
Dehydrogenative Conversions of Aldehydes and Amines to Amides Catalyzed by a Nickel(II) Pincer Complex


 ,
, 
Abstract
:
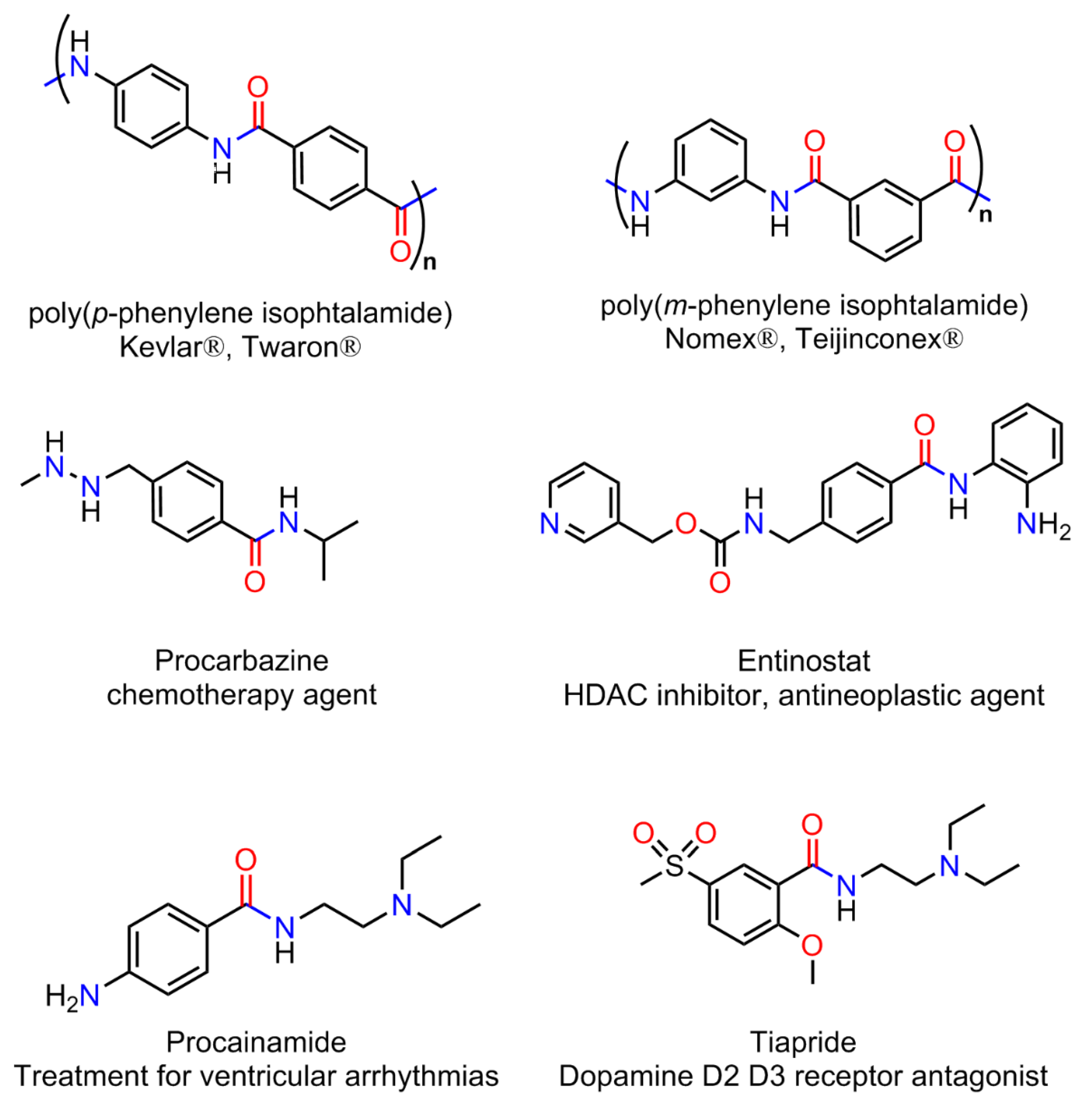
1. Introduction
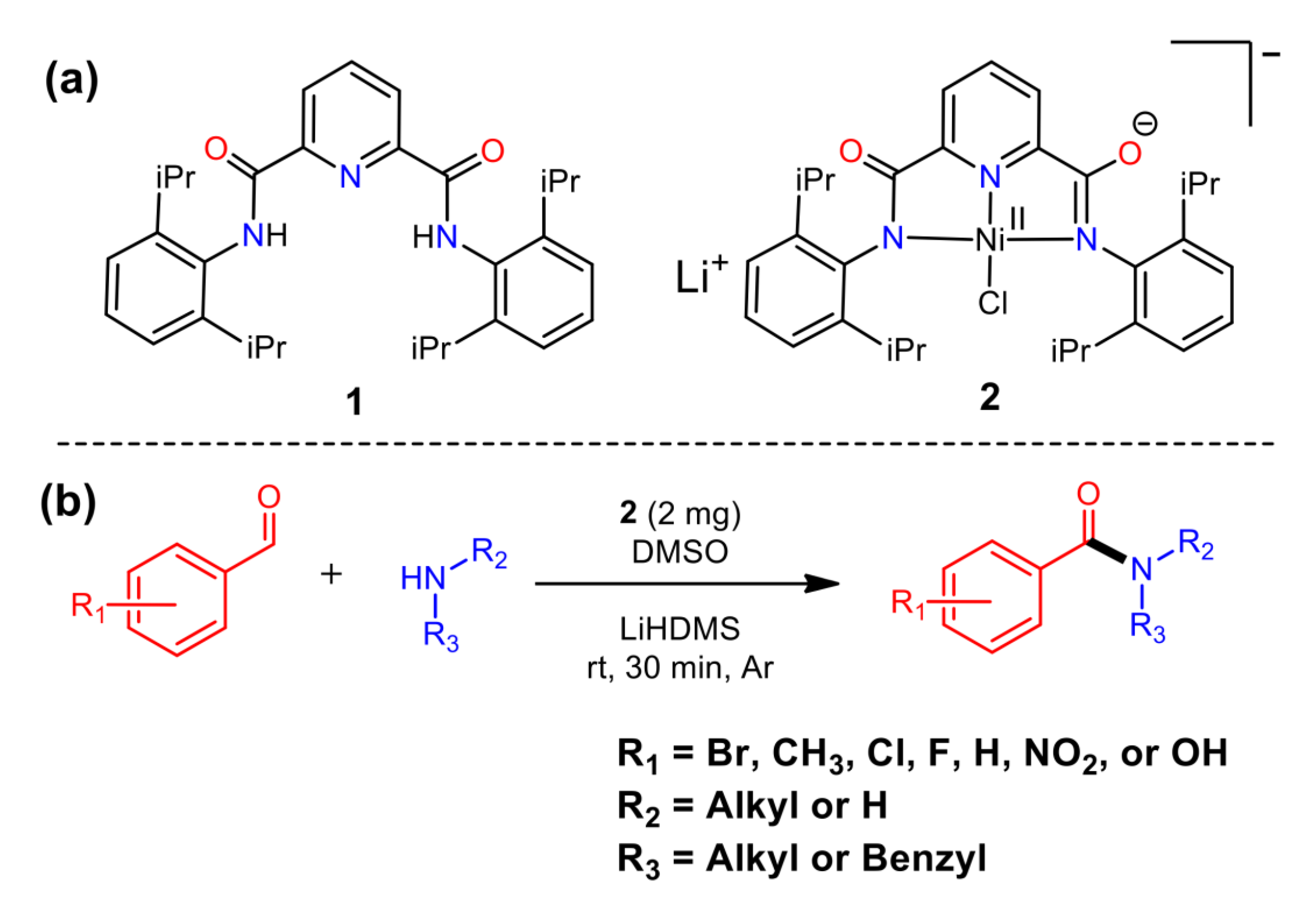
2. Results
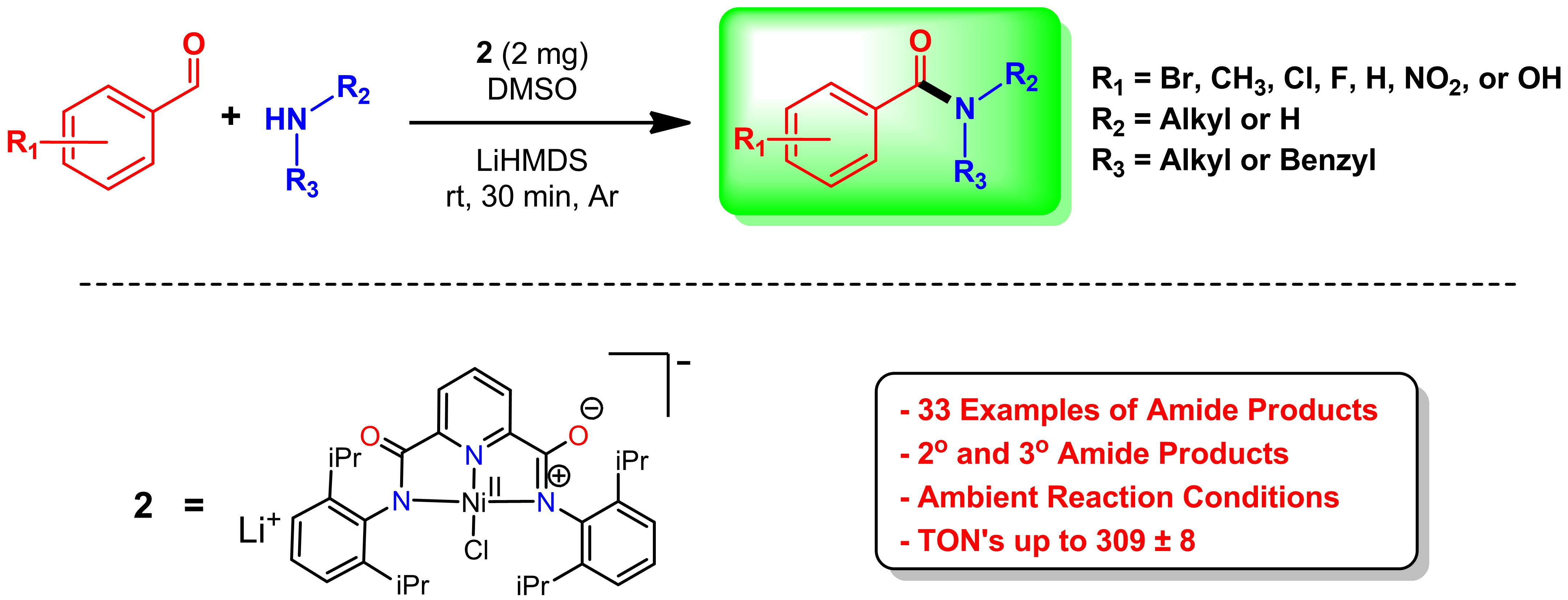
2.1. Optimization of Reaction Atmosphere, Base, and Solvent
2.2. Optimization of Temperature and Time
2.3. Optimization of Starting Reagent Ratios
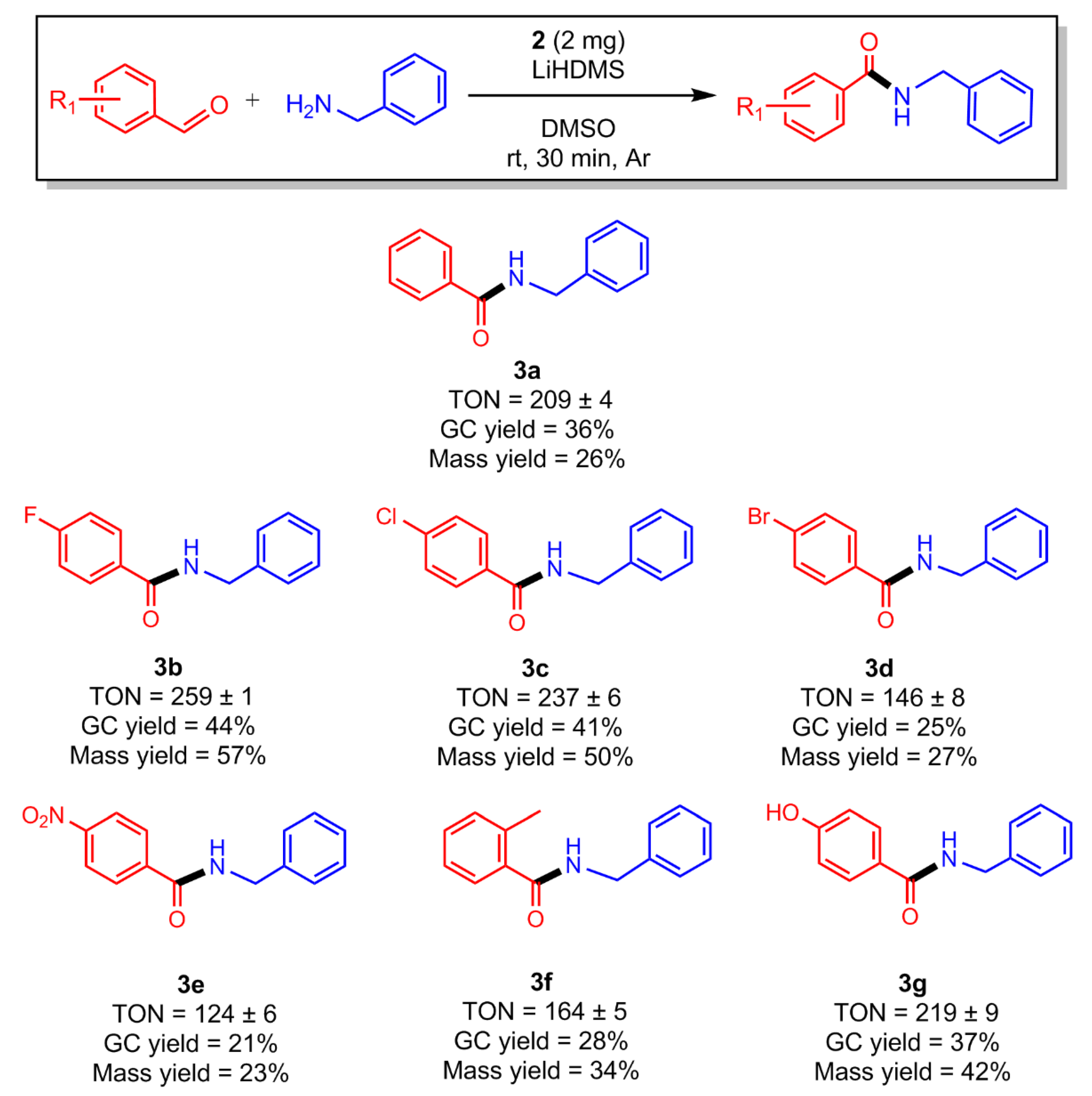
2.4. Amidations in the Scope of Coupling Substituted Aromatic Aldehydes with Benzylamine
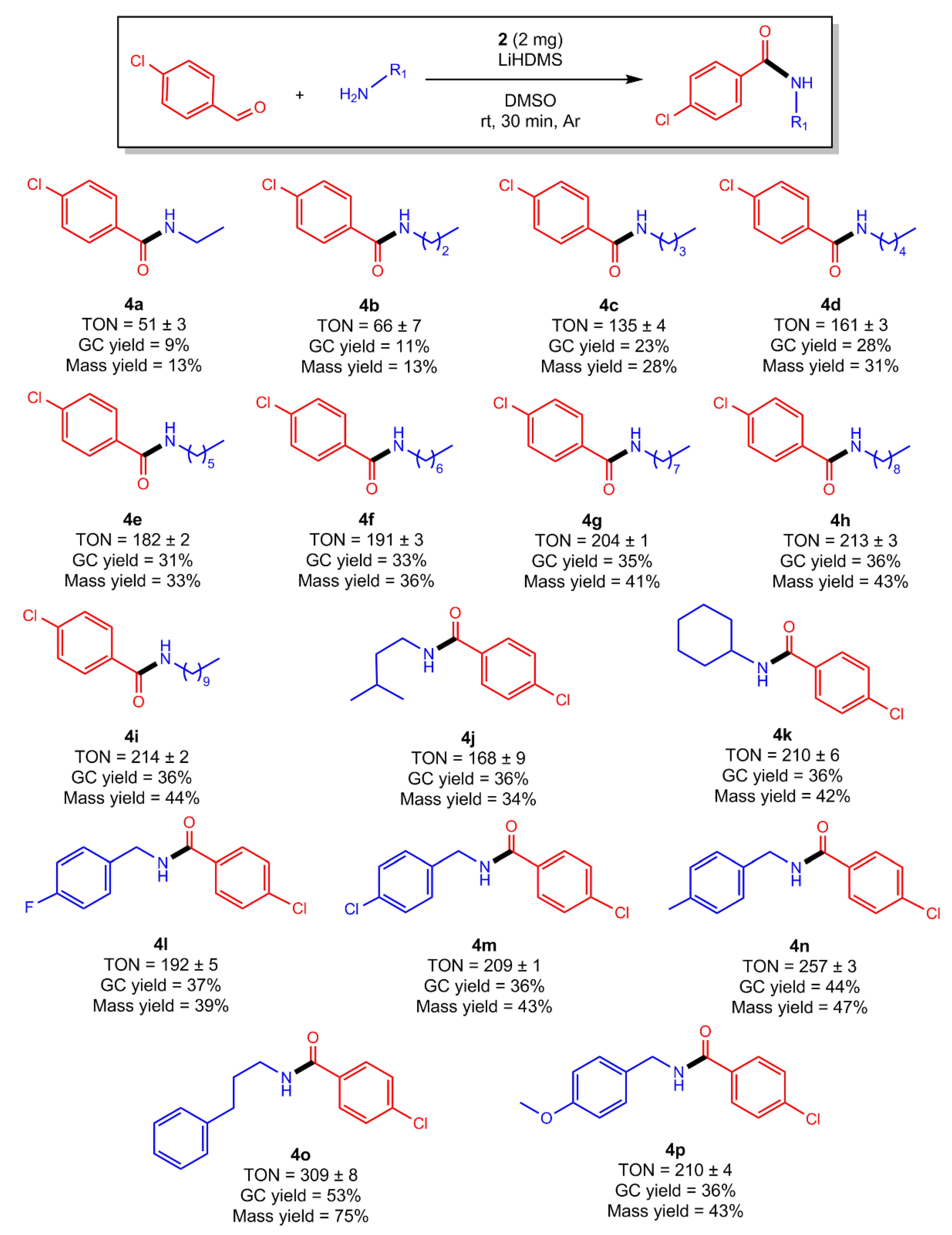
2.5. Amidations in the Scope of Coupling 4-Chlorobenzaldehyde with Various Primary (1°) Amine Partners
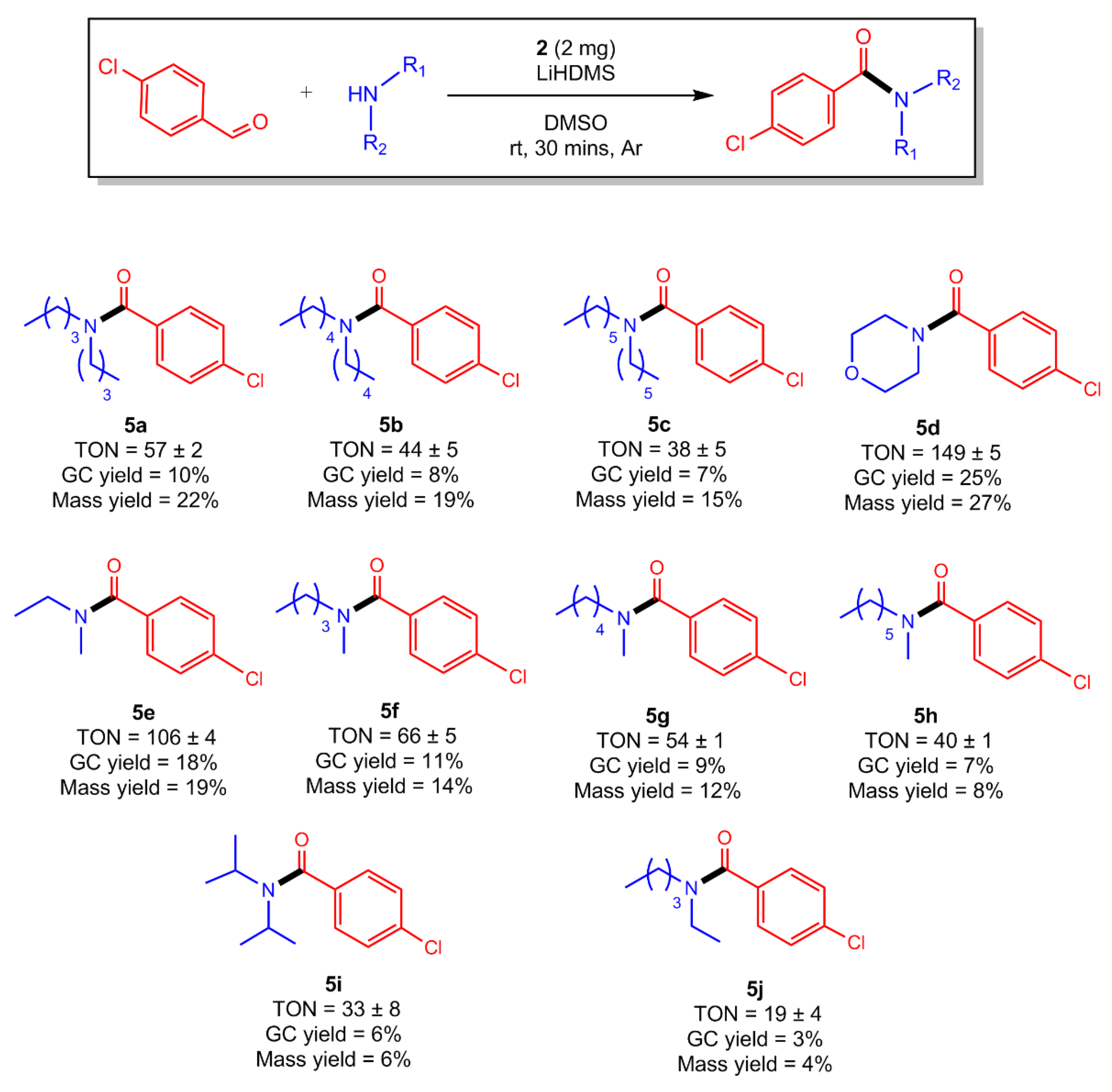
2.6. Amidations in the Scope of Coupling 4-Chlorobenzaldhyde with Various Secondary (2°) Amines
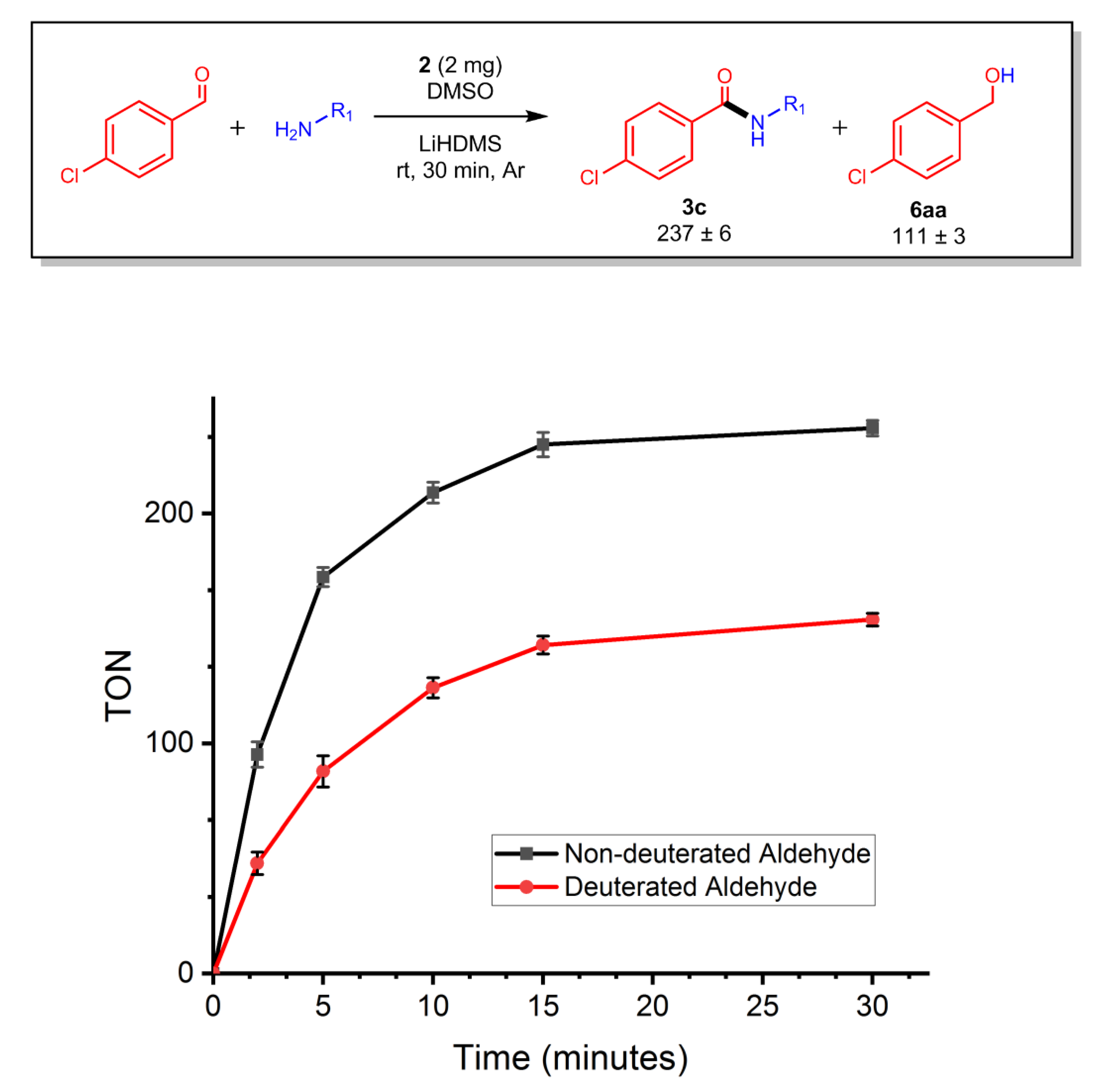
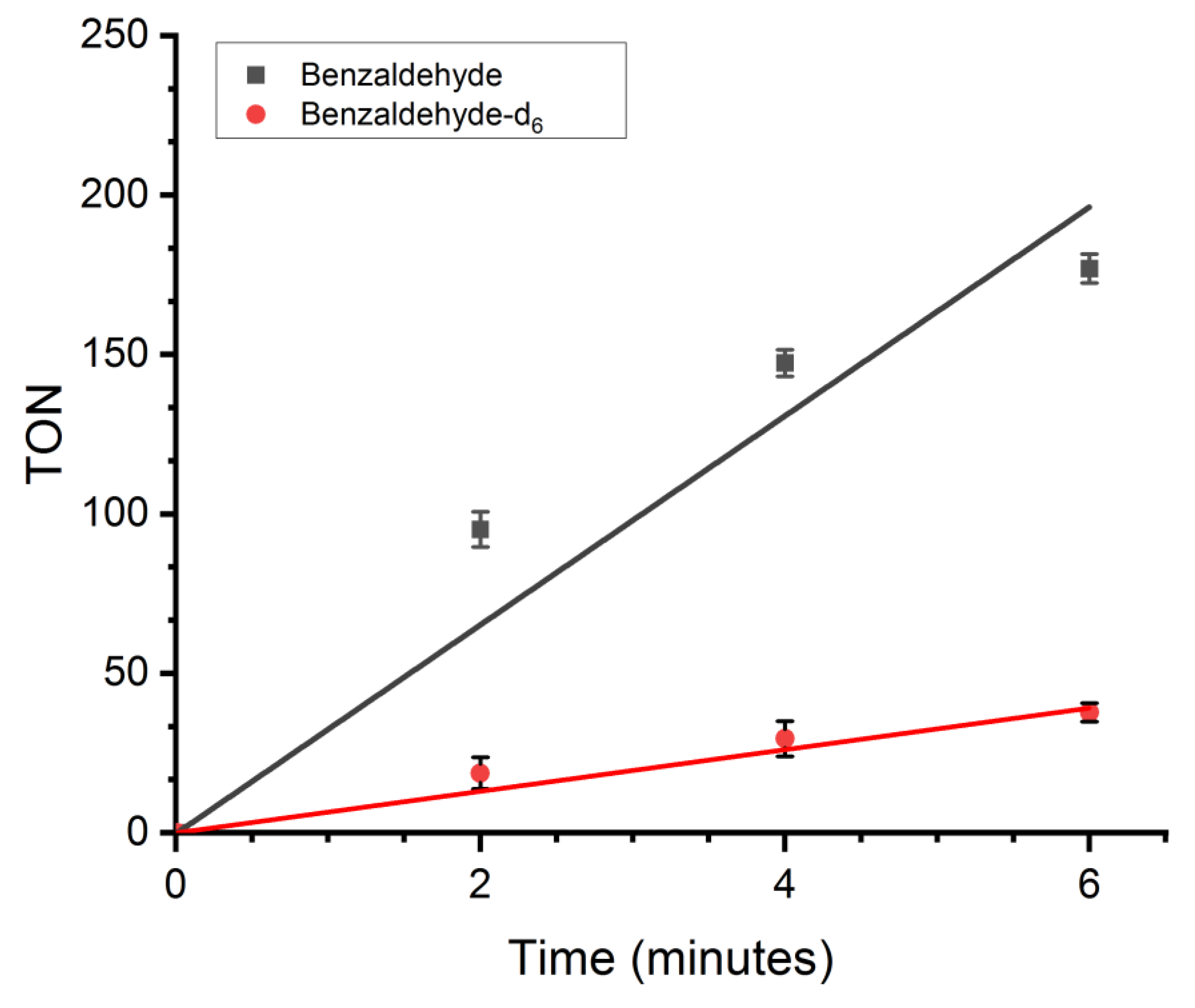
2.7. Evaluation of Side Product Formation and Specificity
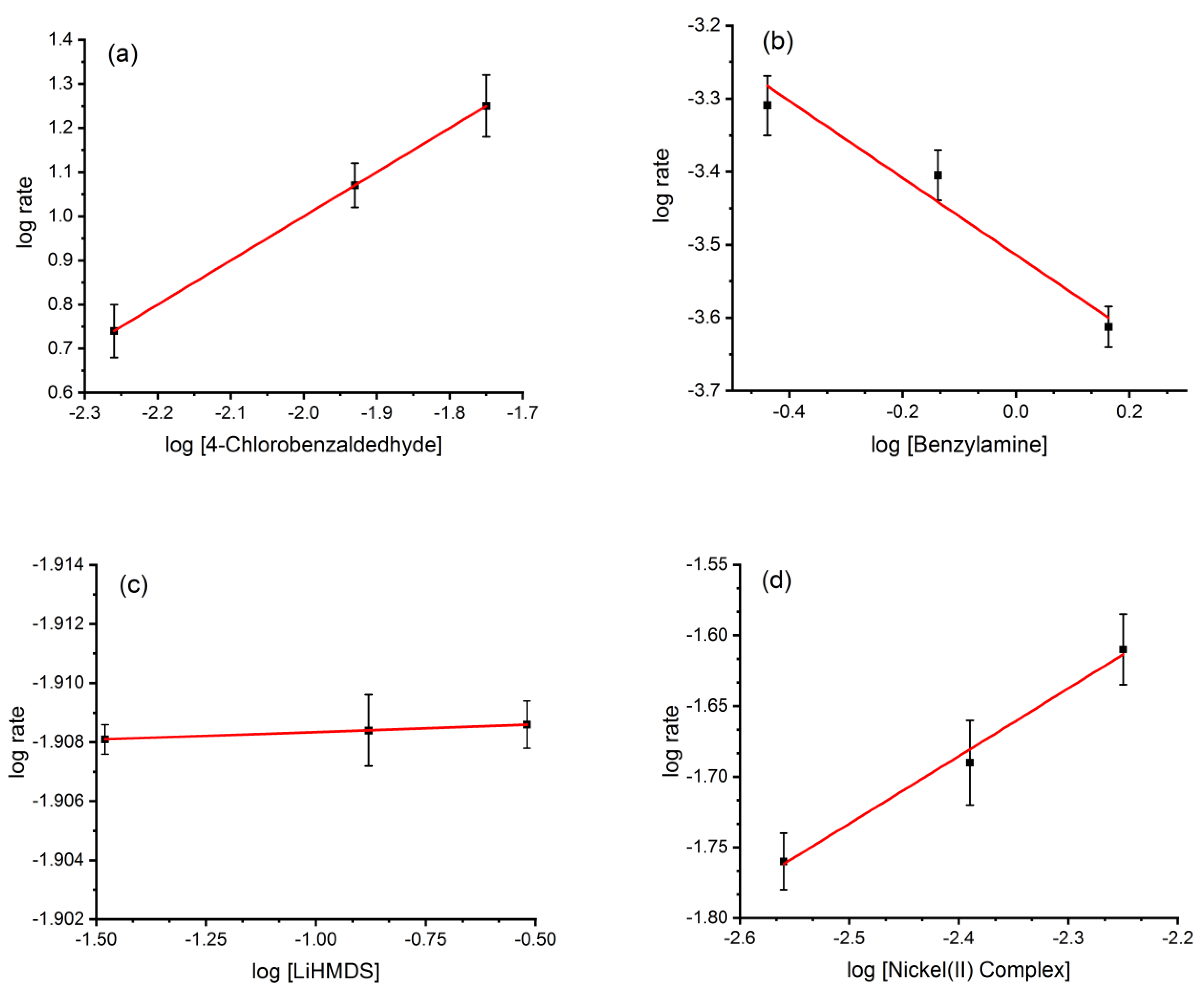
2.8. Reaction Kinetics
2.9. Determination of Thermodynamic Parameters at the Transition State
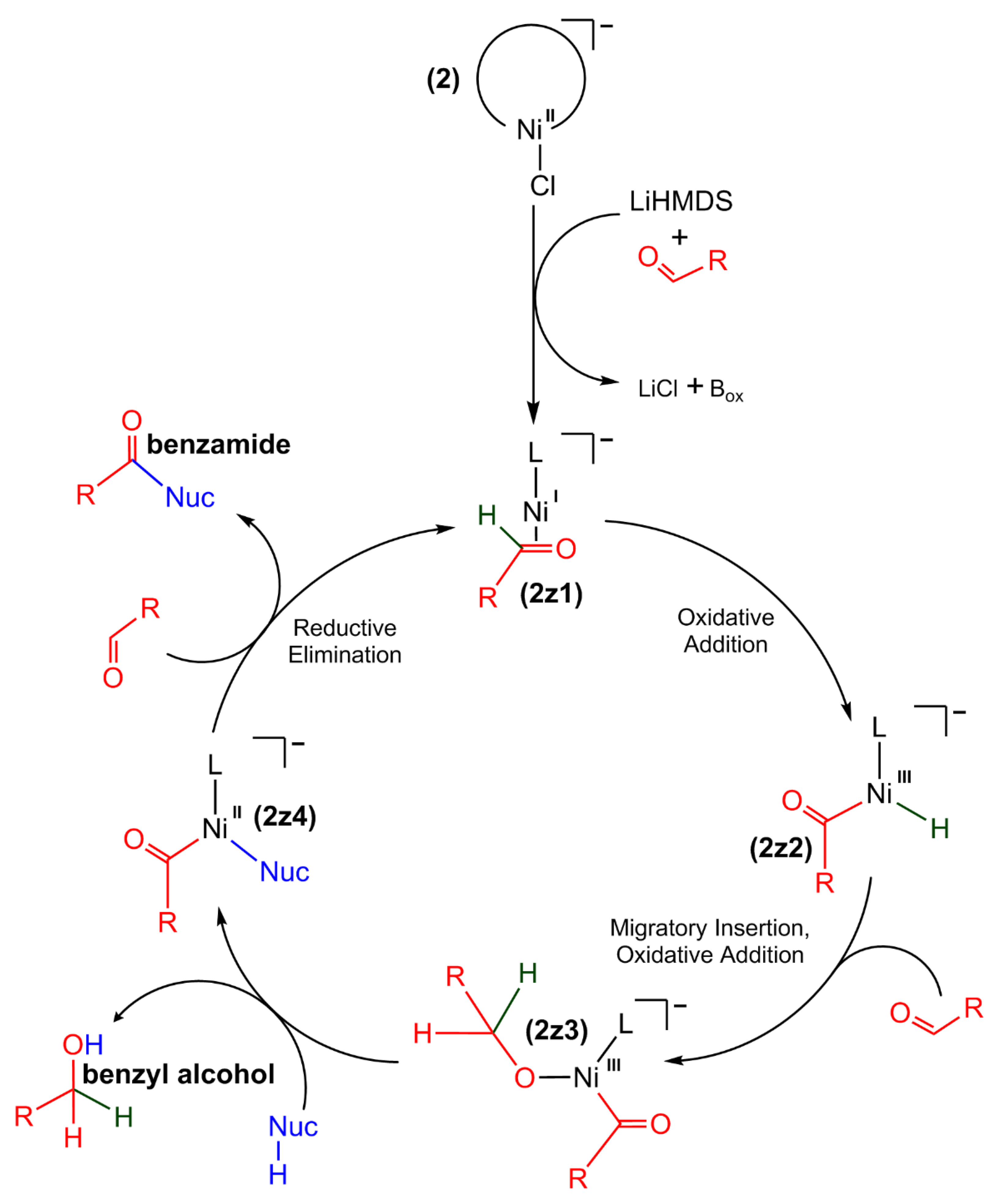
2.10. Proposed Reaction Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis Pincer Ligand (1)
4.3. Synthesis of Ni (II) Complex (2)
4.4. General Synthesis of Benzamides
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar]
- Góbi, S.; Magyarfalvi, G.; Tarczay, G. VCD Robustness of the Amide-I and Amide-II Vibrational Modes of Small Peptide Models. Chirality 2015, 27, 625–634. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Wieland, T.; Bodanszky, M. The World of Peptides; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Arregui, M.C.; Sánchez, D.; Althaus, R.; Scotta, R.R.; Bertolaccini, I. Assessing the risk of pesticide environmental impact in several Argentinian cropping systems with a fuzzy expert indicator. Pest Manag. Sci. 2010, 66, 736–740. [Google Scholar] [CrossRef]
- Hartwig, J.F. Carbon–heteroatom bond formation catalysed by organometallic complexes. Nature 2008, 455, 314–322. [Google Scholar] [CrossRef]
