
Structure and Catalytic Performance of Carbon-Based Solid Acids from Biomass Activated by ZnCl2

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization of Carbon-Based Solid Acid Catalysts
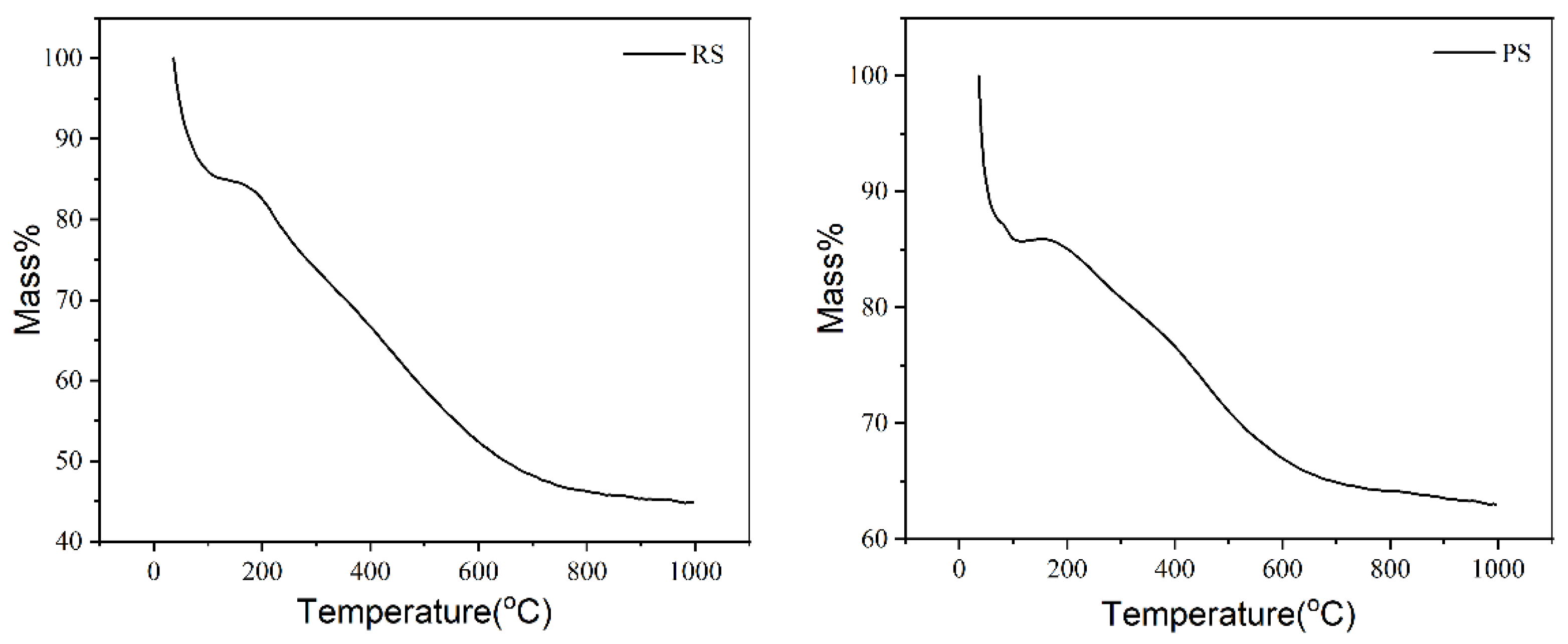
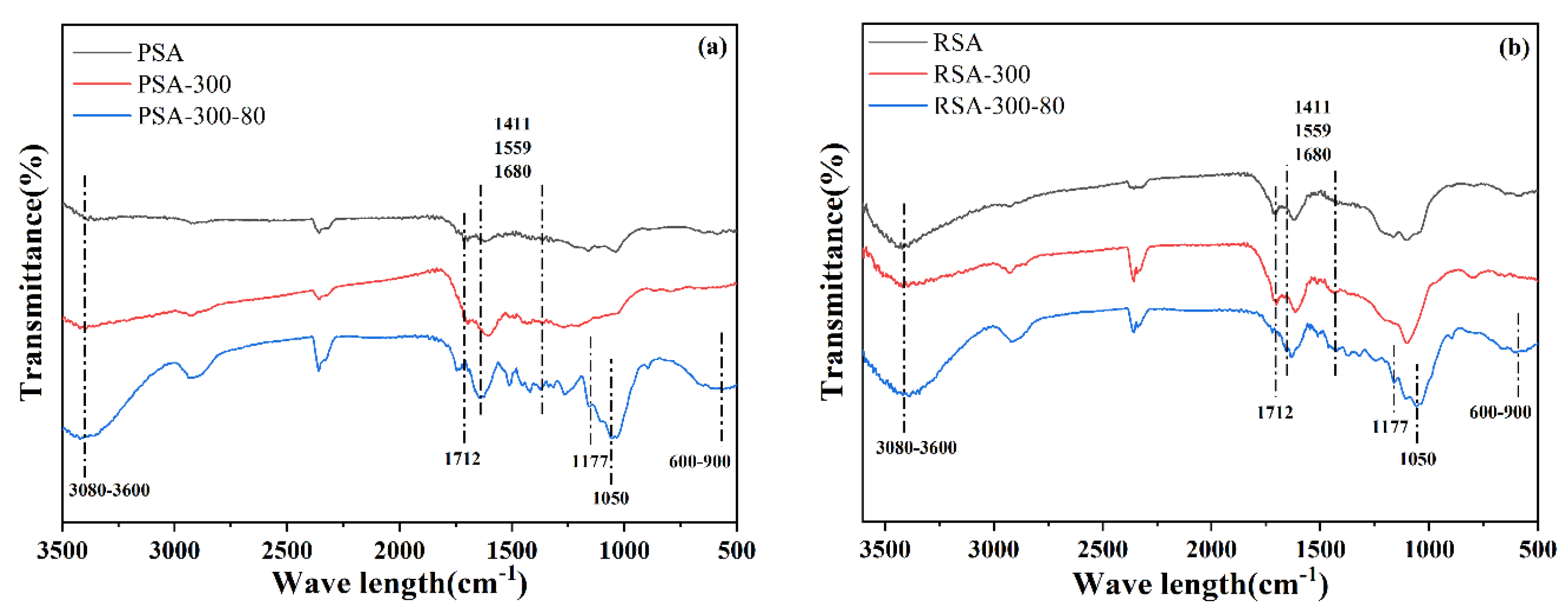
2.1.1. Thermal Stability
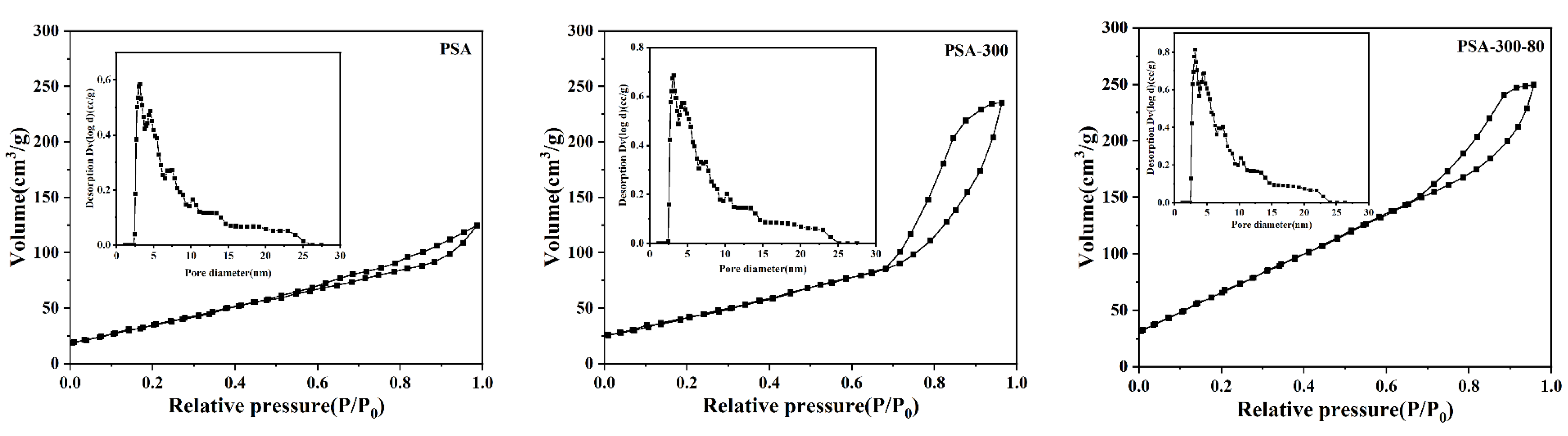
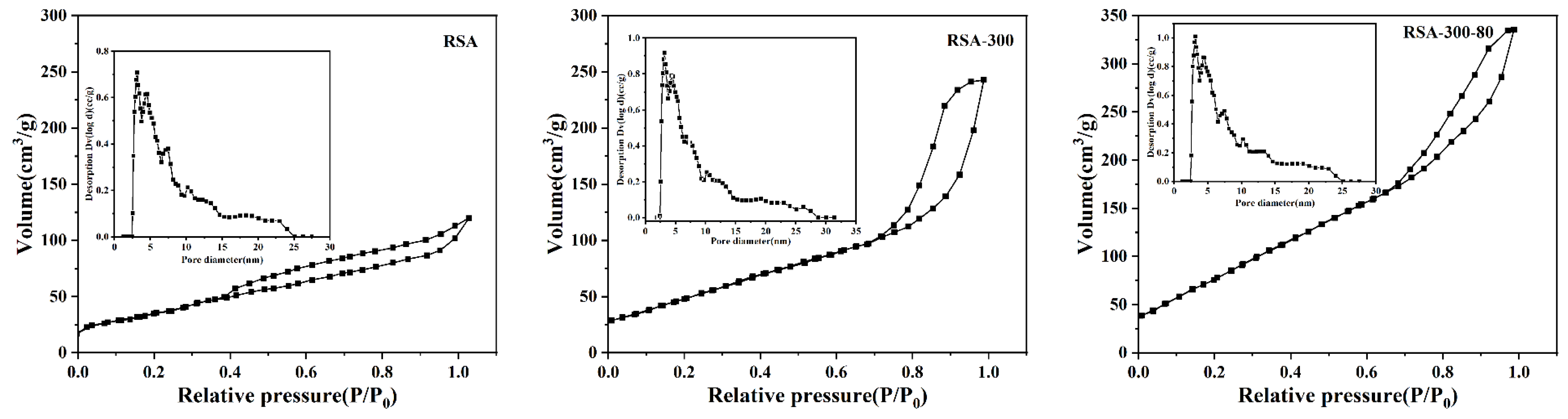
2.1.2. The Structure of Pore and Surface Morphology
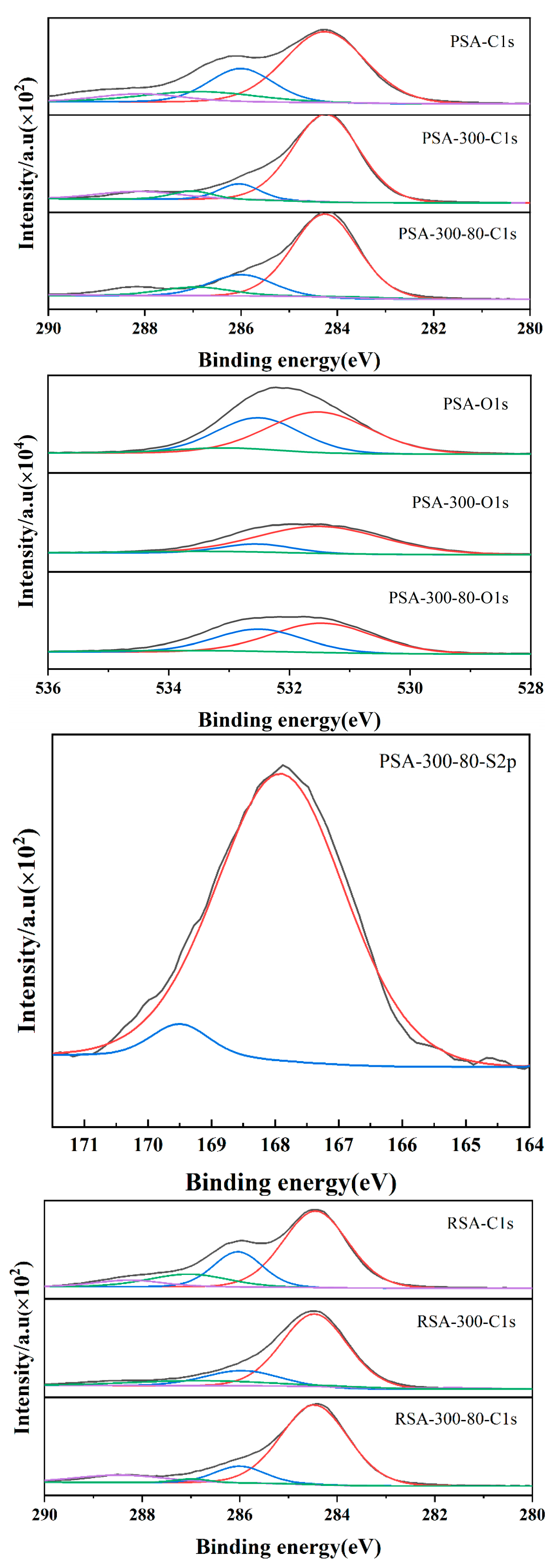
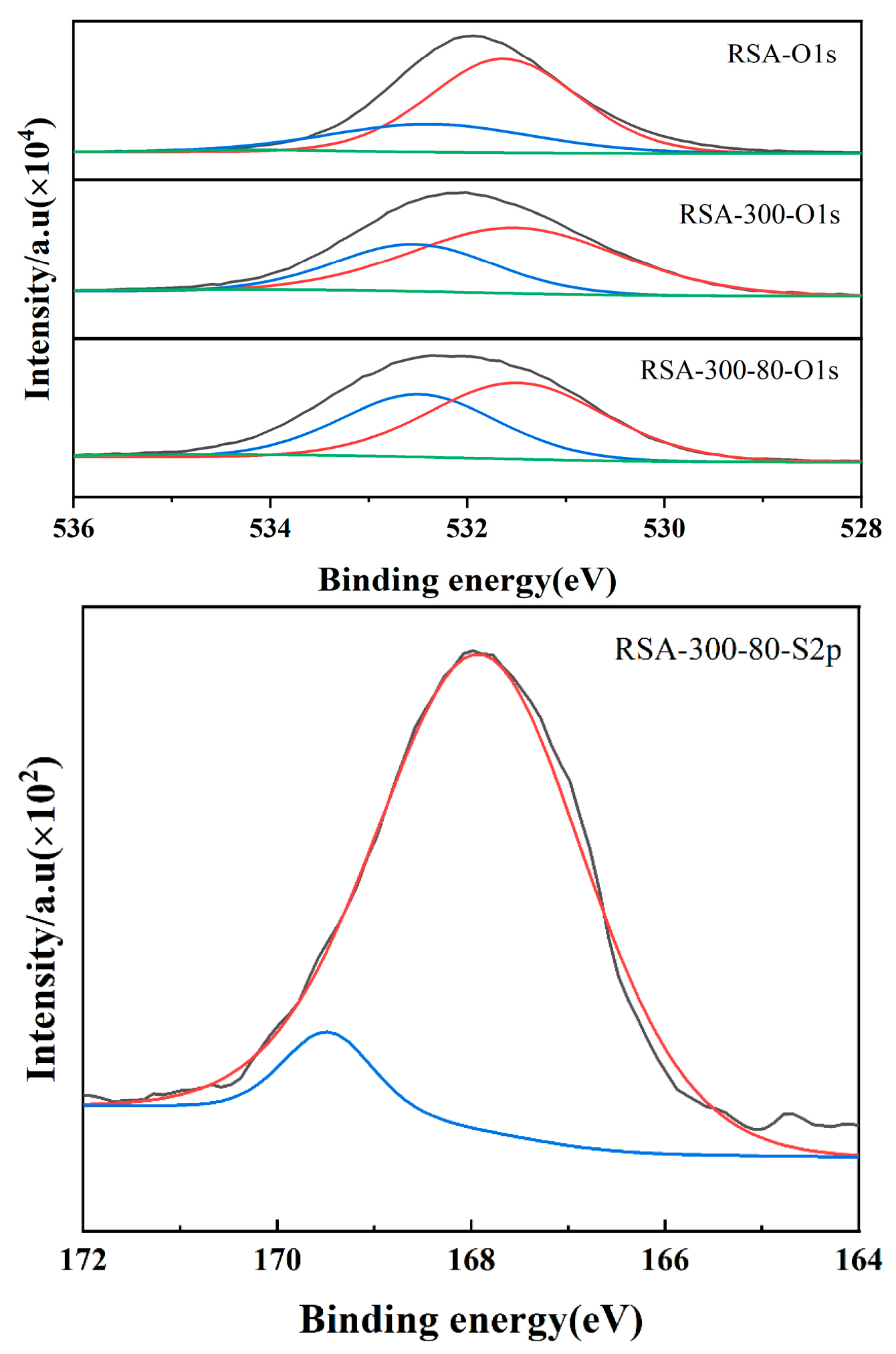
2.1.3. Catalytic Active Center and Acid Density
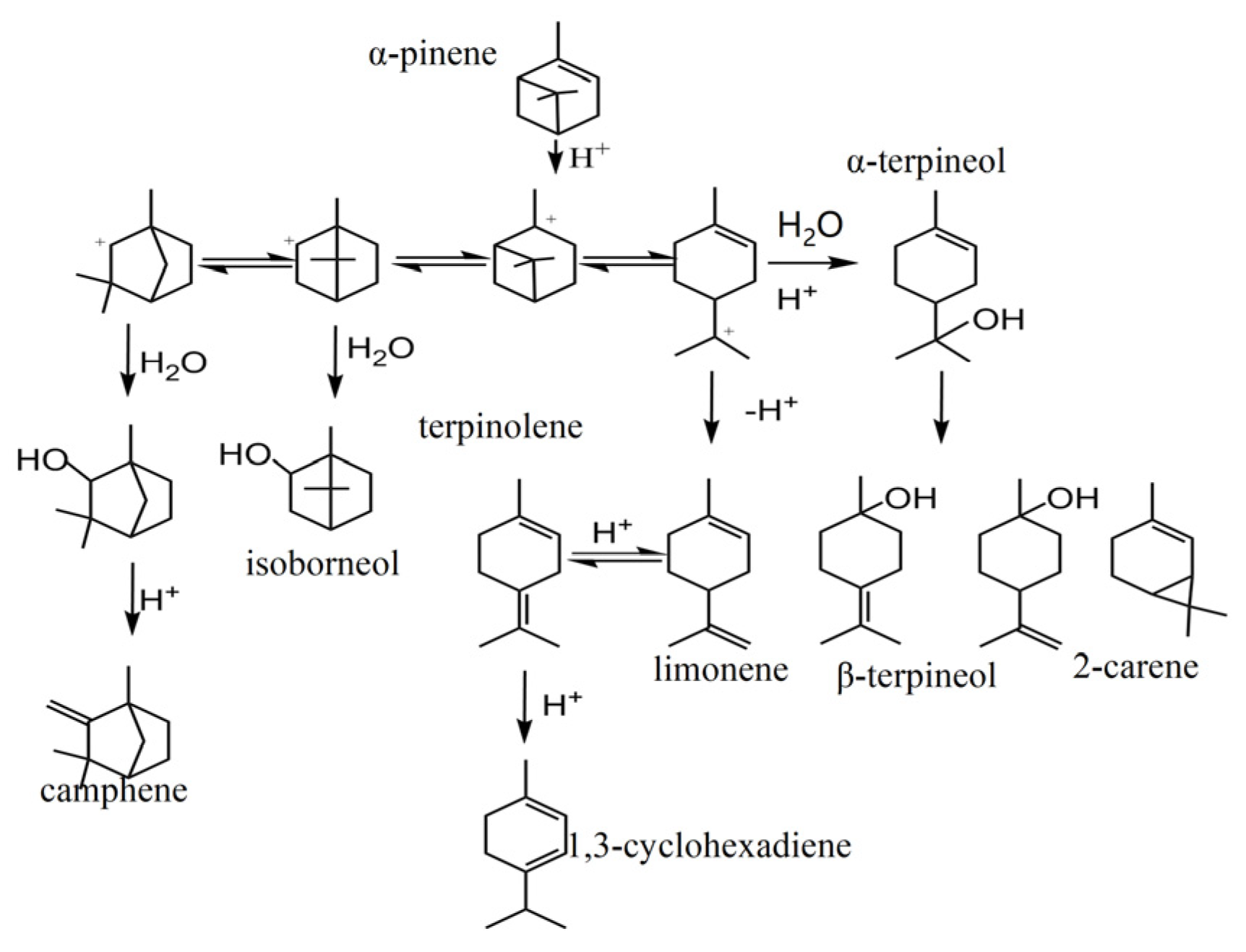
2.2. Catalytic Activity in Hydration of α-Pinene
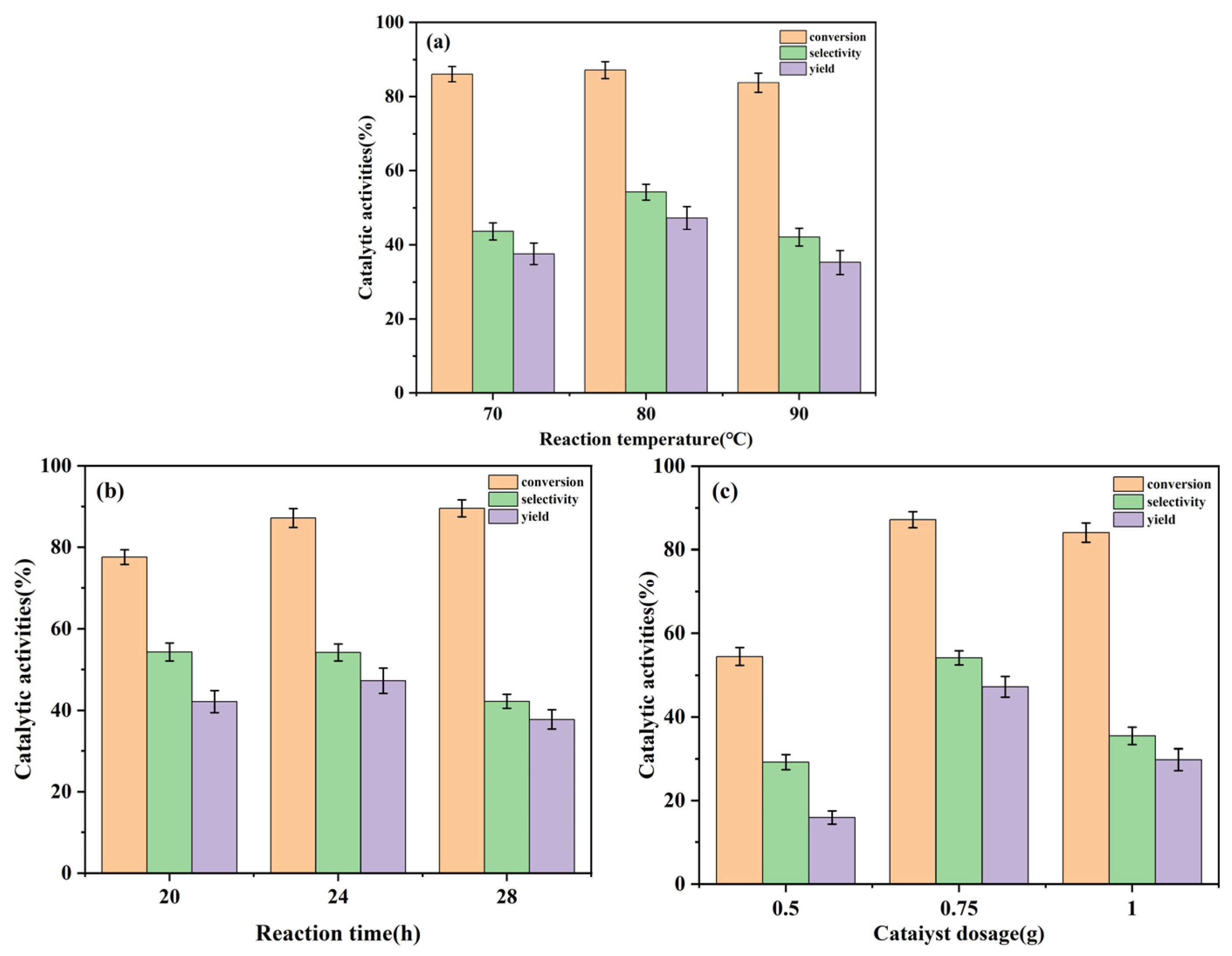
2.2.1. Effect of Reaction Temperature
2.2.2. Effect of Reaction Time
2.2.3. Effect of Catalyst Dosage
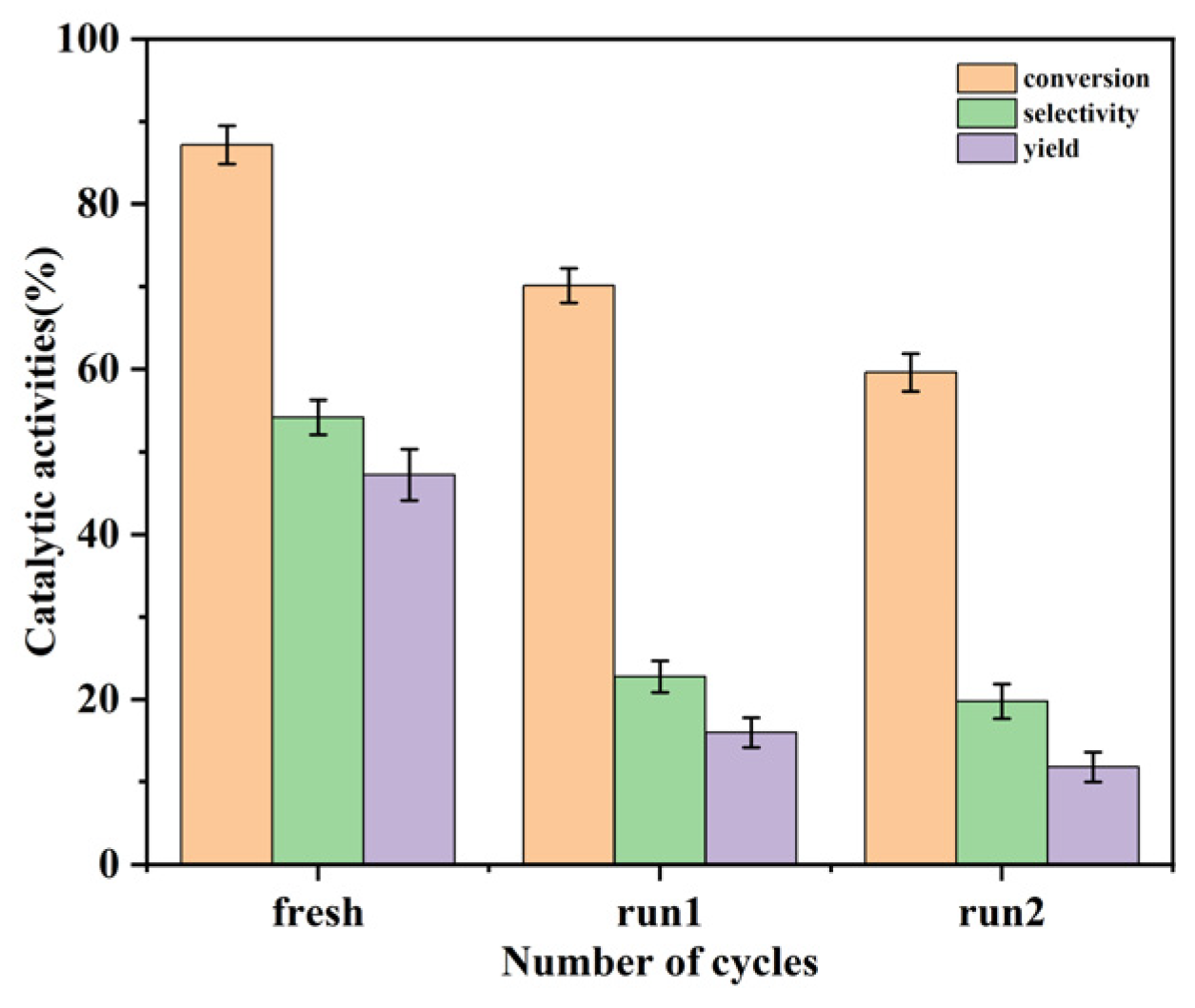
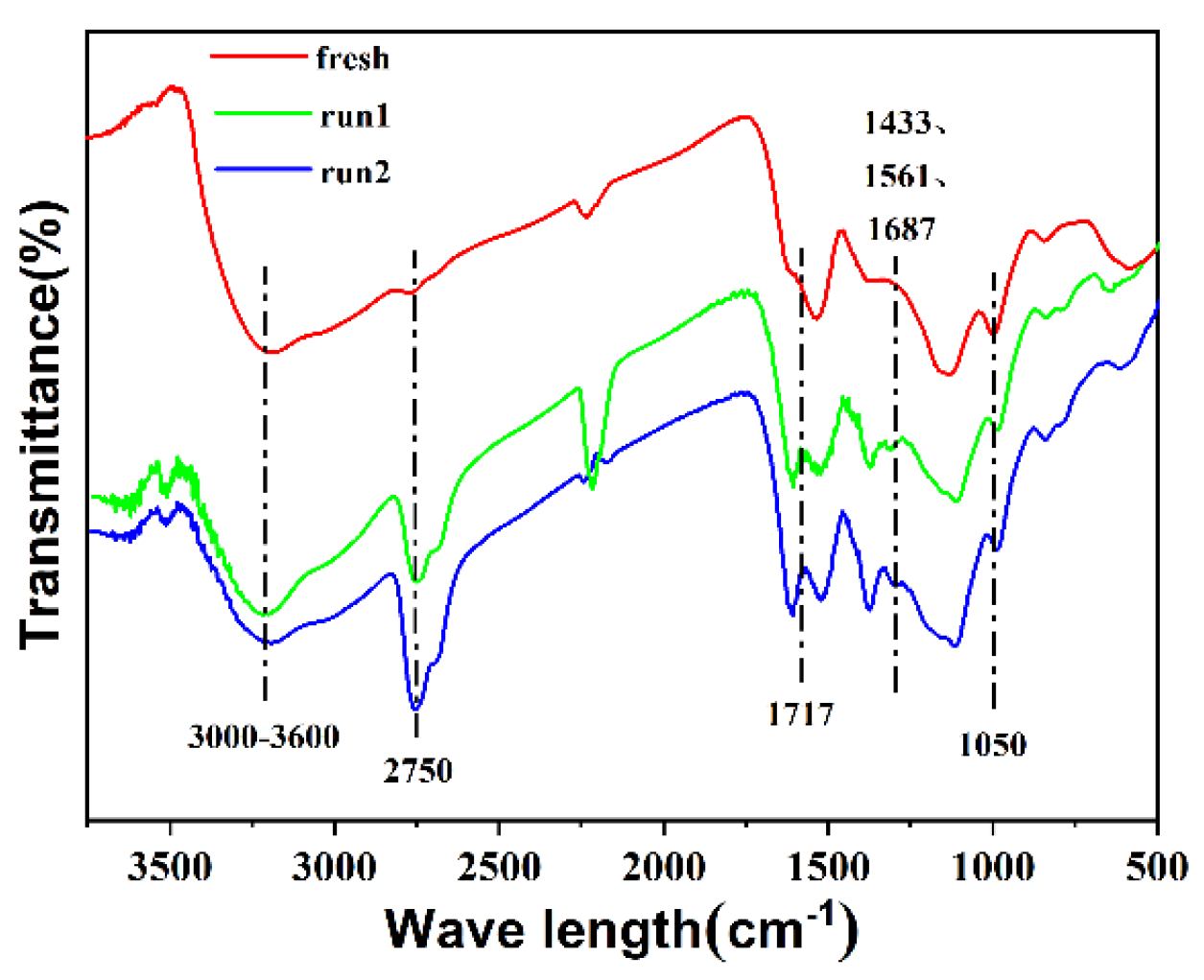
2.3. Reusability of Catalyst
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Characterization of the Catalysts
3.4. Investigation of Catalytic Activity
3.5. Products Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Zhang, L.; Wang, W.; Wang, G. Recent updates on bioactive properties of alpha-terpineol. J. Essent. Oil Res. 2023, 35, 274–288. [Google Scholar] [CrossRef]
- Zheng, G.; Chao, Y.; Liu, M.; Yang, Y.; Zhang, D.; Wang, K.; Tao, Y.; Zhang, J.; Li, Y.; Wei, M. Evaluation of dynamic changes in the bioactive components in Citri Reticulatae Pericarpium (Citrus reticulata ‘Chachi’) under different harvesting and drying conditions. J. Sci. Food Agric. 2021, 101, 3280–3289. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Zhong, H.; Liu, P.; Liu, X.; Xie, C.; Yu, F.; Yu, S.; Zhang, J. Heteropolyacid Bisalt of N-octyl Ethoxylated Octadecylamine: An Efficient and Reusable Catalyst for Carboxylic Acid-Free Hydration of α-Pinene. Catal. Lett. 2016, 146, 929–936. [Google Scholar] [CrossRef]
