Liu, C.; Nauert, S.L.; Alsina, M.A.; Wang, D.; Grant, A.; He, K.; Weitz, E.; Nolan, M.; Gray, K.A.; Notestein, J.M. Role of surface reconstruction on Cu/TiO2 nanotubes for CO2 conversion. Appl. Catal. B Environ. 2019, 255, 117754. [Google Scholar]
- Xie, S.; Wang, Y.; Zhang, Q.; Deng, W.; Wang, Y. MgO- and Pt-Promoted TiO2 as an Efficient Photocatalyst for the Preferential Reduction of Carbon Dioxide in the Presence of Water. ACS Catal. 2014, 4, 3644–3653. [Google Scholar] [CrossRef]
- Jin, L.; Shaaban, E.; Bamonte, S.; Cintron, D.; Shuster, S.; Zhang, L.; Li, G.; He, J. Surface Basicity of Metal@TiO2 to Enhance Photocatalytic Efficiency for CO2 Reduction. ACS Appl. Mater. Interfaces 2021, 13, 38595–38603. [Google Scholar] [CrossRef]
- Li, N.; Liu, M.; Yang, B.; Shu, W.; Shen, Q.; Liu, M.; Zhou, J. Enhanced photocatalytic performance toward CO2 hydrogeneration over nanosized TiO2-loaded Pd under UV irradiation. J. Phys. Chem. C 2017, 121, 2923–2932. [Google Scholar] [CrossRef]
- Zhou, R.; Rui, N.; Fan, Z.; Liu, C.-J. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation. Int. J. Hydrogen Energy 2016, 41, 22017–22025. [Google Scholar] [CrossRef]
- Li, Y.; Rao, Z.; Liu, Z.; Zeng, J.; Bao, W.; Wang, Z.; Li, J.; Yu, F.; Dai, B.; Zhou, Y. Photo-Assisted CO/CO2 Methanation over Ni/TiO2 Catalyst: Experiment and Density Functional Theory Calculation. ChemCatChem 2022, 14, e202200182. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Moioli, E.; Chen, W.; Zhang, M.; Terlingen, B.J.P.; Zijlstra, B.; Filot, I.A.W.; Zuttel, A.; Pidko, E.A.; Hensen, E.J.M. Efficient Base-Metal NiMn/TiO2 Catalyst for CO2 Methanation. ACS Catal. 2019, 9, 7823–7839. [Google Scholar] [CrossRef]
- Kohno, Y.; Hayashi, H.; Takenaka, S.; Tanaka, T.; Funabiki, T.; Yoshida, S. Photo-enhanced reduction of carbon dioxide with hydrogen over Rh/TiO2. J. Photochem. Photobiol. A 1999, 126, 117–123. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, Z.; Xiang, G.; Zhai, T.; Liu, Z.; Zhao, W.; Liang, X.; Wang, L. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 2022, 13, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Yang, X.; Wang, R.; Duan, H.; Shen, Z.; Li, L.; Su, Y.; Yang, R.; Zhang, Y.; et al. Tuning selectivity of CO2 hydrogenation by modulating the strong metal–support interaction over Ir/TiO2 catalysts. Green Chem. 2020, 22, 6855–6861. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, C.; Yang, P.; He, Y.; Feng, J.; Li, D. Synergetic promotional effect of oxygen vacancy-rich ultrathin TiO2 and photochemical induced highly dispersed Pt for photoreduction of CO2 with H2O. Appl. Catal. B Environ. 2019, 244, 919–930. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, L.; Huang, C.; Ren, Z.; Wang, H.; Hu, J.; Zhang, H.; Jiang, Z.; Song, F. Exploring the CO2 reduction reaction mechanism on Pt/TiO2 with the ambient-pressure X-ray photoelectron spectroscopy. Appl. Surf. Sci. 2021, 568, 150933. [Google Scholar] [CrossRef]
- Qiu-ye, L.; Lan-Lan, Z.; Chen, L.; Yu-hui, C.; Xiao-Dong, W.; Jian-Jun, Y. Photocatalytic Reduction of CO2 to Methane on Pt/TiO2 Nanosheet Porous Film. Adv. Condens. Matter Phys. 2014, 316589. [Google Scholar]