- Liu, C.; Szostak, M. Decarbonylative cross-coupling of amides. Org. Biomol. Chem. 2018, 16, 7998–8010. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Sheppard, T.D. Recent Developments in Amide Synthesis: Direct Amidation of Carboxylic Acids and Transamidation Reactions. Eur. J. Org. Chem. 2013, 2013, 7453–7465. [Google Scholar] [CrossRef]
- Capparelli, E.V.; Holland, D.; Okamoto, C.; Gragg, B.; Durelle, J.; Marquie-Beck, J.; Van den Brande, G.; Ellis, R.; Letendre, S. Lopinavir concentrations in cerebrospinal fluid exceed the 50% inhibitory concentration for HIV. AIDS 2005, 19, 949–952. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef]
- Lu, B.; Xiao, W.-J.; Chen, J.-R. Recent Advances in Visible-Light-Mediated Amide Synthesis. Molecules 2022, 27, 517. [Google Scholar] [CrossRef]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef]
- Bates, R. Organic Synthesis Using Transition Metals; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Y.; Zhu, L.; Liang, Y.; Man, X.; Bi, S. Mechanism of Amide Bond Formation from Carboxylic Acids and Amines Promoted by 9-Silafluorenyl Dichloride Derivatives. J. Org. Chem. 2017, 82, 9087–9096. [Google Scholar] [CrossRef]
- Trost, B.M. Atom Economy—A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. In Angewandte Chemie International Edition in English; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1995; Volume 34, pp. 259–281. [Google Scholar]
- Brennführer, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Compounds. Angew. Chem. Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Ohara, S.; Yamamoto, H. 3,4,5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst. J. Org. Chem. 1996, 61, 4196–4197. [Google Scholar] [CrossRef]
- Ali, A.; Siddiki, S.M.A.H.; Kon, K.; Shimizu, K.-I. A Heterogeneous Niobium(V) Oxide Catalyst for the Direct Amidation of Esters. ChemCatChem 2015, 7, 2705–2710. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Direct Amide Coupling of Non-activated Carboxylic Acids and Amines Catalysed by Zirconium(IV) Chloride. Chem.-A Eur. J. 2012, 18, 3822–3826. [Google Scholar] [CrossRef]
- Lundberg, H.; Adolfsson, H. Hafnium-Catalyzed Direct Amide Formation at Room Temperature. ACS Catal. 2015, 5, 3271–3277. [Google Scholar] [CrossRef]