- ávila, M.C.; Comelli, N.A.; Rodríguez-Castellón, E.; Jiménez-López, A.; Carrizo Flores, R.; Ponzi, E.N.; Ponzi, M.I. Study of solid acid catalysis for the hydration of α-pinene. J. Mol. Catal. A Chem. 2010, 322, 106–112. [Google Scholar] [CrossRef]
- Yang, G.; Liu, Y.; Zhou, Z.; Zhang, Z. Kinetic study of the direct hydration of turpentine. Chem. Eng. J. 2011, 168, 351–358. [Google Scholar] [CrossRef]
- Vital, J.; Ramos, A.M.; Silva, I.F.; Valente, H.; Castanheiro, J.E. Hydration of alpha-pinene over zeolites and activated carbons dispersed in polymeric membranes. Catal. Today 2000, 56, 167–172. [Google Scholar] [CrossRef]
- Mochida, T.; Ohnishi, R.; Horita, N.; Kamiya, Y.; Okuhara, T. Hydration of alpha-pinene over hydrophobic zeolites in 1,4-dioxane-water and in water. Microporous Mesoporous Mater. 2007, 101, 176–183. [Google Scholar] [CrossRef]
- Dal, A.E.B. Refining of Crude Sulfate Turpentine Obtained from a Kraft Pulp Mill: A Pilot Scale Treatment. Bioresources 2021, 16, 8098–8110. [Google Scholar]
- Wang, C.; Xie, Q.; Dou, X.; Zhang, L.; Yang, X. Conversion of carbonaceous materials into solid acids for tylosin mitigation: Effect of preprocessing methods on the reactivity of sulfonation reaction. Biochar 2023, 5, 32. [Google Scholar] [CrossRef]
- Lam, E.; Luong, J.H.T. Carbon Materials as Catalyst Supports and Catalysts in the Transformation of Biomass to Fuels and Chemicals. ACS Catal. 2014, 4, 3393–3410. [Google Scholar] [CrossRef]
