
Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
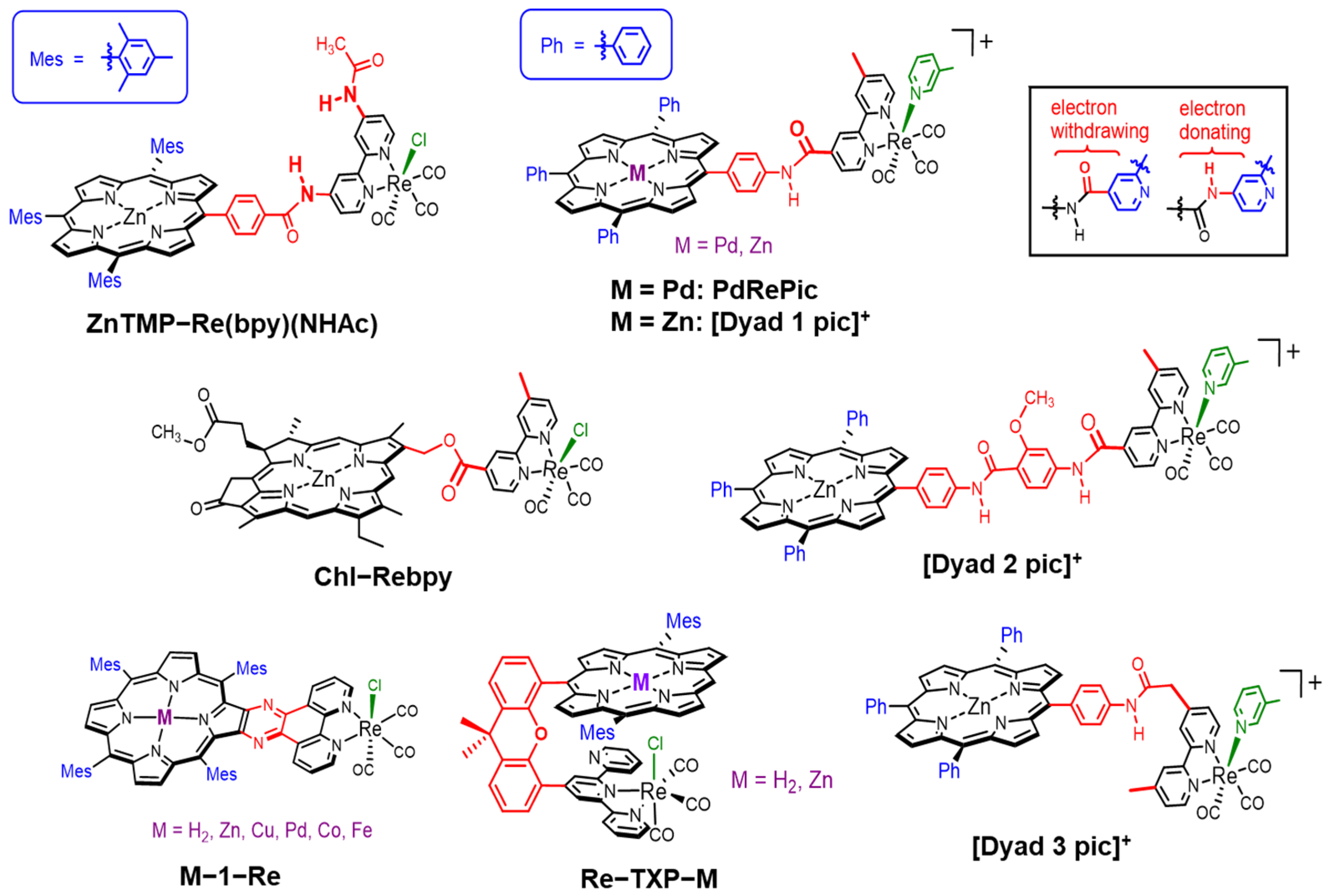
2. Dyad Systems Based on a Porphyrin–Re Complex
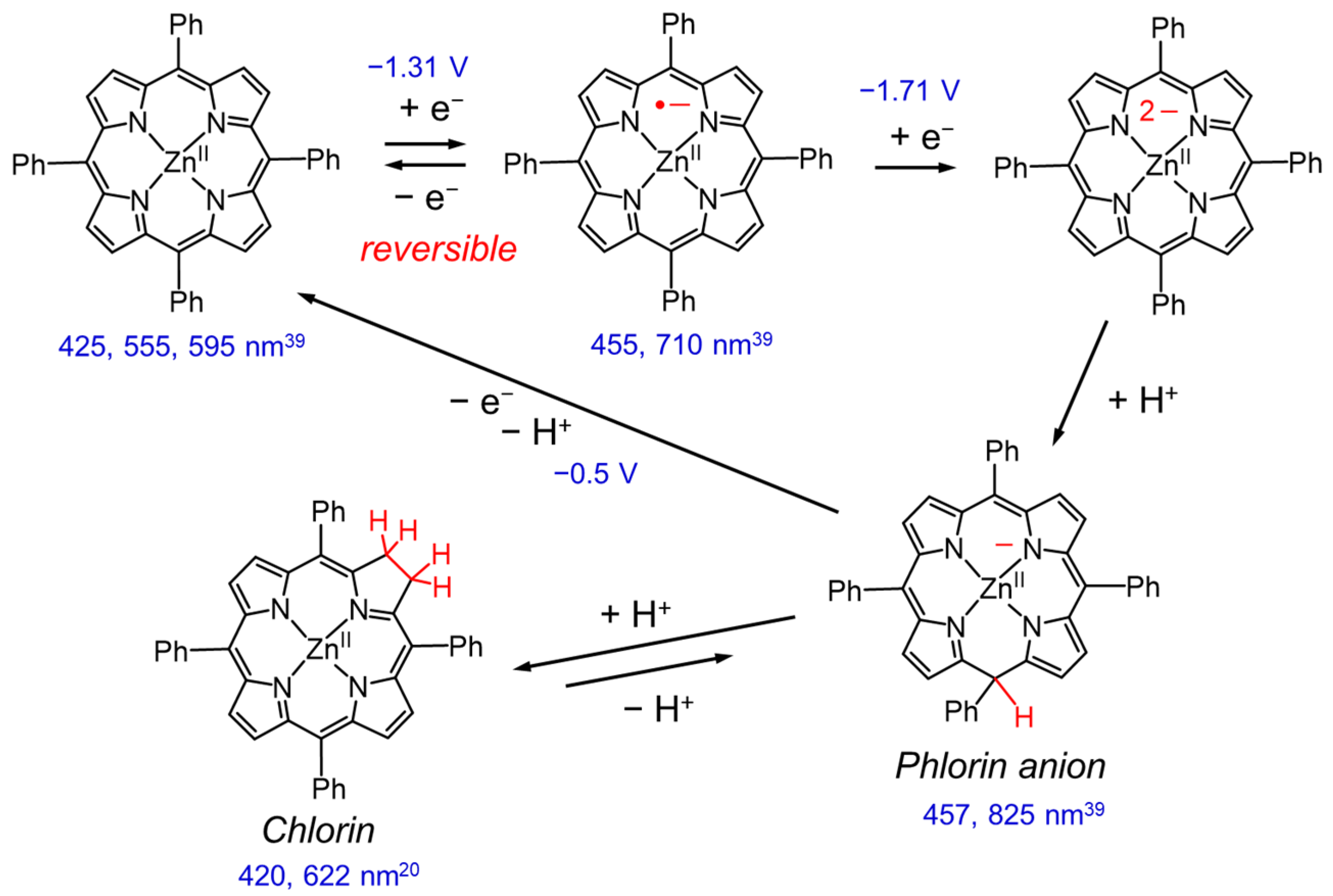
3. Behavior of Electrochemical Reduction of Zn Porphyrin
4. Porphyrin–Re Complex Dyads with Direct and Orthogonal Connection
5. Special Pair Mimic Porphyrin Connected with Re Complex(es)
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Friedlingstein, P.; O’Sullivan, M.; Jones, M.W.; Andrew, R.M.; Gregor, L.; Hauck, J.; Le Quéré, C.; Luijkx, I.T.; Olsen, A.; Peters, G.P.; et al. Global carbon budget 2022. Earth Syst. Sci. Data 2022, 14, 4811–4900. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Dong, L.-Z.; Liu, J.; Li, S.-L.; Lan, Y.Q. From molecular metal complex to metal-organic framework: The CO2 reduction photocatalysts with clear and tunable structure. Coord. Chem. Rev. 2019, 390, 86–126. [Google Scholar] [CrossRef]
- Nakada, A.; Kumagai, H.; Robert, M.; Ishitani, O.; Maeda, K. Molecule/Semiconductor Hybrid Materials for Visible-Light CO2 Reduction: Design Principles and Interfacial Engineering. Acc. Mater. Res. 2021, 2, 458–470. [Google Scholar] [CrossRef]
- Son, H.-J.; Pac, C.; Kang, S.O. Inorganometallic Photocatalyst for CO2 Reduction. Acc. Chem. Res. 2021, 54, 4530–4544. [Google Scholar] [CrossRef]
- Pirzada, B.M.; Dar, A.H.; Shaikh, M.N.; Qurashi, A. Reticular-Chemistry-Inspired Supramolecule Design as a Tool to Achieve Efficient Photocatalysts for CO2 Reduction. ACS Omega 2021, 6, 29291–29324. [Google Scholar] [CrossRef]
- Fujita, E.; Grills, D.C.; Manbeck, G.F.; Polyansky, D.E. Understanding the Role of Inter- and Intramolecular Promoters in Electro- and Photochemical CO2 Reduction Using Mn, Re, and Ru Catalysts. Acc. Chem. Res. 2022, 55, 616–628. [Google Scholar] [CrossRef]