- Shu, W.; Liu, M.S. Catalytic, metal-free amide synthesis from aldehydes and imines enabled by a dual-catalyzed umpolung strategy under redox-neutral conditions. ACS Catal. 2020, 10, 12960–12966. [Google Scholar]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Rutjes, F.P.J.T.; Mecinović, J. Triphenylphosphine-catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 2014, 50, 5763–5766. [Google Scholar] [CrossRef]
- Hamstra, D.F.J.; Lenstra, D.C.; Koenders, T.J.; Rutjes, F.P.J.T.; Mecinović, J. Poly(methylhydrosiloxane) as a green reducing agent in organophosphorus-catalysed amide bond formation. Org. Biomol. Chem. 2017, 15, 6426–6432. [Google Scholar] [CrossRef]
- Akondi, S.M.; Gangireddy, P.; Pickel, T.C.; Liebeskind, L.S. Aerobic, Diselenide-Catalyzed Redox Dehydration: Amides and Peptides. Org. Lett. 2018, 20, 538–541. [Google Scholar] [CrossRef]
- Handoko, N.; Panigrahi, R.; Arora, P.S. Two-Component Redox Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2022, 144, 3637–3643. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Lyons, D.J.M. A novel aromatic carbocation-based coupling reagent for esterification and amidation reactions. Chem. Commun. 2015, 51, 3131–3134. [Google Scholar] [CrossRef]
- Nordstrøm, L.U.; Vogt, H.; Madsen, R. Amide Synthesis from Alcohols and Amines by the Extrusion of Dihydrogen. J. Am. Chem. Soc. 2008, 130, 17672–17673. [Google Scholar] [CrossRef]
- Tillack, A.; Rudloff, I.; Beller, M. Catalytic Amination of Aldehydes to Amides. Eur. J. Org. Chem. 2001, 3, 523–528. [Google Scholar] [CrossRef]
- Gunanathan, C.; Ben-David, Y.; Milstein, D. Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2. Science 2007, 317, 790–792. [Google Scholar] [CrossRef]
- Capeness, M.; Edmundson, M.; Horsfall, L. Nickel and platinum group metal nanoparticle production by Desulfovibrio alaskensis G20. New Biotechnol. 2015, 32, 727–731. [Google Scholar] [CrossRef]
- Chirik, P.; Morris, R. Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res. 2015, 48, 2495. [Google Scholar] [CrossRef]