- Thushari, I.; Babel, S.; Samart, C. Biodiesel production in an autoclave reactor using waste palm oil and coconut coir husk derived catalyst. Renew. Energy 2019, 134, 125–134. [Google Scholar] [CrossRef]
- Zuo, P.; Duan, J.; Fan, H.; Qu, S.; Shen, W. Facile synthesis high nitrogen-doped porous carbon nanosheet from pomelo peel and as catalyst support for nitrobenzene hydrogenation. Appl. Surf. Sci. 2018, 435, 1020–1028. [Google Scholar] [CrossRef]
- Mo, X.; Lotero, E.; Lu, C.; Liu, Y.; Goodwin, J.G. A Novel Sulfonated Carbon Composite Solid Acid Catalyst for Biodiesel Synthesis. Catal. Lett. 2008, 123, 1–6. [Google Scholar] [CrossRef]
- Li, R.; Yang, G.; Wang, Q.; Chen, J.; Dong, J.; Zhang, X. Preparation of Carbon-based Solid Acid from Corncob Residual and its Performance for Acid-Catalyzed Hydrolysis. Bioresources 2017, 12, 7439–7451. [Google Scholar] [CrossRef]
- Chen, G.; Fang, B. Preparation of solid acid catalyst from glucose-starch mixture for biodiesel production. Bioresour. Technol. 2011, 102, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- Mardhiah, H.H.; Ong, H.C.; Masjuki, H.H.; Lim, S.; Pang, Y.L. Investigation of carbon-based solid acid catalyst from Jatropha curcas biomass in biodiesel production. Energy Convers. Manag. 2017, 144, 10–17. [Google Scholar] [CrossRef]
- Fedie, R.L.; Mcneff, C.; Mcneff, L.C.; Greuel, P.G.; Yan, B.; Jenkins, J.A.; Brethorst, J.T.; Frost, G.B.; Hoye, T.R. Hydrothermal catalysis of waste greases into green gasoline, jet, and diesel biofuels in continuous flow supercritical water. Biofuels Bioprod. Biorefin. Biofpr 2022, 16, 349–369. [Google Scholar] [CrossRef]
- Alvarez, M.L.; Mendez, A.; Rodriguez-Pacheco, R.; Paz-Ferreiro, J.; Gasco, G. Recovery of Zinc and Copper from Mine Tailings by Acid Leaching Solutions Combined with Carbon-Based Materials. Appl. Sci. Basel 2021, 11, 5166. [Google Scholar] [CrossRef]