- Permporn, D.; Khunphonoi, R.; Wilamat, J.; Khemthong, P.; Chirawatkul, P.; Butburee, T.; Sangkhun, W.; Wantala, K.; Grisdanurak, N.; Santatiwongchai, J.; et al. Insight into the Roles of Metal Loading on CO2 Photocatalytic Reduction Behaviors of TiO2. Nanomaterials 2022, 12, 474. [Google Scholar] [CrossRef]
- Neatu, S.; Maciá-Agulló, J.A.; Concepción, P.; Garcia, H. Gold–Copper Nanoalloys Supported on TiO2 as Photocatalysts for CO2 Reduction by Water. J. Am. Chem. Soc. 2014, 136, 15969–15976. [Google Scholar] [CrossRef]
- Reñones, P.; Collado, L.; Iglesias-Juez, A.; Oropeza, F.E.; Fresno, F.; de la Peña-O’Shea, V.A. Silver–Gold Bimetal-Loaded TiO2 Photocatalysts for CO2 Reduction. Ind. Eng. Chem. Res. 2020, 59, 9440–9450. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, U. Noble metal modified TiO2: Selective photoreduction of CO2 to hydrocarbons. Mol. Catal. 2017, 439, 91–99. [Google Scholar] [CrossRef]
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlögl, R. CO2 Hydrogenation with Cu/ZnO/Al2O3: A Benchmark Study. ChemCatChem 2020, 12, 3216–3222. [Google Scholar] [CrossRef]
- Kattel, S.; Ramirez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Nie, R.; Wu, G.; Hou, Z. Hydrogenation of CO2 to CH3OH over Cu/ZnO catalysts with different ZnO morphology. Fuel 2015, 154, 161–166. [Google Scholar] [CrossRef]
- Mahapatra, M.; Gutiérrez, R.A.; Kang, J.; Rui, N.; Hamlyn, R.; Liu, Z.; Orozco, I.; Ramírez, P.J.; Senanayake, S.D.; Rodriguez, J.A. The behavior of inverse oxide/metal catalysts: CO oxidation and water-gas shift reactions over ZnO/Cu(111) surfaces. Surf. Sci. 2019, 681, 116–121. [Google Scholar] [CrossRef]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu–ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu+ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Marcos, F.C.F.; Lin, L.; Betancourt, L.E.; Senanayake, S.D.; Rodriguez, J.A.; Assaf, J.M.; Giudici, R.; Assaf, E.M. Insights into the methanol synthesis mechanism via CO2 hydrogenation over Cu-ZnO-ZrO2 catalysts: Effects of surfactant/Cu-Zn-Zr molar ratio. J. CO2 Util. 2020, 41, 101215. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C.; Liao, F.; Huang, Y.; et al. Morphology-dependent interactions of ZnO with Cu nanoparticles at the materials' interface in selective hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Phongprueksathat, N.; Bansode, A.; Toyao, T.; Urakawa, A. Greener and facile synthesis of Cu/ZnO catalysts for CO2 hydrogenation to methanol by urea hydrolysis of acetates. RSC Adv. 2021, 11, 14323–14333. [Google Scholar] [CrossRef]
- Guzmán, H.; Salomone, F.; Bensaid, S.; Castellino, M.; Russo, N.; Hernández, S. CO2 Conversion to Alcohols over Cu/ZnO Catalysts: Prospective Synergies between Electrocatalytic and Thermocatalytic Routes. ACS Appl. Mater. Interfaces 2022, 14, 517–530. [Google Scholar] [CrossRef]
- Sakurai, H.; Tsubota, S.; Haruta, M. Hydrogenation of CO2 over gold supported on metal oxides. Appl. Catal. A 1993, 102, 125–136. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Hauble, A.; Ishida, T.; Murayama, T.; Parlinska-Wojtan, M.; Behm, R.J. Performance of Au/ZnO catalysts in CO2 reduction to methanol: Varying the Au loading / Au particle size. Appl. Catal. A Gen. 2021, 624, 118318. [Google Scholar] [CrossRef]