- Bizzarri, C. Homogeneous Systems Containing Earth-Abundant Metal Complexes for Photoactivated CO2 Reduction: Recent Advances. Eur. J. Org. Chem. 2022, 24, e202200185. [Google Scholar]
- Rudolph, M.; Dautz, S.; Jäger, E.-G. Macrocyclic [N42−] Coordinated Nickel Complexes as Catalysts for the Formation of Oxalate by Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2000, 122, 10821–10830. [Google Scholar] [CrossRef]
- Rao, H.; Schmidt, L.C.; Bonin, J.; Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74–77. [Google Scholar] [CrossRef]
- Sato, S.; Arai, T.; Morikawa, T.; Uemura, K.; Suzuki, T.M.; Tanaka, H.; Kajino, T. Selective CO2 conversion to formate conjugated with H2O oxidation utilizing semiconductor/complex hybrid photocatalysts. J. Am. Chem. Soc. 2011, 133, 15240–15243. [Google Scholar] [CrossRef]
- Sahara, G.; Kumagai, H.; Maeda, K.; Kaeffer, N.; Artero, V.; Higashi, M.; Abe, R.; Ishitani, O. Photoelectrochemical reduction of CO2 Coupled to water oxidation using a photocathode with a Ru(II)–Re (I) complex photocatalyst and a CoOx/TaON photoanode. J. Am. Chem. Soc. 2016, 138, 14152–14158. [Google Scholar] [CrossRef]
- Suzuki, T.M.; Yoshino, S.; Takayama, T.; Iwase, A.; Kudo, A.; Morikawa, T. Z-Schematic and visible-light-driven CO2 reduction using H2O as an electron donor by a particulate mixture of a Ru complex/(CuGa)1−xZn2xS2 hybrid catalyst, BiVO4 and an electron mediator. Chem. Commun. 2018, 54, 10199–10202. [Google Scholar] [CrossRef]
- Kamata, R.; Kumagai, H.; Yamazaki, Y.; Sahara, G.; Ishitani, O. Photoelectrochemical CO2 reduction using a Ru(II)-Re(I) supramolecular photocatalyst connected to a vinyl polymer on a NiO electrode. ACS Appl. Mater. Interfaces 2019, 11, 5632–5641. [Google Scholar] [CrossRef]