- Soulé, J.F.; Miyamura, H.; Kobayashi, S. Powerful amide synthesis from alcohols and amines under aerobic conditions catalyzed by gold or gold/iron, -nickel or-cobalt nanoparticles. J. Am. Chem. Soc. 2011, 133, 18550–18553. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, A.M.; Dong, V.M. Nickel-Catalyzed Dehydrogenative Cross-Coupling: Direct Transformation of Aldehydes into Esters and Amides. Angew. Chem. Int. Ed. 2015, 54, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Alandini, N.; Buzzetti, L.; Favi, G.; Schulte, T.; Candish, L.; Collins, K.D.; Melchiorre, P. Amide Synthesis by Nickel/Photoredox-Catalyzed Direct Carbamoylation of (Hetero)Aryl Bromides. Angew. Chem. 2020, 132, 5286–5291. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, F.; Wang, X.; Qiang, Q.; Li, Y.; Zhang, Y.; Rong, Z.-Q. Redox-neutral dehydrogenative cross-coupling of alcohols and amines enabled by nickel catalysis. Org. Chem. Front. 2022, 9, 1703–1710. [Google Scholar] [CrossRef]
- Sang, J.W.; Li, Q.; Zhang, C.; Zhang, Y.; Wang, J.; Zhang, W.D. Nickel/Photoredox-Catalyzed Direct Amidation of Aldehydes with Nitroarenes via Fully Catalytic Process. Org. Lett. 2023, 25, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Goel, B.; Vyas, V.; Tripathi, N.; Singh, A.K.; Menezes, P.W.; Indra, A.; Jain, S.K. Amidation of Aldehydes with Amines under Mild Conditions Using Metal-Organic Framework Derived NiO@Ni Mott-Schottky Catalyst. ChemCatChem 2020, 12, 5743–5749. [Google Scholar] [CrossRef]
- Mennen, S.M.; Alhambra, C.; Allen, C.L.; Barberis, M.; Berritt, S.; Brandt, T.A.; Campbell, A.D.; Castañón, J.; Cherney, A.H.; Chritensen, M.; et al. The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org. Process Res. Dev. 2019, 23, 1213–1242. [Google Scholar] [CrossRef]
- Gartia, Y.; Ramidi, P.; Jones, D.E.; Pulla, S.; Ghosh, A. Nickel Complex Catalyzed Efficient Activation of sp3 and sp2 C–H Bonds for Alkylation and Arylation of Oxygen Containing Heterocyclic Molecules. Catal. Lett. 2014, 144, 507–515. [Google Scholar] [CrossRef]
- Gartia, Y.; Pulla, S.; Ramidi, P.; Farris, C.C.; Nima, Z.; Jones, D.E.; Biris, A.S.; Ghosh, A. A Novel Iron Complex for Cross-Coupling Reactions of Multiple C–Cl Bonds in Polychlorinated Solvents with Grignard Reagents. Catal. Lett. 2012, 142, 1397–1404. [Google Scholar] [CrossRef]
- Albkuri, Y.M.; RanguMagar, A.B.; Brandt, A.; Wayland, H.A.; Chhetri, B.P.; Parnell, C.M.; Szwedo, P.; Parameswaran-Thankam, A.; Ghosh, A. C–N Cross-coupling Reactions of Amines with Aryl Halides Using Amide-Based Pincer Nickel(II) Catalyst. Catal. Lett. 2020, 150, 1669–1678. [Google Scholar] [CrossRef]