- Hartadi, Y.; Widmann, D.; Behm, R.J. Methanol synthesis via CO2 hydrogenation over a Au/ZnO catalyst: An isotope labelling study on the role of CO in the reaction process. Phys. Chem. Chem. Phys. 2016, 18, 10781–10791. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mageed, A.M.; Klyushin, A.; Rezvani, A.; Knop-Gericke, A.; Schlögl, R.; Behm, R.J. Negative Charging of Au Nanoparticles during Methanol Synthesis from CO2 /H2 on a Au/ZnO Catalyst: Insights from Operando IR and Near-Ambient-Pressure XPS and XAS Measurements. Angew. Chem. Int. Ed. 2019, 58, 10325–10329. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Mochizuki, C.; Ishida, T.; Murayama, T.; Rabeah, J.; Parlinska-Wojtan, M.; Brückner, A.; Behm, R.J. Formation and Performance of Au/ZnO Catalysts in CO2 Reduction to Methanol by the ZnO Particle Size. ACS Catal. 2021, 11, 9022–9033. [Google Scholar] [CrossRef]
- Liao, W.; Tang, C.; Zheng, H.; Ding, J.; Zhang, K.; Wang, H.; Lu, J.; Huang, W.; Zhang, Z. Tuning activity and selectivity of CO2 hydrogenation via metal-oxide interfaces over ZnO-supported metal catalysts. J. Catal. 2022, 407, 126–140. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Khai Beh, G.; Zhang, R.; Sit, P.H.-L.; Yang Teoh, W. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal. B Environ. 2017, 219, 715–726. [Google Scholar] [CrossRef]
- Wu, D.; Deng, K.; Hu, B.; Lu, Q.; Liu, G.; Hong, X. Plasmon-Assisted Photothermal Catalysis of Low-Pressure CO2 Hydrogenation to Methanol over Pd/ZnO Catalyst. ChemCatChem 2019, 11, 1598–1601. [Google Scholar] [CrossRef]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based heterogeneous catalysts toward catalytic conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Ebrahimi, P.; Kumar, A.; Khraisheh, M. A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction. Catalysts 2022, 12, 1101. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Fernández Sanz, J.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef]
- Wang, M.; Shen, M.; Jin, X.; Tian, J.; Li, M.; Zhou, Y.; Zhang, L.; Li, Y.; Shi, J. Oxygen Vacancy Generation and Stabilization in CeO2–x by Cu Introduction with Improved CO2 Photocatalytic Reduction Activity. ACS Catal. 2019, 9, 4573–4581. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castañeda, R.; Lin, J.; Senanayake, S.D.; et al. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Figueiredo, W.T.; Escudero, C.; Pérez-Dieste, V.; Ospina, C.A.; Bernardi, F. Determining the Surface Atomic Population of CuxNi1–x/CeO2 (0 < x ≤ 1) Nanoparticles during the Reverse Water–Gas Shift (RWGS) Reaction. J. Phys. Chem. C 2020, 124, 16868–16878. [Google Scholar]
- Yan, Y.; Wong, R.J.; Ma, Z.; Donat, F.; Xi, S.; Saqline, S.; Fan, Q.; Du, Y.; Borgna, A.; He, Q.; et al. CO2 hydrogenation to methanol on tungsten-doped Cu/CeO2 catalysts. Appl. Catal. B Environ. 2022, 306, 121098. [Google Scholar] [CrossRef]
- Li, M.; Pham, T.H.M.; Ko, Y.; Zhao, K.; Zhong, L.; Luo, W.; Züttel, A. Support-Dependent Cu–In Bimetallic Catalysts for Tailoring the Activity of Reverse Water Gas Shift Reaction. ACS Sustain. Chem. Eng. 2022, 10, 1524–1535. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Villora-Pico, J.J.; Sepúlveda-Escribano, A.; Tian, F.; Zhu, M.; Han, Y.-F.; Ramirez Reina, T. Highly Active and Selective Multicomponent Fe–Cu/CeO2–Al2O3 Catalysts for CO2 Upgrading via RWGS: Impact of Fe/Cu Ratio. ACS Sustain. Chem. Eng. 2021, 9, 12155–12166. [Google Scholar] [CrossRef]