- Tamaki, Y.; Ishitani, O. Supramolecular photocatalysts for the reduction of CO2. ACS Catal. 2017, 7, 3394–3409. [Google Scholar] [CrossRef]
- Tamaki, Y.; Koike, K.; Morimoto, T.; Ishitani, O. Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor. J. Catal. 2013, 304, 22–28. [Google Scholar] [CrossRef]
- Kuttassery, F.; Kumagai, H.; Kamata, R.; Ebato, Y.; Higashi, M.; Suzuki, H.; Abe, R.; Ishitani, O. Supramolecular photocatalysts fixed on the inside of the polypyrrole layer in dye sensitized molecular photocathodes: Application to photocatalytic CO2 reduction coupled with water oxidation. Chem. Sci. 2021, 12, 13216–13232. [Google Scholar] [CrossRef]
- Taniguchi, M.; Lindsey, J.S. Synthetic chlorins, possible surrogates for chlorophylls, prepared by derivatization of porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef]
- Otsuki, J. Supramolecular approach towards light-harvesting materials based on porphyrins and chlorophylls. J. Mater. Chem. A 2018, 6, 6710–6753. [Google Scholar] [CrossRef]
- Nagata, N.; Kuramochi, Y.; Kobuke, Y. Energy transfer among light-harvesting macrorings incorporated into a bilayer membrane. J. Am. Chem. Soc. 2009, 131, 10–11. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Sandanayaka, A.S.D.; Satake, A.; Araki, Y.; Ogawa, K.; Ito, O.; Kobuke, Y. Energy transfer followed by electron transfer in a porphyrin macrocycle and central acceptor ligand: A model for a photosynthetic composite of the light-harvesting complex and reaction center. Chem.-Eur. J. 2009, 15, 2317–2327. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Satake, A.; Kobuke, Y. Light harvesting macroring accommodating a tetrapodal ligand based on complementary and cooperative coordinations. J. Am. Chem. Soc. 2004, 126, 8668–8669. [Google Scholar] [CrossRef]
