



Prominent COF, g-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction

 , ,
, ,  , , ,
, , , 

Abstract
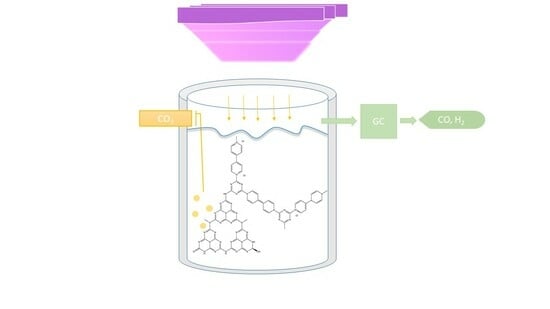
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
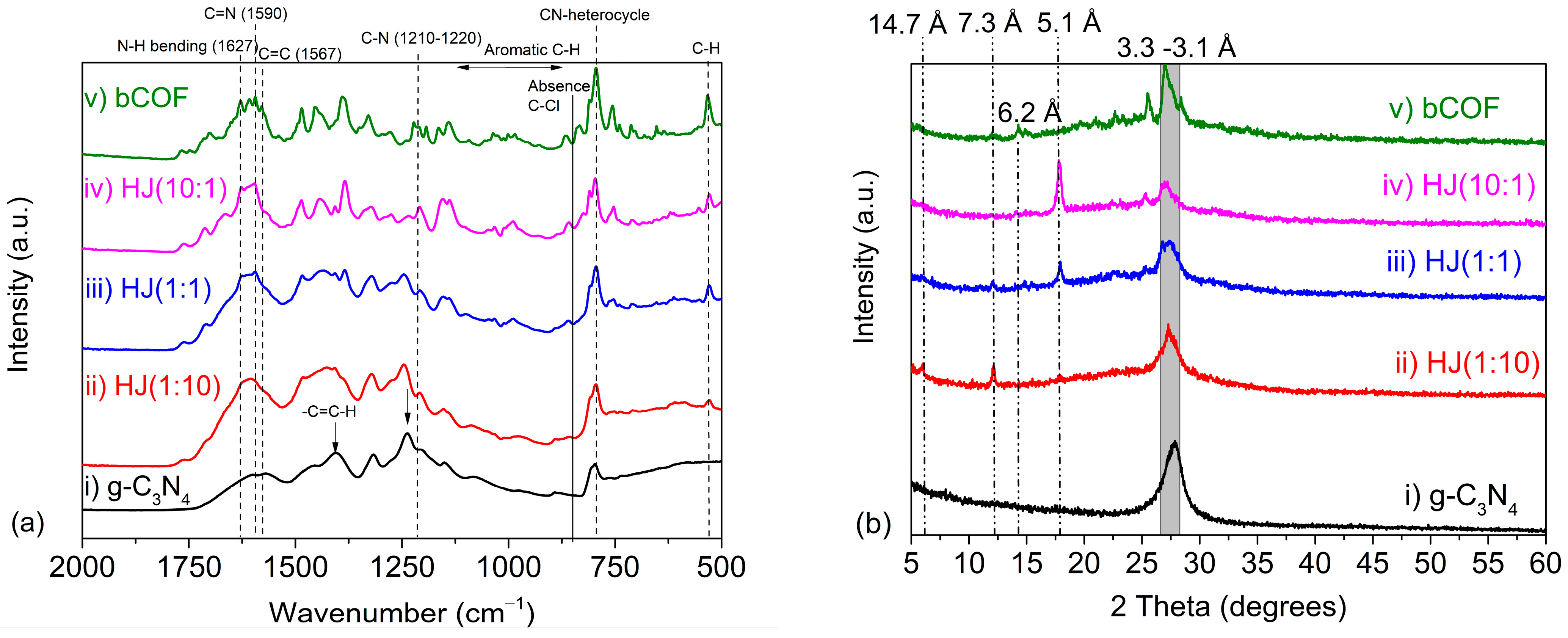
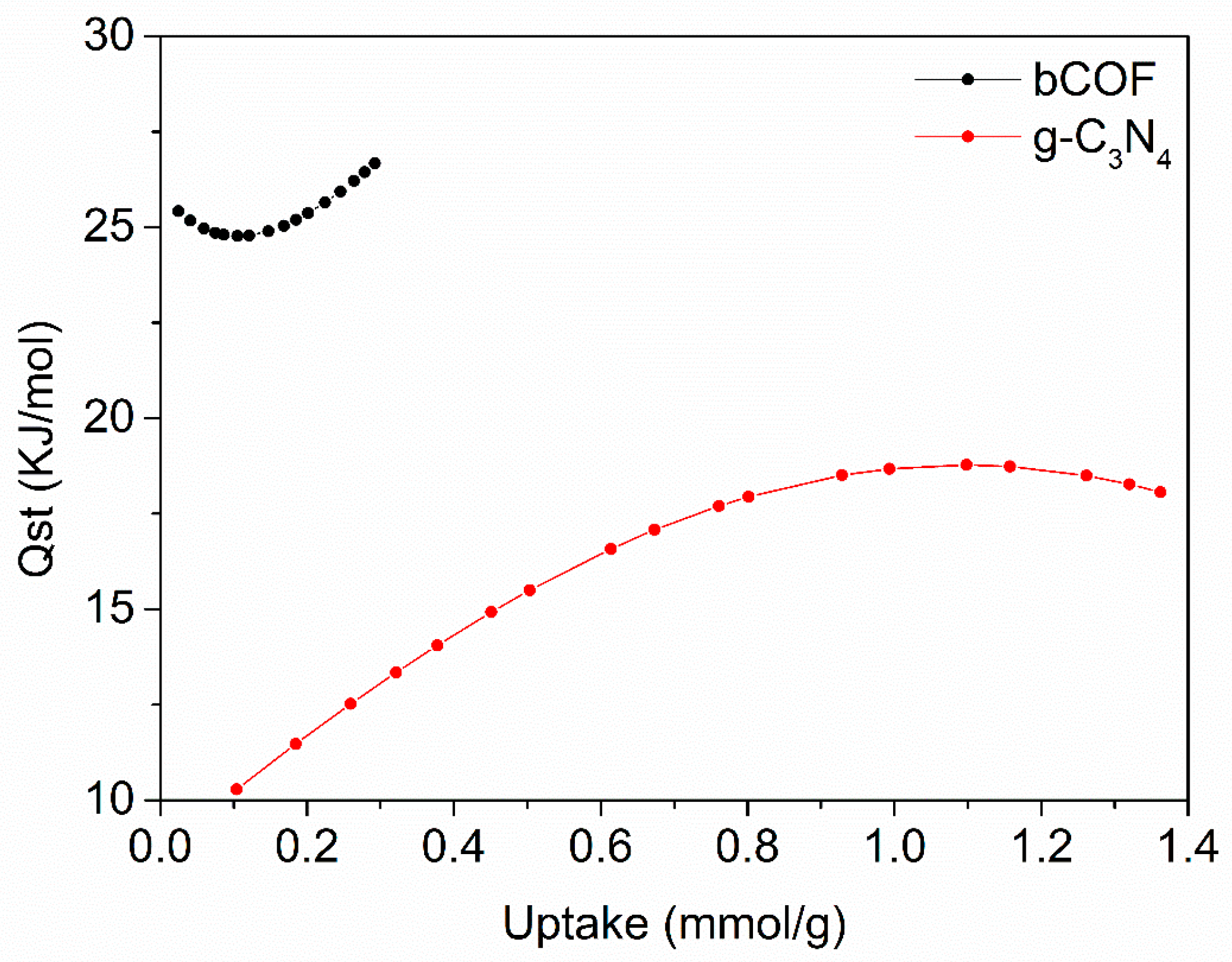