- Wang, L.-C.; Khazaneh, M.T.; Widmann, D.; Behm, R.J. TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst—A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Wang, L.; Widmann, D.; Behm, R.J. Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction. Catal. Sci. Technol. 2015, 5, 925–941. [Google Scholar] [CrossRef]
- Lu, B.; Quan, F.; Sun, Z.; Jia, F.; Zhang, L. Photothermal reverse-water-gas-shift over Au/CeO2 with high yield and selectivity in CO2 conversion. Catal. Commun. 2019, 129, 105724. [Google Scholar] [CrossRef]
- Rezvani, A.; Abdel-Mageed, A.M.; Ishida, T.; Murayama, T.; Parlinska-Wojtan, M.; Behm, R.J. CO2 Reduction to Methanol on Au/CeO2 Catalysts: Mechanistic Insights from Activation/Deactivation and SSITKA Measurements. ACS Catal. 2020, 10, 3580–3594. [Google Scholar] [CrossRef]
- Yang, X.; Kattel, S.; Senanayake, S.D.; Boscoboinik, J.A.; Nie, X.; Graciani, J.; Rodríguez, J.A.; Liu, P.; Stacchiola, D.J.; Chen, J.G. Low Pressure CO2 Hydrogenation to Methanol over Gold Nanoparticles Activated on a CeOx/TiO2 Interface. J. Am. Chem. Soc. 2015, 137, 10104–10107. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, J.; Chu, M.; Yue, J.; Cui, Y.; Xu, G. Cooperation between active metal and basic support in Ni-based catalyst for low-temperature CO2 methanation. Catal. Lett. 2020, 150, 1418–1426. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293-294, 89–96. [Google Scholar] [CrossRef]
- Rui, N.; Zhang, X.; Zhang, F.; Liu, Z.; Cao, X.; Xie, Z.; Zou, R.; Senanayake, S.D.; Yang, Y.; Rodriguez, J.A.; et al. Highly active Ni/CeO2 catalyst for CO2 methanation: Preparation and characterization. Appl. Catal. B Environ. 2021, 282, 119581. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology dependence of catalytic properties of Ni/CeO2 for CO2 methanation: A kinetic and mechanism study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Jomjaree, T.; Sintuya, P.; Srifa, A.; Koo-amornpattana, W.; Kiatphuengporn, S.; Assabumrungrat, S.; Sudoh, M.; Watanabe, R.; Fukuhara, C.; Ratchahat, S. Catalytic performance of Ni catalysts supported on CeO2 with different morphologies for low-temperature CO2 methanation. Catal. Today 2021, 375, 234–244. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Binas, V.; Marnellos, G.E.; Konsolakis, M. Deciphering the role of Ni particle size and nickel-ceria interfacial perimeter in the low-temperature CO2 methanation reaction over remarkably active Ni/CeO2 nanorods. Appl. Catal. B Environ. 2021, 297, 120401. [Google Scholar] [CrossRef]
- Lin, L.; Gerlak, C.A.; Liu, C.; Llorca, J.; Yao, S.; Rui, N.; Zhang, F.; Liu, Z.; Zhang, S.; Deng, K.; et al. Effect of Ni particle size on the production of renewable methane from CO2 over Ni/CeO2 catalyst. J. Energy Chem. 2021, 61, 602–611. [Google Scholar] [CrossRef]
- Winter, L.R.; Gomez, E.; Yan, B.; Yao, S.; Chen, J.G. Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation. Appl. Catal. B Environ. 2018, 224, 442–450. [Google Scholar] [CrossRef]