- Borah, K.D.; Bhuyan, J. Magnesium porphyrins with relevance to chlorophylls. Dalton Trans. 2017, 46, 6497–6509. [Google Scholar] [CrossRef]
- Chen, L.X.; Zhang, X.; Wasinger, E.C.; Attenkofer, K.; Jennings, G.; Muresan, A.Z.; Lindsey, J.S. Tracking Electrons and Atoms in a Photoexcited Metalloporphyrin by X-ray Transient Absorption Spectroscopy. J. Am. Chem. Soc. 2007, 129, 9616–9618. [Google Scholar] [CrossRef]
- Nikoloudakis, E.; López-Duarte, I.; Charalambidis, G.; Ladomenou, K.; Ince, M.; Coutsolelos, A.G. Porphyrins and phthalocyanines as biomimetic tools for photocatalytic H2 production and CO2 reduction. Chem. Soc. Rev. 2022, 51, 6965–7045. [Google Scholar] [CrossRef]
- Kiyosawa, K.; Shiraishi, N.; Shimada, T.; Masui, D.; Tachibana, H.; Takagi, S.; Ishitani, O.; Tryk, D.A.; Inoue, H. Electron transfer from the porphyrin S2 State in a zinc porphyrin-rhenium bipyridyl dyad having carbon dioxide reduction activity. J. Phys. Chem. C 2009, 113, 11667–11673. [Google Scholar] [CrossRef]
- Schneider, J.; Vuong, K.Q.; Calladine, J.A.; Sun, X.-Z.; Whitwood, A.C.; George, M.W.; Perutz, R.N. Photochemistry and photophysics of a Pd(II) metalloporphyrin: Re(I) tricarbonyl bipyridine molecular dyad and its activity toward the photoreduction of CO2 to CO. Inorg. Chem. 2011, 50, 11877–11889. [Google Scholar] [CrossRef]
- Windle, C.D.; Campian, M.V.; Duhme-Klair, A.-K.; Gibson, E.A.; Perutz, R.N.; Schneider, J. CO2 photoreduction with long-wavelength light: Dyads and monomers of zinc porphyrin and rhenium bipyridine. Chem. Commun. 2012, 48, 8189–8191. [Google Scholar] [CrossRef]