- Ma, Z.; Xing, X.; Qu, Z.; Sun, Y.; Sun, G.; Wang, X.; Han, Y. Activity of microporous lignin-derived carbon-based solid catalysts used in biodiesel production. Int. J. Biol. Macromol. 2020, 164, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.F.; Abo El Naga, A.O.; El Saied, M.; Abubaker, M.M.; Shaban, S.A.; El Kady, F.Y. Potato peel waste-derived carbon-based solid acid for the esterification of oleic acid to biodiesel. Environ. Technol. Innov. 2021, 21, 101355. [Google Scholar] [CrossRef]
- Xie, J.; Han, Q.; Feng, B.; Liu, Z. Preparation of amphiphilic mesoporous carbon-based solid acid from kraft lignin activated by phosphoric acid and its catalytic performance for hydration of α-pinene. Bioresources 2019, 14, 4284–4303. [Google Scholar] [CrossRef]
- Li, Y.; Shen, S.; Wang, C.; Peng, X.; Yuan, S. The effect of difference in chemical composition between cellulose and lignin on carbon based solid acids applied for cellulose hydrolysis. Cellulose 2018, 25, 1851–1863. [Google Scholar] [CrossRef]
- Fang, J.; Jin, L.; Meng, Q.; Shan, S.; Wang, D.; Lin, D. Biochar effectively inhibits the horizontal transfer of antibiotic resistance genes via transformation. J. Hazard. Mater. 2022, 423, 127150. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Hara, M. Amorphous Carbon with SO3H Groups as a Solid Bronsted Acid Catalyst. ACS Catal. 2012, 2, 1296–1304. [Google Scholar] [CrossRef]
- Arce-Saldana, L.A.; Espinoza-Mosso, E.; Castro-Rodriguez, B.; Portillo-Lopez, A.; Munoz-Munoz, F.; Dominguez, D.; Farias, M.H.; Soto, G. Plasma synthesis of carbon powder with embedded Fe3C nanoparticles for magnetic separation of biomolecules. Adv. Powder Technol. 2018, 29, 1035–1041. [Google Scholar] [CrossRef]