- Sun, C.; Beaunier, P.; La Parola, V.; Liotta, L.F.; Da Costa, P. Ni/CeO2 Nanoparticles Promoted by Yttrium Doping as Catalysts for CO2 Methanation. ACS Appl. Nano Mater. 2020, 3, 12355–12368. [Google Scholar] [CrossRef]
- Alvarez-Galvan, C.; Lustemberg, P.G.; Oropeza, F.E.; Bachiller-Baeza, B.; Dapena Ospina, M.; Herranz, M.; Cebollada, J.; Collado, L.; Campos-Martin, J.M.; de la Peña-O’Shea, V.A.; et al. Highly Active and Stable Ni/La-Doped Ceria Material for Catalytic CO2 Reduction by Reverse Water-Gas Shift Reaction. ACS Appl. Mater. Interfaces 2022, 14, 50739–50750. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: Effect of selected transition metal promoters. Appl. Catal. B Environ. 2018, 232, 464471. [Google Scholar] [CrossRef]
- Xie, F.; Xu, S.; Deng, L.; Xie, H.; Zhou, G. CO2 hydrogenation on Co/CeO2-δ catalyst: Morphology effect from CeO2 support. Int. J. Hydrogen Energy 2020, 45, 26938–26952. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Kim, H.B.; Park, E.D. CO and CO2 Methanation over CeO2-Supported Cobalt Catalysts. Catalysts 2022, 12, 212. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; Bueno-López, A. Effect of Ru loading on Ru/CeO2 catalysts for CO2 methanation. Mol. Catal. 2021, 515, 111911. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; Herrera, F.C.; Pellegrin, E.; Escudero, C.; García-Melchor, M.; Bueno-López, A. Elucidating the Role of the Metal Catalyst and Oxide Support in the Ru/CeO2-Catalyzed CO2 Methanation Mechanism. J. Phys. Chem. C 2021, 125, 25533–25544. [Google Scholar] [CrossRef]
- Guo, Y.; Mei, S.; Yuan, K.; Wang, D.-J.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Low-Temperature CO2 Methanation over CeO2-Supported Ru Single Atoms, Nanoclusters, and Nanoparticles Competitively Tuned by Strong Metal–Support Interactions and H-Spillover Effect. ACS Catal. 2018, 8, 6203–6215. [Google Scholar] [CrossRef]
- Panaritis, C.; Edake, M.; Couillard, M.; Einakchi, R.; Baranova, E.A. Insight towards the role of ceria-based supports for reverse water gas shift reaction over RuFe nanoparticles. J. CO2 Util. 2018, 26, 350–358. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Villora-Pico, J.J.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. CO2 valorisation via reverse water-gas shift reaction using promoted Fe/CeO2-Al2O3 catalysts: Showcasing the potential of advanced catalysts to explore new processes design. Appl. Catal. A Gen. 2020, 593, 117442. [Google Scholar] [CrossRef]
- Lou, Y.; Jiang, F.; Zhu, W.; Wang, L.; Yao, T.; Wang, S.; Yang, B.; Yang, B.; Zhu, Y.; Liu, X. CeO2 supported Pd dimers boosting CO2 hydrogenation to ethanol. Appl. Catal. B Environ. 2021, 291, 120122. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, S.; Liu, B.; Liu, J.; Wang, L.; Xiao, Y.; Xu, Y.; Liu, X. Insights into the Influence of CeO2 Crystal Facet on CO2 Hydrogenation to Methanol over Pd/CeO2 Catalysts. ACS Catal. 2020, 10, 11493–11509. [Google Scholar] [CrossRef]
- Zhang, F.; Gutiérrez, R.A.; Lustemberg, P.G.; Liu, Z.; Rui, N.; Wu, T.; Ramírez, P.J.; Xu, W.; Idriss, H.; Ganduglia-Pirovano, M.V.; et al. Metal–Support Interactions and C1 Chemistry: Transforming Pt-CeO2 into a Highly Active and Stable Catalyst for the Conversion of Carbon Dioxide and Methane. ACS Catal. 2021, 11, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Goguet, A.; Meunier, F.; Breen, J.P.; Burch, R.; Petch, M.I.; Ghenciu, A.F. Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (RWGS) reaction. J. Catal. 2004, 226, 382–392. [Google Scholar] [CrossRef]