- Brandt, A.; RanguMagar, A.B.; Szwedo, P.; Wayland, H.A.; Parnell, C.M.; Munshi, P.; Ghosh, A. Highly economical and direct amination of sp3 carbon using low-cost nickel pincer catalyst. RSC Adv. 2021, 11, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Fraser, R.R.; Mansour, T.S. Acidity measurements with lithiated amines: Steric reduction and electronic enhancement of acidity. J. Org. Chem. 1984, 49, 3442–3443. [Google Scholar] [CrossRef]
- Fraser, R.R.; Mansour, T.S.; Savard, S. Acidity measurements on pyridines in tetrahydrofuran using lithiated silylamines. J. Org. Chem. 1985, 50, 3232–3234. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Algrim, D.; Vanier, N.R. Acidities of anilines and toluenes. J. Org. Chem. 1977, 42, 1817–1819. [Google Scholar] [CrossRef]
- Munshi, P.; Main, A.D.; Linehan, J.C.; Tai, C.-C.; Jessop, P.G. Hydrogenation of Carbon Dioxide Catalyzed by Ruthenium Trimethylphosphine Complexes: The Accelerating Effect of Certain Alcohols and Amines. J. Am. Chem. Soc. 2002, 124, 7963–7971. [Google Scholar] [CrossRef]
- Zhang, T.; Zhong, K.; Lin, Z.K.; Niu, L.; Li, Z.Q.; Bai, R.; Engle, K.M.; Lan, Y. Revised Mechanism of C(sp3)-C(sp3) Reductive Elimination from Ni(II) with the Assistance of a Z-Type Metalloligand. J. Am. Chem. Soc. 2023, 145, 2207–2218. [Google Scholar] [CrossRef]
- Reyes, A.; Scott, R.M. Specific effects of dimethyl sulfoxide on the relative basicities of aliphatic amines. J. Phys. Chem. 1980, 84, 3600–3603. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. “Turning Over” Definitions in Catalytic Cycles. Acc. Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Lente, G. Comment on “Turning Over” Definitions in Catalytic Cycles. Acc. Catal. 2013, 3, 381–382. [Google Scholar] [CrossRef]
- Wheeler, S.E. Understanding Substituent Effects in Noncovalent Interactions Involving Aromatic Rings. Accounts Chem. Res. 2013, 46, 1029–1038. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. THEOCHEM 2008, 895, 86–91. [Google Scholar] [CrossRef]
- Rosenthal, J.; Schuster, D.I. The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena. J. Chem. Educ. 2003, 80, 679–690. [Google Scholar] [CrossRef]