- Windle, C.D.; George, M.W.; Perutz, R.N.; Summers, P.A.; Sun, X.Z.; Whitwood, A.C. Comparison of rhenium–porphyrin dyads for CO2 photoreduction: Photocatalytic studies and charge separation dynamics studied by time-resolved IR spectroscopy. Chem. Sci. 2015, 6, 6847–6864. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Takeda, H.; Ohashi, K.; Asatani, T.; Kosumi, D.; Hashimoto, H.; Ishitani, O.; Tamiaki, H. Photochemical reduction of CO2 with red light using synthetic chlorophyll–rhenium bipyridine dyad. Chem. Lett. 2014, 43, 1383–1385. [Google Scholar] [CrossRef]
- Matlachowski, C.; Braun, B.; Tschierlei, S.; Schwalbe, M. Photochemical CO2 reduction catalyzed by phenanthroline extended tetramesityl porphyrin complexes linked with a rhenium-(I) tricarbonyl unit. Inorg. Chem. 2015, 54, 10351–10360. [Google Scholar] [CrossRef]
- Lang, P.; Pfrunder, M.; Quach, G.; Braun-Cula, B.; Moore, E.G.; Schwalbe, M. Sensitized photochemical CO2 reduction by hetero–Pacman compounds linking a ReI tricarbonyl with a porphyrin unit. Chem.-Eur. J. 2019, 25, 4509–4519. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, Y.; Fukaya, K.; Yoshida, M.; Ishida, H. Trans-(Cl)-[Ru(5,5′-diamide-2,2′-bipyridine)(CO)2Cl2]: Synthesis, structure, and photocatalytic CO2 reduction activity. Chem.-Eur. J. 2015, 21, 10049–10060. [Google Scholar] [CrossRef]
- Gabrielsson, A.; Lindsay Smith, J.R.; Perutz, R.N. Remote site photosubstitution in metalloporphyrin–rhenium tricarbonylbipyridine assemblies: Photo-reactions of molecules with very short lived excited states. Dalton Trans. 2008, 4259–4269. [Google Scholar] [CrossRef]
- Gabrielsson, A.; Hartl, F.; Zhang, H.; Lindsay Smith, J.R.; Towrie, M.; Vlcek, A.; Perutz, R.N. Ultrafast charge separation in a photoreactive rhenium-appended porphyrin assembly monitored by picosecond transient infrared spectroscopy. J. Am. Chem. Soc. 2006, 128, 4253–4266. [Google Scholar] [CrossRef]