- Mei, S.; Wang, F.; Hu, X.; Yang, K.; Xie, D.; Yang, L.; Wu, Z.; Wei, J. Construction of a hierarchical micro & nanoporous surface for loading genistein on the composite of polyetheretherketone/tantalum pentoxide possessing antibacterial activity and accelerated osteointegration. Biomater. Sci. 2021, 9, 167–185. [Google Scholar] [PubMed]
- El, M.; El, A.M.A.; Elhosiny, F.I.; El, F.Y. A Comprehensive Investigation on Biomass Solid Waste Conversion to a Novel Catalyst for Hydrothermal Production of Bio-Fuel Feedstock. J. Clean. Prod. 2019, 218, 157–166. [Google Scholar]
- Yang, L.; Yuan, H.; Wang, S. Preparation and application of ordered mesoporous carbon-based solid acid catalysts for transesterification and epoxidation. J. Porous Mater. 2019, 26, 1435–1445. [Google Scholar] [CrossRef]
- Feng, B. Preparation of Lignin Carbon-Based Solid Acid and Its Catalytic Synthesis of α-Terpineol from α-Pinene. Master’s Thesis, Guangxi University for Nationalities, Nanning, China, 2019. [Google Scholar]
- Inagaki, M. Pores in carbon materials-importance of their control. New Carbon Mater. 2009, 24, 193–222. [Google Scholar] [CrossRef]
- Hong, D.; Zhou, J.; Hu, C.; Zhou, Q.; Mao, J.; Qin, Q. Mercury removal mechanism of AC prepared by one-step activation with ZnCl2. Fuel 2019, 235, 326–335. [Google Scholar] [CrossRef]
- Ceyhan, A.A.; Sahin, O.; Baytar, O.; Saka, C. Surface and porous characterization of activated carbon prepared from pyrolysis of biomass by two-stage procedure at low activation temperature and it’s the adsorption of iodine. J. Anal. Appl. Pyrolysis 2013, 104, 378–383. [Google Scholar] [CrossRef]