- Matulis, D.; Bloomfield, V.A. Thermodynamics of the hydrophobic effect. I. Coupling of aggregation and pKa shifts in solutions of aliphatic amines. Biophys. Chem. 2001, 93, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Brotzel, F.; Ying, C.C.; Mayr, H. Nucleophilicities of primary and secondary amines in water. J. Org. Chem. 2007, 72, 3679–3688. [Google Scholar] [CrossRef]
- Lanigan, R.M.; Starkov, P.; Sheppard, T.D. Direct synthesis of amides from carboxylic acids and amines using B(OCH2CF3)3. J. Org. Chem. 2013, 78, 4512–4523. [Google Scholar] [CrossRef]
- Schuhmacher, A.; Shiro, T.; Ryan, S.J.; Bode, J.W. Synthesis of secondary and tertiary amides without coupling agents from amines and potassium acyltrifluoroborates (KATs). Chem. Sci. 2020, 11, 7609–7614. [Google Scholar] [CrossRef]
- Henderson, W.A.; Schultz, C.J. The Nucleophilicity of Amines. J. Org. Chem. 1962, 27, 4643–4646. [Google Scholar] [CrossRef]
- Nystrom, R.F.; Brown, W.G. Reduction of Organic Compounds by Lithium Aluminum Hydride. I. Aldehydes, Ketones, Esters, Acid Chlorides and Acid Anhydrides. J. Am. Chem. Soc. 1947, 69, 1197–1199. [Google Scholar] [CrossRef]
- Brown, H.C.; Wheeler, O.H.; Ichikawa, K. Chemical effects of steric strains—XIII: Kinetics of the reaction of sodium borohydride with carbonyl groups—A convenient tool for investigating the reactivities of aldehydes and ketones. Tetrahedron 1957, 1, 214–220. [Google Scholar] [CrossRef]
- Ge, S.; Green, R.A.; Hartwig, J.F. Controlling First-Row Catalysts: Amination of Aryl and Heteroaryl Chlorides and Bromides with Primary Aliphatic Amines Catalyzed by a BINAP-Ligated Single-Component Ni0 Complex. J. Am. Chem. Soc. 2014, 136, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Chang, R. Physical Chemistry for the Biosciences; University Science Books: Herndon, VA, USA, 2005. [Google Scholar]
- Nicolas, E.; Ohleier, A.F.; D’Accriscio; Pécharman, A.F.; Demange, M.; Ribagnac, J.; Ballester, J.; Gosmini, C.; Mézailles, N. ‘(Diphosphine)Nickel’-Catalyzed Negishi Cross-Coupling: An Experimental and Theoretical Study. Chem.-A Eur. J. 2015, 21, 7690–7694. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.; Kampf, J.W.; Ariafard, A.; Canty, A.J.; Sanford, M.S. Oxidatively Induced C–H Activation at High Valent Nickel. J. Am. Chem. Soc. 2017, 139, 6058–6061. [Google Scholar] [CrossRef] [PubMed]