- Zheng, K.; Li, Y.; Liu, B.; Jiang, F.; Xu, Y.; Liu, X. Ti-doped CeO2 Stabilized Single-Atom Rhodium Catalyst for Selective and Stable CO2 Hydrogenation to Ethanol. Angew. Chem. Int. Ed. 2022, 61, e202210991. [Google Scholar] [CrossRef] [PubMed]
- Toth, L.E. Transition Metal Carbides and Nitrides; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Oyama, S.T. The Chemistry of Transition Metal Carbides and Nitrides; Blackie Academic and Professional: Scotland, UK, 1996. [Google Scholar]
- Levy, R.B.; Boudart, M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef]
- Woo, H.C.; Park, K.Y.; Kim, Y.G.; Nam, I.S.; Chung, J.S.; Lee, J.S. Mixed alcohol synthesis from carbon monoxide and dihydrogen over potassium-promoted molybdenum carbide catalysts. Appl. Catal. 1991, 75, 267–280. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Trends in the chemical properties in early transition metal carbide surfaces: A density functional study. Catal. Today 2005, 105, 66–73. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Viñes, F.; Illas, F.; Liu, P.; Takahashi, Y.; Nakamura, K. Adsorption of gold on TiC(001): Au-C interactions and charge polarization. J. Chem. Phys. 2007, 127, 211102. [Google Scholar] [CrossRef]
- Ono, L.K.; Sudfeld, D.; Roldan-Cuenya, B. In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation. Surf. Sci. 2006, 600, 5041. [Google Scholar] [CrossRef]
- Ono, L.K.; Roldan-Cuenya, B. Effect of interparticle interaction on the low temperature oxidation of CO over size-selected Au nanocatalysts supported on ultrathin TiC films. Catal. Lett. 2007, 113, 86–94. [Google Scholar] [CrossRef]
- Gomez, T.; Florez, E.; Rodriguez, J.A.; Illas, F. Reactivity of Transition Metals (Pd, Pt, Cu, Ag, Au) toward Molecular Hydrogen Dissociation: Extended Surfaces versus Particles Supported on TiC(001) or Small Is Not Always Better and Large Is Not Always Bad. J. Phys. Chem. C 2011, 115, 11666–11672. [Google Scholar] [CrossRef]
- Asara, G.G.; Feria, L.; Florez, E.; Ricart, J.M.; Liu, P.; Rodriguez, J.A.; Illas, F. Theoretical Study of the Interaction of CO on TiC(001) and Au Nanoparticles Supported on TiC(001): Probing the Nature of the Au/TiC Interface. J. Phys. Chem. C 2011, 115, 22495–22504. [Google Scholar] [CrossRef]
- Vidal, A.B.; Feria, L.; Evans, J.; Takahashi, Y.; Liu, P.; Nakamura, K.; Illas, F.; Rodriguez, J.A. CO2 Activation and Methanol Synthesis on Novel Au/TiC and Cu/TiC Catalysts. J. Phys. Chem. Lett. 2012, 3, 2275–2280. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Lozano-Reis, P.; Sayós, R.; Rodriguez, J.A.; Illas, F. Structural, electronic, and magnetic properties of Ni nanoparticles supported on the TiC(001) surface. Phys. Chem. Chem. Phys. 2020, 22, 26145–26154. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Reis, P.; Prats, H.; Sayós, R.; Rodriguez, J.A.; Illas, F. Assessing the Activity of Ni Clusters Supported on TiC(001) toward CO2 and H2 Dissociation. J. Phys. Chem. C 2021, 125, 12019–12027. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramirez, P.J.; Gutierrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Evans, J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Cu/δ-MoC catalysts for the conversion of CO2: The metal/C ratio as a key factor defining activity, selectivity, and stability. J. Am. Chem. Soc. 2016, 138, 8269–8278. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Ramirez, P.J.; Vidal, A.B.; Rodriguez, J.A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Fundamentals of methanol synthesis on metal carbide based catalysts: Activation of CO2 and H2. Top. Catal. 2015, 58, 159–173. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Structure and electronic properties of Cu nanoclusters supported on Mo2C(001) and MoC(001) surfaces. J. Chem. Phys. 2015, 143, 114704. [Google Scholar] [CrossRef] [PubMed]
- Heracleous, E.; Koidi, V.; Lappas, A.A. CO2 conversion over Cu–Mo2C catalysts: Effect of the Cu promoter and preparation method. Catal. Sci. Technol. 2021, 11, 1467–1480. [Google Scholar] [CrossRef]
- Zhang, R.; Wei, C.; Guo, W.; Li, Z.; Wang, B.; Ling, L.; Li, D. Syngas Conversion to C2 Oxygenates over the Cu/β-Mo2C Catalyst: Probing into the Effect of the Interface between Cu and β-Mo2C on Catalytic Performance. J. Phys. Chem. C 2019, 123, 21022–21030. [Google Scholar] [CrossRef]
- Jing, H.; Li, Q.; Wang, J.; Liu, D.; Wu, K. Enhanced N2-Fixation by Engineering the Edges of Two-Dimensional Transition-Metal Disulfides. J. Phys. Chem. C 2019, 123, 1235–1251. [Google Scholar] [CrossRef]
- Zhang, Q.; Pastor-Pérez, L.; Jin, W.; Gu, S.; Reina, T.R. Understanding the promoter effect of Cu and Cs over highly effective β-Mo2C catalysts for the reverse water-gas shift reaction. Appl. Catal. B Environ. 2019, 244, 889–898. [Google Scholar] [CrossRef]
- Delporte, P.; Meunier, F.; Pham-Huu, C.; Vennegues, P.; Ledoux, M.J.; Guille, J. Physical characterization of molybdenum oxycarbide catalyst; TEM, XRD and XPS. Catal. Today 1995, 23, 251–267. [Google Scholar] [CrossRef]
- Dixit, M.; Peng, X.; Porosoff, M.D.; Willauer, H.D.; Mpourmpakis, G. Elucidating the role of oxygen coverage in CO2 reduction on Mo2C. Catal. Sci. Technol. 2017, 7, 5521–5529. [Google Scholar] [CrossRef]
- Yao, L.; Wang, Y.; Galvez, M.E.; Hu, C.; da Costa, P. g-Alumina-Supported Ni-Mo Carbides as Promising Catalysts for CO2 Methanation. Modern Res. Catal. 2017, 6, 135–145. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Asakura, T.; Gomes, J.; Nakamura, K. Optimization and Application of Lithium Parameters for the Reactive Force Field, ReaxFF. J. Phys. Chem. B 2005, 109, 4575–4582. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Water-Gas-Shift Reaction on Molybdenum Carbide Surfaces: Essential Role of the Oxycarbide. J. Phys. Chem. B 2006, 110, 19418–19425. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, C.; Viñes, F.; Illas, F. Transition metal carbides as novel materials for CO2 capture storage, and activation. Energy Environ. Sci. 2016, 9, 141–144. [Google Scholar] [CrossRef]
- Len, T.; Luque, R. Addressing the CO2 challenge through thermocatalytic hydrogenation to carbon monoxide, methanol and methane. Green Chem. 2023, 25, 490–521. [Google Scholar] [CrossRef]