- Sandbrink, L.; Lazaridis, T.; Rose, M.; Palkovits, R. Ambient temperature gas phase sulfonation: A mild route towards acid functionalized ordered mesoporous organosilica. Microporous Mesoporous Mater. 2018, 267, 198–202. [Google Scholar] [CrossRef]
- Paddison, S.J.; Elliott, J.A. Molecular Modeling of the short-side-chain perfluorosulfonic acid membrane. J. Phys. Chem. A 2005, 109, 7583–7593. [Google Scholar] [CrossRef]
- Grosmaire, L.; Castagnoni, S.; Huguet, P.; Sistat, P.; Boucher, M.; Bouchard, P.; Bebin, P.; Deabate, S. Probing proton dissociation in ionic polymers by means of in situ ATR-FTIR spectroscopy. Phys. Chem. Chem. Phys. 2008, 10, 1577–1583. [Google Scholar] [CrossRef]
- Hao, H.; Shen, F.; Yang, J.; Qiu, M.; Guo, H.; Qi, X. Synthesis of Sulfonated Carbon from Discarded Masks for Effective Production of 5-Hydroxymethylfurfural. Catalysts 2022, 12, 1567. [Google Scholar] [CrossRef]
- Toda, M.; Takagaki, A.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Green chemistry-Biodiesel made with sugar catalyst. Nature 2005, 438, 178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, B.; Chen, Z.; Zhang, S. A bifunctional carbon catalyst with -Cl and -SO3H groups for hydrolyzing furfural residue to levulinic acid. J. Chem. Technol. Biotechnol. 2022, 97, 2805–2814. [Google Scholar] [CrossRef]
- Wei, Z.; Xiong, D.; Duan, P.; Ding, S.; Li, Y.; Li, L.; Niu, P.; Chen, X. Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene. Catalysts 2020, 10, 213. [Google Scholar] [CrossRef]
- Raut, E.R.; Thakur, M.A.B.; Chaudhari, A.R. Study of synthesis and characterization of raw bagasse, its char and activated carbon prepared using chemical additive. Water Sci. Technol. 2023, 87, 2233–2249. [Google Scholar] [CrossRef]