- Weires, N.A.; Caspi, D.D.; Garg, N.K. Kinetic Modeling of the Nickel-Catalyzed Esterification of Amides. ACS Catal. 2017, 7, 4381–4385. [Google Scholar] [CrossRef]
- Dander, J.E.; Garg, N.K. Breaking Amides using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef]
- Beromi, M.M.; Nova, A.; Balcells, D.; Brasacchio, A.M.; Brudvig, G.W.; Guard, L.M.; Hazari, N.; Vinyard, D.J. Mechanistic Study of an Improved Ni Precatalyst for Suzuki–Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc. 2017, 139, 922–936. [Google Scholar] [CrossRef]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of Biaryls through Nickel-Catalyzed Suzuki-Miyaura Coupling of Amides by Carbon-Nitrogen Bond Cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef]
- Wasilke, J.C.; Wu, G.; Bu, X.; Kehr, G.; Erker, G. Ruthenium carbene complexes featuring a tridentate pincer-type ligand. Organometallics 2005, 24, 4289–4297. [Google Scholar] [CrossRef]
- Gartia, Y.; Ramidi, P.; Cheerla, S.; Felton, C.M.; Jones, D.E.; Das, B.C.; Ghosh, A. Activation of sp3 and sp2 CH bonds of oxygen containing heterocyclic molecules for alkylation and arylation reactions catalyzed by an iron complex. J. Mol. Catal. A Chem. 2014, 392, 253–259. [Google Scholar] [CrossRef]
- Zheng, B.; Tang, F.; Luo, J.; Schultz, J.W.; Rath, N.P.; Mirica, L.M. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon–Heteroatom Bond Formation Reactions. J. Am. Chem. Soc. 2014, 136, 6499–6504. [Google Scholar] [CrossRef]
- Kozuch, S.; Lee, S.E.; Shaik, S. Theoretical Analysis of the Catalytic Cycle of a Nickel Cross-Coupling Process: Application of the Energetic Span Model. Organometallics 2009, 28, 1303–1308. [Google Scholar] [CrossRef]