- Nagai, M.; Matsuda, K. Low-temperature water–gas shift reaction over cobalt–molybdenum carbide catalyst. J. Catal. 2006, 238, 489–496. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ramírez, P.J.; Gutierrez, R.A. Highly Active Pt/MoC and Pt/TiC Catalysts for the Low-Temperature Water-Gas Shift Reaction: Effects of the Carbide Metal/Carbon Ratio on the Catalyst Performance. Catal. Today 2017, 289, 47–52. [Google Scholar] [CrossRef]
- Wan, W.; Tackett, B.M.; Chen, J.G. Reactions of water and C1 molecules on carbide and metal-modified carbide surfaces. Chem. Soc. Rev. 2017, 46, 1807–1823. [Google Scholar] [CrossRef]
- Kelly, T.G.; Stottlemyer, A.L.; Ren, H.; Chen, J.G. Comparison of O-H, C-H, and C-O bond scission sequence of methanol on tungsten carbide surfaces modified by Ni, Rh, and Au. J. Phys. Chem. C 2011, 115, 6644–6650. [Google Scholar] [CrossRef]
- Mehdizadeh, S.; Sadjadi, A.; Poater, A.; Mansouri, A.; Bahri-Laleh, N. Molecular modelling aided catalyst design for PAO oils hydrofinishing. J. Mol. Liq. 2022, 352, 118675. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Lang, R.; Du, X.; Huang, Y.; Jiang, X.; Zhang, Q.; Guo, Y.; Liu, K.; Qiao, B.; Wang, A.; Zhang, T. Single-Atom Catalysts Based on the Metal–Oxide Interaction. Chem. Rev. 2020, 120, 11986–12043. [Google Scholar] [CrossRef]
- He, H.; Wang, H.H.; Liu, J.; Liu, X.; Li, W.; Wang, Y. Research Progress and Application of Single-Atom Catalysts: A Review. Molecules 2021, 26, 6501. [Google Scholar] [CrossRef] [PubMed]
- Hannagan, R.T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Single-Atom Alloy Catalysis. Chem. Rev. 2020, 120, 12044–12088. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Zhang, L.; Doyle-Davis, K.; Sun, X. Single-Atom Catalysts: From Design to Application. Electrochem. Energy Rev. 2019, 2, 539–573. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, Z.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Guo, L.W.; Du, P.P.; Fu, X.P.; Ma, C.; Zeng, J.; Si, R.; Huang, Y.Y.; Jia, C.J.; Zhang, Y.W.; Yan, C.H. Contributions of Distinct Gold Species to Catalytic Reactivity for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 13481. [Google Scholar] [CrossRef]
- Chen, J.; Iyemperumal, S.K.; Fenton, T.; Carl, A.; Grimm, R.; Li, G.; Deskins, N.A. Synergy between Defects, Photoexcited Electrons, and Supported Single Atom Catalysts for CO2 Reduction. ACS Catal. 2018, 8, 10464–10478. [Google Scholar] [CrossRef]
- Morales-García, A.; Calle-Vallejo, F.; Illas, F. MXenes: New Horizons in Catalysis. ACS Catal. 2020, 10, 13487–13503. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, Z.; Yang, W.; Liu, S.; Zhang, X.; Yu, Y.; Cheong, W.-C.; Zheng, L.; Ren, F.; Ying, G.; et al. MXene (Ti3C2) Vacancy-Confined Single-Atom Catalyst for Efficient Functionalization of CO2. J. Am. Chem. Soc. 2019, 141, 4086–4093. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef]
- Jurado, L.; Esvan, J.; Luque-Álvarez, L.A.; Bobadilla, L.F.; Odriozola, J.A.; Posada-Pérez, S.; Poater, A.; Comas-Vives, A.; Axet, M.R. Highly dispersed Rh single atoms over graphitic carbon nitride as a robust catalyst for the hydroformylation reaction. Catal. Sci. Technol. 2023. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).