- Koike, K.; Hori, H.; Ishizuka, M.; Westwell, J.R.; Takeuchi, K.; Ibusuki, T.; Enjouji, K.; Konno, H.; Sakamoto, K.; Ishitani, O. Key process of the photocatalytic reduction of CO2 using [Re(4,4′-X2-bipyridine)(CO)3PR3]+ (X = CH3, H, CF3; PR3 = phosphorus ligands): Dark reaction of the one-electron-reduced complexes with CO2. Organometallics 1997, 16, 5724–5729. [Google Scholar] [CrossRef]
- Lanese, J.G.; Wilson, G.S. Electrochemical studies of zinc tetraphenylporphin. J. Electrochem. Soc. 1972, 119, 1039–1043. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Soeta, A.; Toeda, H.; Itoh, K. Raman scattering study on electrochemical reduction products of magnesium, zinc and copper tetraphenylporphines. J. Electroanal. Chem. Interfacial Electrochem. 1983, 159, 347–359. [Google Scholar] [CrossRef]
- Closs, G.L.; Closs, L.E. Negative ions of porphin metal complexes. J. Am. Chem. Soc. 1963, 85, 818–819. [Google Scholar] [CrossRef]
- Whitlock, H.W.; Oester, M.Y. Behavior of di- and tetrahydroporphyrins under alkaline conditions. Direct observation of the chlorin-phlorin equilibrium. J. Am. Chem. Soc. 1973, 95, 5738–5741. [Google Scholar] [CrossRef] [PubMed]
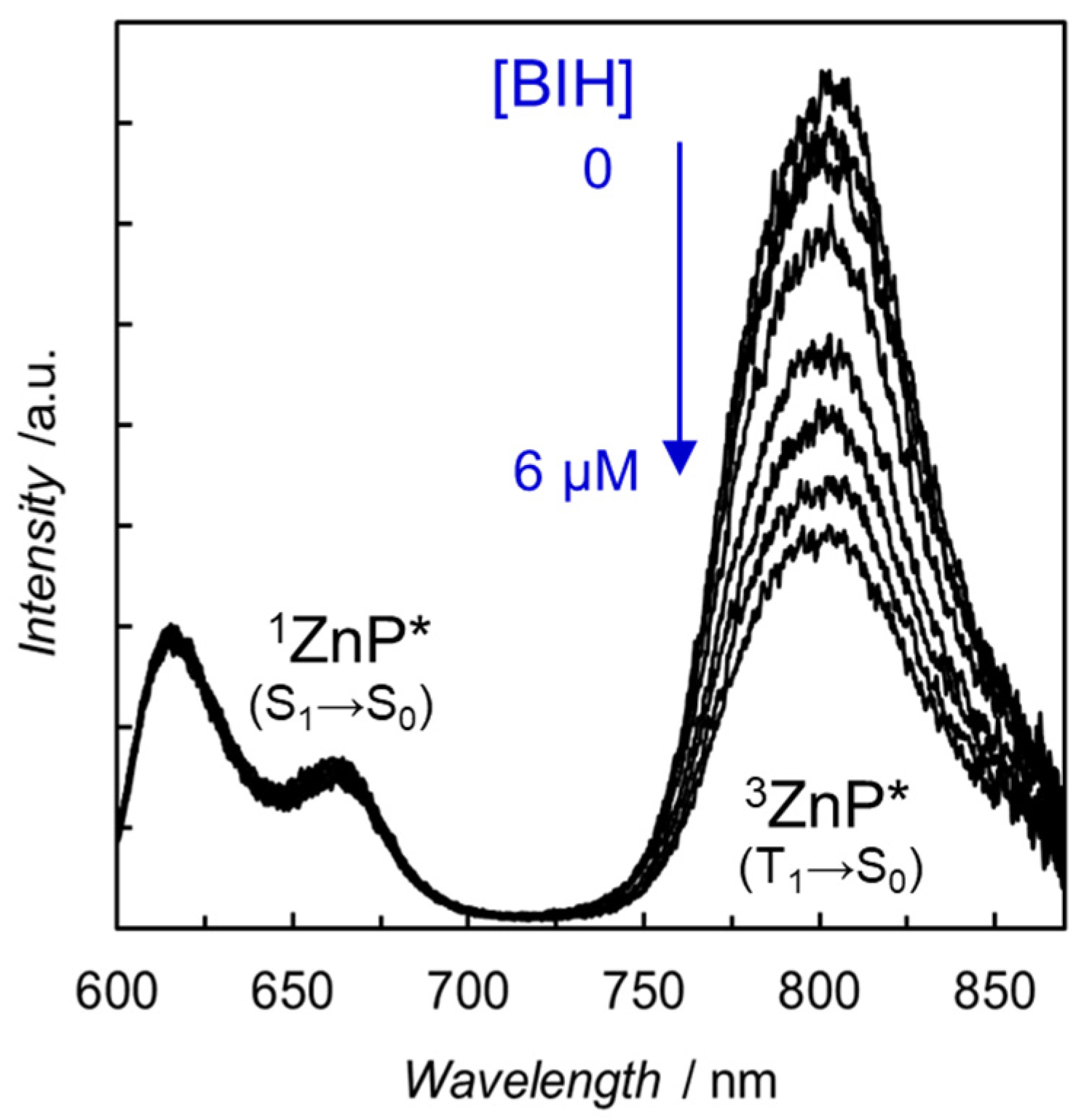
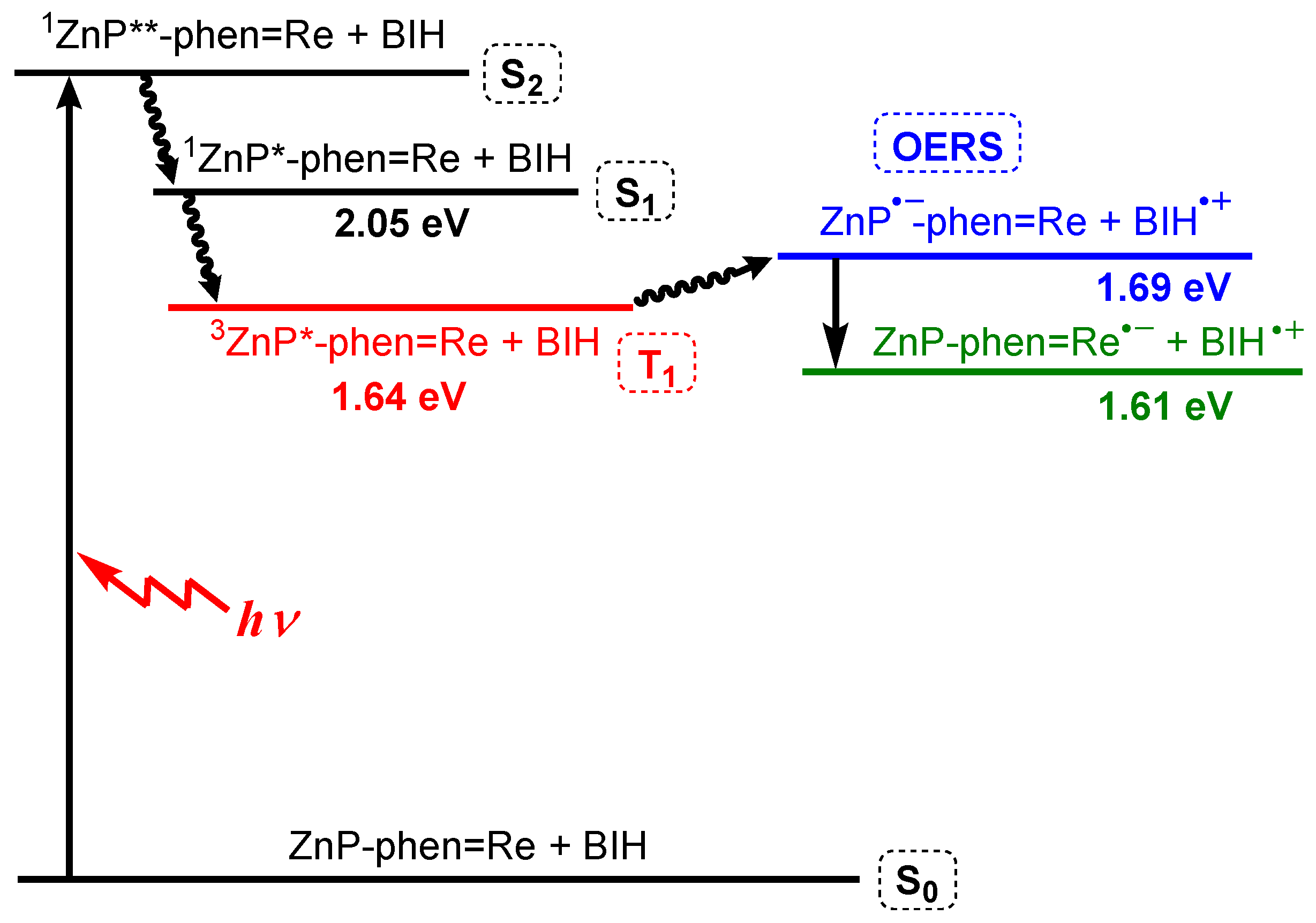
- Kuramochi, Y.; Fujisawa, Y.; Satake, A. Photocatalytic CO2 reduction mediated by electron transfer via the excited triplet state of Zn(II) porphyrin. J. Am. Chem. Soc. 2020, 142, 705–709. [Google Scholar] [CrossRef]
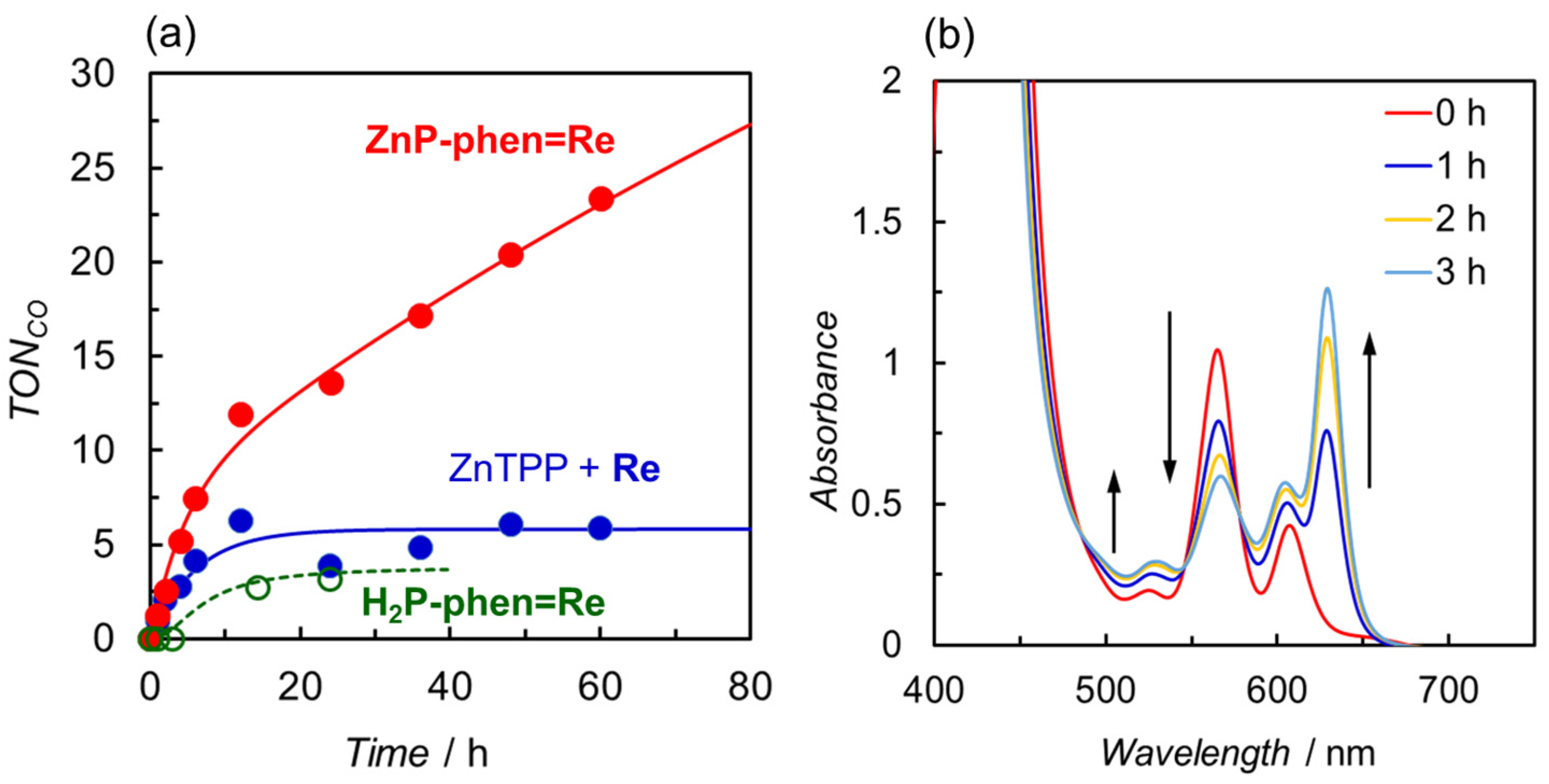
- Kuramochi, Y.; Satake, A. Photocatalytic CO2 reductions catalyzed by meso-(1,10-Phenanthrolin-2-yl)-porphyrins having a rhenium(I) tricarbonyl complex. Chem.-Eur. J. 2020, 26, 16365–16373. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Kamiya, M.; Ishida, H. Photocatalytic CO2 reduction in N,N-dimethylacetamide/water as an alternative solvent system. Inorg. Chem. 2014, 53, 3326–3332. [Google Scholar] [CrossRef] [PubMed]