- Liu, H.; Jia, X.; Wang, F.; Dai, Q.; Wang, B.; Bi, J.; Zhang, C.; Zhao, L.; Bai, C.; Hu, Y.; et al. Synthesis of bis(N-arylcarboximidoylchloride)pyridine cobalt(ii) complexes and their catalytic behavior for 1,3-butadiene polymerization. Dalt. Trans. 2013, 42, 13723. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Holm, R.H. Reactions of the terminal Ni(II)-OH group in substitution and electrophilic reactions with carbon dioxide and other substrates: Structural definition of binding modes in an intramolecular Ni(II)Fe(II) bridged site. J. Am. Chem. Soc. 2010, 132, 4693–4701. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, X.; Spasyuk, D.M.; Zargarian, D. New POCOP-type pincer complexes of Nickel(II). J. Organomet. Chem. 2011, 696, 864–870. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Ar Atmosphere | Base | Solvent | TON | GC Yield |
| 1 | a 0 min | LiHMDS | DMSO | 60 ± 1 | 10% |
| 2 | b 0 min | LiHMDS | DMSO | 60 ± 2 | 10% |
| 3 | 5 min | LiHMDS | DMSO | 132 ± 6 | 23% |
| 4 | 30 min | LiHMDS | DMSO | 197 ± 4 | 34% |
| 5 | 30 min | LiHMDS | DMF | 143 ± 2 | 24% |
| 6 | 30 min | LiHMDS | 1,4-dioxane | 158 ± 6 | 27% |
| 7 | 30 min | LiHMDS | THF | 137 ± 1 | 23% |
| 8 | 30 min | KOH | DMSO | 0 ± 0 | 0% |
| 9 | 30 min | Cs2CO3 | DMSO | 11 ± 2 | 2% |
| 10 | 30 min | KOtBu | DMSO | 32 ± 4 | 5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szwedo, P.; Jumper, T.; Sanford, K.; Arnold, T.; Coffman, S.; Hokes, D.; Munshi, P.; Walker, B.; Ghosh, A. Dehydrogenative Conversions of Aldehydes and Amines to Amides Catalyzed by a Nickel(II) Pincer Complex. Catalysts 2023, 13, 1423. https://doi.org/10.3390/catal13111423
Szwedo P, Jumper T, Sanford K, Arnold T, Coffman S, Hokes D, Munshi P, Walker B, Ghosh A. Dehydrogenative Conversions of Aldehydes and Amines to Amides Catalyzed by a Nickel(II) Pincer Complex. Catalysts. 2023; 13(11):1423. https://doi.org/10.3390/catal13111423
Chicago/Turabian StyleSzwedo, Peter, Travis Jumper, Karie Sanford, Taylor Arnold, Sarah Coffman, Davonte Hokes, Pradip Munshi, Brian Walker, and Anindya Ghosh. 2023. "Dehydrogenative Conversions of Aldehydes and Amines to Amides Catalyzed by a Nickel(II) Pincer Complex" Catalysts 13, no. 11: 1423. https://doi.org/10.3390/catal13111423
APA StyleSzwedo, P., Jumper, T., Sanford, K., Arnold, T., Coffman, S., Hokes, D., Munshi, P., Walker, B., & Ghosh, A. (2023). Dehydrogenative Conversions of Aldehydes and Amines to Amides Catalyzed by a Nickel(II) Pincer Complex. Catalysts, 13(11), 1423. https://doi.org/10.3390/catal13111423