- Wang, J.; Feng, B.; Che, J.; Luo, J.; Liu, Z. Preparation of lignin carbon-based solid acid by sol-gel method and its catalytic performance for α-pinene hydration reaction. Chin. J. Papermak. 2021, 36, 9–17. [Google Scholar]
- Prakoso, T.; Hanley, J.; Soebianta, M.N.; Soerawidjaja, T.H.; Indarto, A. Synthesis of Terpineol from α-Pinene Using Low-Price Acid Catalyst. Catal. Lett. 2018, 148, 725–731. [Google Scholar] [CrossRef]
- Kitano, M.; Yamaguchi, D.; Suganuma, S.; Nakajima, K.; Kato, H.; Hayashi, S.; Hara, M. Adsorption-enhanced hydrolysis of beta-1,4-glucan on graphene-based amorphous carbon bearing SO3H, COOH, and OH groups. Langmuir 2009, 25, 5068–5075. [Google Scholar] [CrossRef]
- Wisniewska, M.; Pawlak, N.; Sternik, D.; Pietrzak, R.; Nowicki, P. Production of Activated Carbons from Food/Storage Waste. Materials 2023, 16, 1349. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2/g) | VTotal (cm3/g) | Dpore (nm) |
|---|---|---|---|
| PS | 365.1 | 0.33 | 3.69 |
| PSA | 306.3 | 0.28 | 3.63 |
| PSA-300 | 362.3 | 0.34 | 3.72 |
| PSA-300-80 | 425.9 | 0.39 | 3.63 |
| PS300-80 | 490.1 | 0.46 | 3.79 |
| RS | 390.6 | 0.39 | 3.56 |
| RSA | 378.9 | 0.35 | 3.66 |
| RSA-300 | 457.7 | 0.46 | 3.98 |
| RSA-300-80 | 527.0 | 0.49 | 3.73 |
| RS300-80 | 420.9 | 0.41 | 3.84 |
| Catalysts | ATotal (mmol/g) | A–SO3H (mmol/g) | A–COOH (mmol/g) | A–OH (mmol/g) |
|---|---|---|---|---|
| PS300-80 | 4.32 | 1.30 | 1.78 | 1.24 |
| PSA-300-80 | 4.20 | 1.37 | 1.05 | 1.78 |
| RS300-80 | 3.80 | 1.37 | 1.07 | 1.36 |
| RSA-300-80 | 3.93 | 1.28 | 1.37 | 1.28 |
| Catalysts | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|
| PS300-80 | 85.70 | 37.35 | 32.01 |