- Quimby, D.J.; Longo, F.R. Luminescence studies on several tetraarylporphins and their zinc derivatives. J. Am. Chem. Soc. 1975, 97, 5111–5117. [Google Scholar] [CrossRef]
- Smieja, J.M.; Benson, E.E.; Kumar, B.; Grice, K.A.; Seu, C.S.; Miller, A.J.M.; Mayer, J.M.; Kubiak, C.P. Kinetic and structural studies, origins of selectivity, and interfacial charge transfer in the artificial photosynthesis of CO. Proc. Natl. Acad. Sci. USA 2012, 109, 15646–15650. [Google Scholar] [CrossRef]
- Kobuke, Y.; Miyaji, H. Supramolecular organization of imidazolyl-porphyrin to a slipped cofacial dimer. J. Am. Chem. Soc. 1994, 116, 4111–4112. [Google Scholar] [CrossRef]
- Satake, A.; Kobuke, Y. Artificial photosynthetic systems: Assemblies of slipped cofacial porphyrins and phthalocyanines showing strong electronic coupling. Org. Biomol. Chem. 2007, 5, 1679–1691. [Google Scholar] [CrossRef]
- Ozeki, H.; Nomoto, A.; Ogawa, K.; Kobuke, Y.; Murakami, M.; Hosoda, K.; Ohtani, M.; Nakashima, S.; Miyasaka, H.; Okada, T. Role of the special pair in the charge-separating event in photosynthesis. Chem.-Eur. J. 2004, 10, 6393–6401. [Google Scholar] [CrossRef]
- Ito, F.; Ishibashi, Y.; Khan, S.R.; Miyasaka, H.; Kameyama, K.; Morisue, M.; Satake, A.; Ogawa, K.; Kobuke, Y. Photoinduced electron transfer and excitation energy transfer in directly linked zinc porphyrin/zinc phthalocyanine composite. J. Phys. Chem. A 2006, 110, 12734–12742. [Google Scholar] [CrossRef] [PubMed]
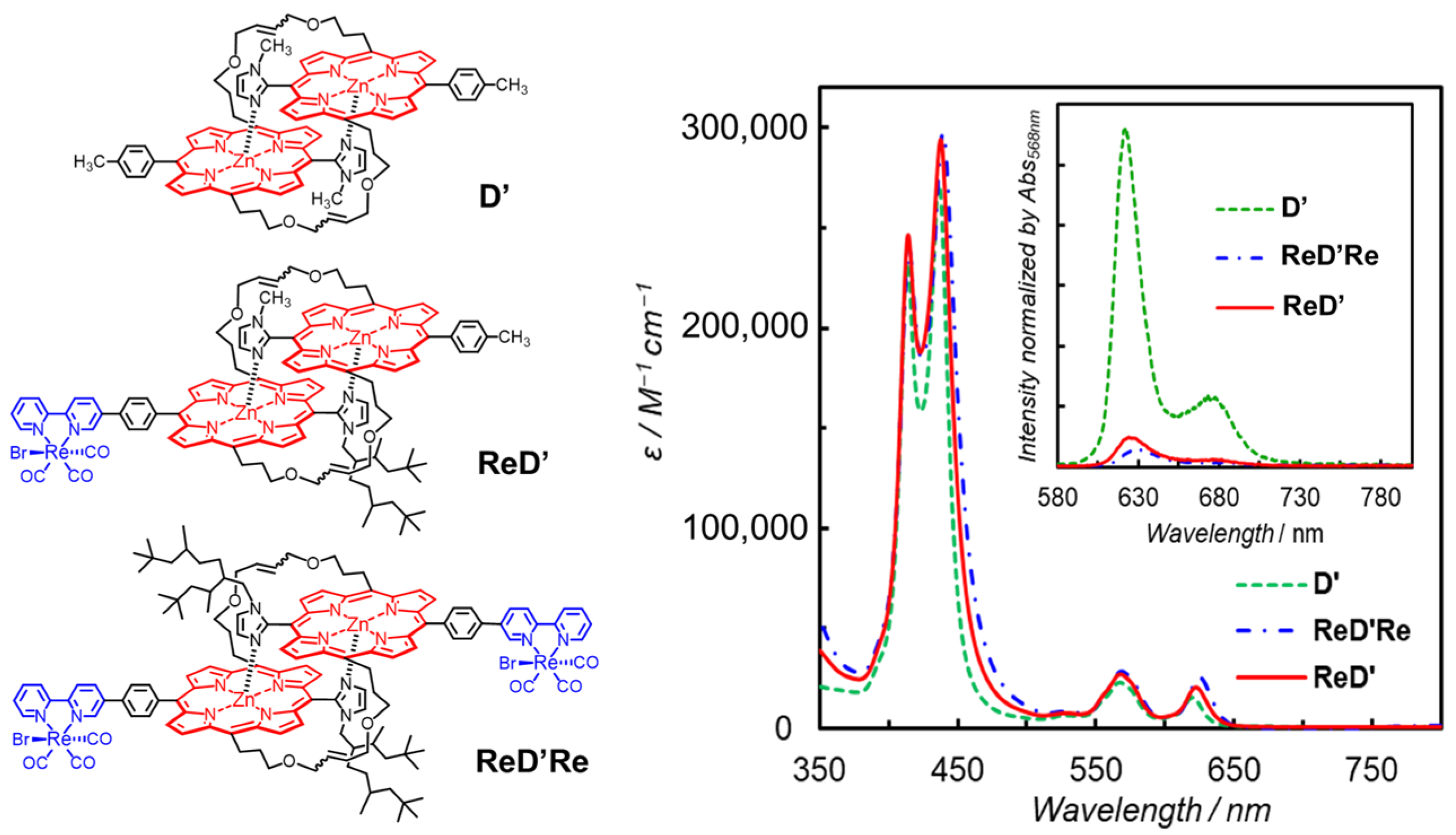
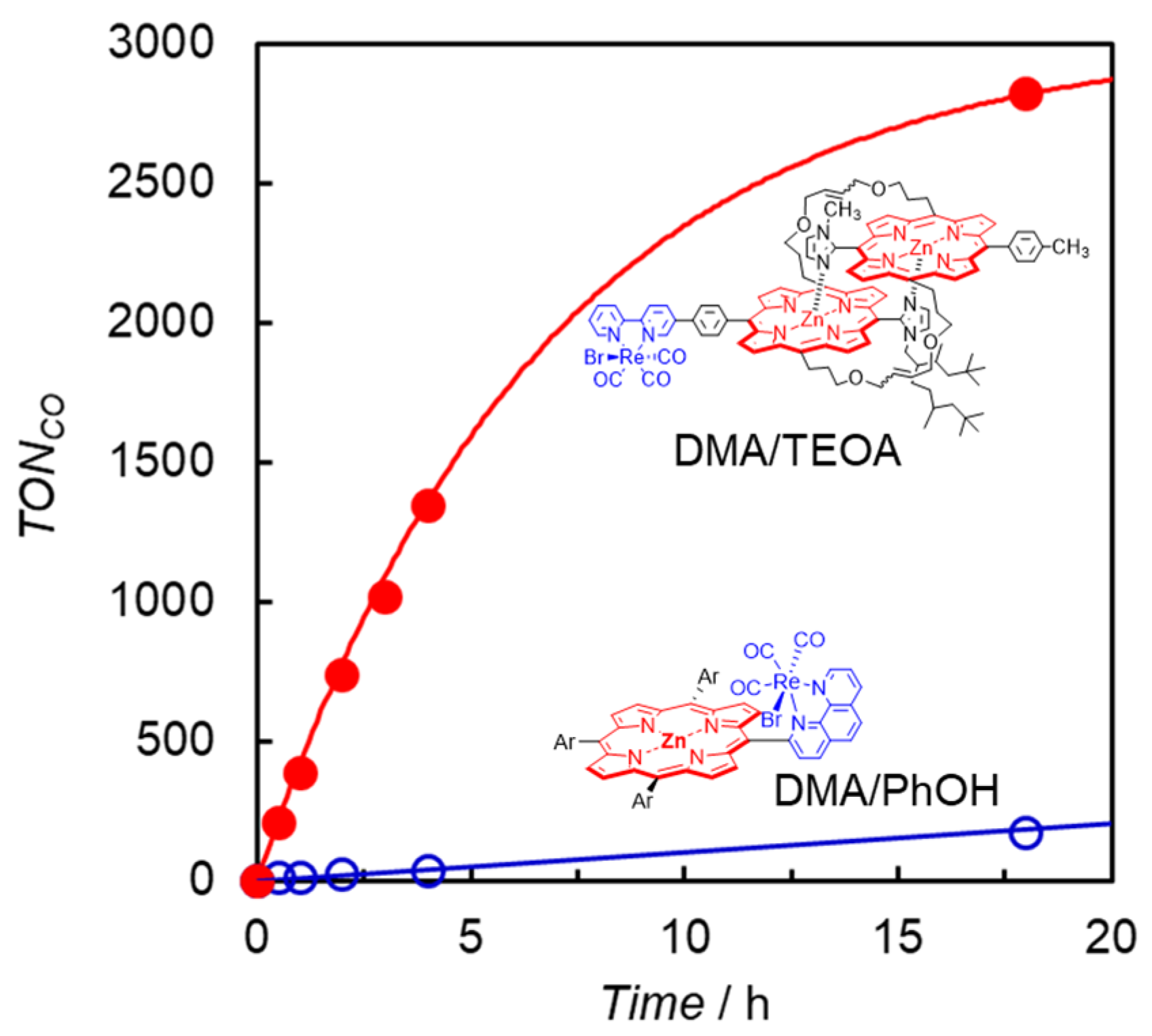
- Kuramochi, Y.; Sato, R.; Sakuma, H.; Satake, A. Photocatalytic CO2 reduction sensitized by a special-pair mimic porphyrin connected with a rhenium(i) tricarbonyl complex. Chem. Sci. 2022, 13, 9861–9879. [Google Scholar] [CrossRef]
- Morimoto, T.; Nakajima, T.; Sawa, S.; Nakanishi, R.; Imori, D.; Ishitani, O. CO2 capture by a rhenium(I) complex with the aid of triethanolamine. J. Am. Chem. Soc. 2013, 135, 16825–16828. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O. Development of an efficient photocatalytic system for CO2 reduction using rhenium(I) complexes based on mechanistic studies. J. Am. Chem. Soc. 2008, 130, 2023–2203. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Nabetani, Y.; Nakazato, R.; Pratheesh, N.V.; Sato, T.; Nozawa, S.; Adachi, S.; Tachibana, H.; Inoue, H. Mechanism of the photoreduction of carbon dioxide catalyzed by the benchmarking rhenium dimethylbipyridine complexes; Operando measurements by XAFS and FT-IR. J. Catal. 2022, 405, 508–519. [Google Scholar] [CrossRef]
- Kamogawa, K.; Shimoda, Y.; Miyata, K.; Onda, K.; Yamazaki, Y.; Tamaki, Y.; Ishitani, O. Mechanistic study of photocatalytic CO2 reduction using a Ru(II)-Re(I) supramolecular photocatalyst. Chem. Sci. 2021, 12, 9682–9693. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuramochi, Y.; Satake, A. Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction. Catalysts 2023, 13, 282. https://doi.org/10.3390/catal13020282
Kuramochi Y, Satake A. Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction. Catalysts. 2023; 13(2):282. https://doi.org/10.3390/catal13020282
Chicago/Turabian StyleKuramochi, Yusuke, and Akiharu Satake. 2023. "Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction" Catalysts 13, no. 2: 282. https://doi.org/10.3390/catal13020282
APA StyleKuramochi, Y., & Satake, A. (2023). Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction. Catalysts, 13(2), 282. https://doi.org/10.3390/catal13020282