| PSA-300-80 | 92.45 | 24.39 | 22.54 |
| RS300-80 | 89.63 | 24.42 | 21.89 |
| RSA-300-80 | 87.15 | 54.19 | 47.23 |
| Number | Retention Time (min) | Name | Comparative Content (%) | Similarity (%) |
|---|---|---|---|---|
| 1 | 6.73 | (+)-Camphene | 21.44 | 89 |
| 2 | 7.12 | (-)-Camphene | 9.86 | 94 |
| 3 | 7.85 | β-Pinene | 0.19 | 94 |
| 4 | 8.64 | α-Phellandrene | 0.21 | 95 |
| 5 | 8.95 | α-Terpinene | 1.28 | 95 |
| 6 | 9.37 | Limonene | 10.39 | 89 |
| 7 | 10.16 | γ-Terpinene | 1.40 | 97 |
| 8 | 11.04 | 2-Carene | 7.62 | 94 |
| 9 | 12.18 | Fenchol | 6.48 | 94 |
| 10 | 13.44 | Isoborneol | 7.26 | 96 |
| 11 | 13.96 | 4-Terpineol | 3.90 | 96 |
| 12 | 14.71 | α-Terpineol | 29.97 | 89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhang, H.; Wei, Z.; Xiong, D.; Bai, S.; Tong, M.; Ma, P. Structure and Catalytic Performance of Carbon-Based Solid Acids from Biomass Activated by ZnCl2. Catalysts 2023, 13, 1436. https://doi.org/10.3390/catal13111436
Wu Y, Zhang H, Wei Z, Xiong D, Bai S, Tong M, Ma P. Structure and Catalytic Performance of Carbon-Based Solid Acids from Biomass Activated by ZnCl2. Catalysts. 2023; 13(11):1436. https://doi.org/10.3390/catal13111436
Chicago/Turabian StyleWu, Yao, Hao Zhang, Zhaozhou Wei, Deyuan Xiong, Songbai Bai, Menglong Tong, and Pengcheng Ma. 2023. "Structure and Catalytic Performance of Carbon-Based Solid Acids from Biomass Activated by ZnCl2" Catalysts 13, no. 11: 1436. https://doi.org/10.3390/catal13111436
APA StyleWu, Y., Zhang, H., Wei, Z., Xiong, D., Bai, S., Tong, M., & Ma, P. (2023). Structure and Catalytic Performance of Carbon-Based Solid Acids from Biomass Activated by ZnCl2. Catalysts, 13(11), 1436. https://doi.org/10.3390/catal13111436