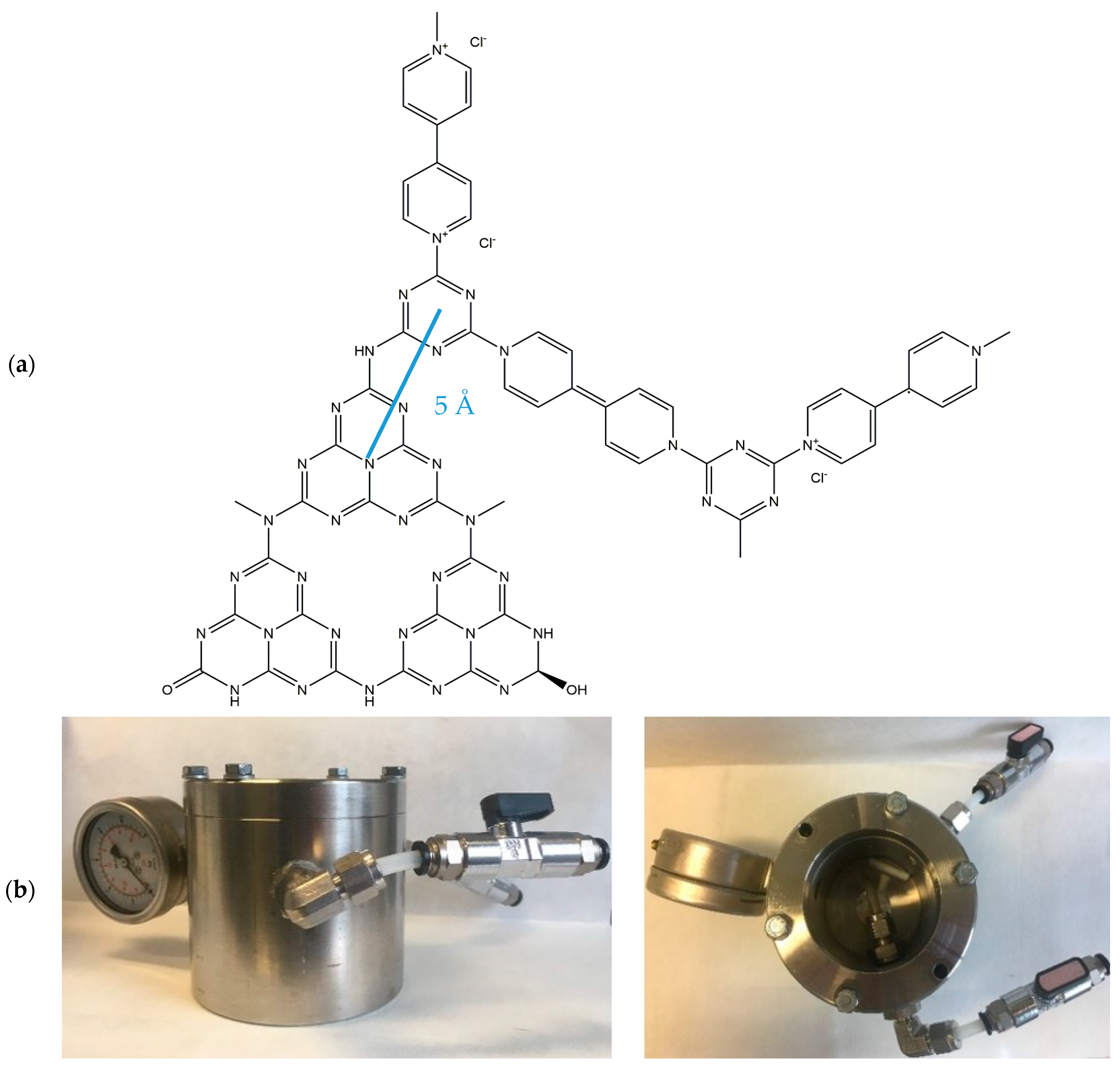
2.1. Chemical and Structural Characterisation
2.2. Morphology before and after CO2 Reduction
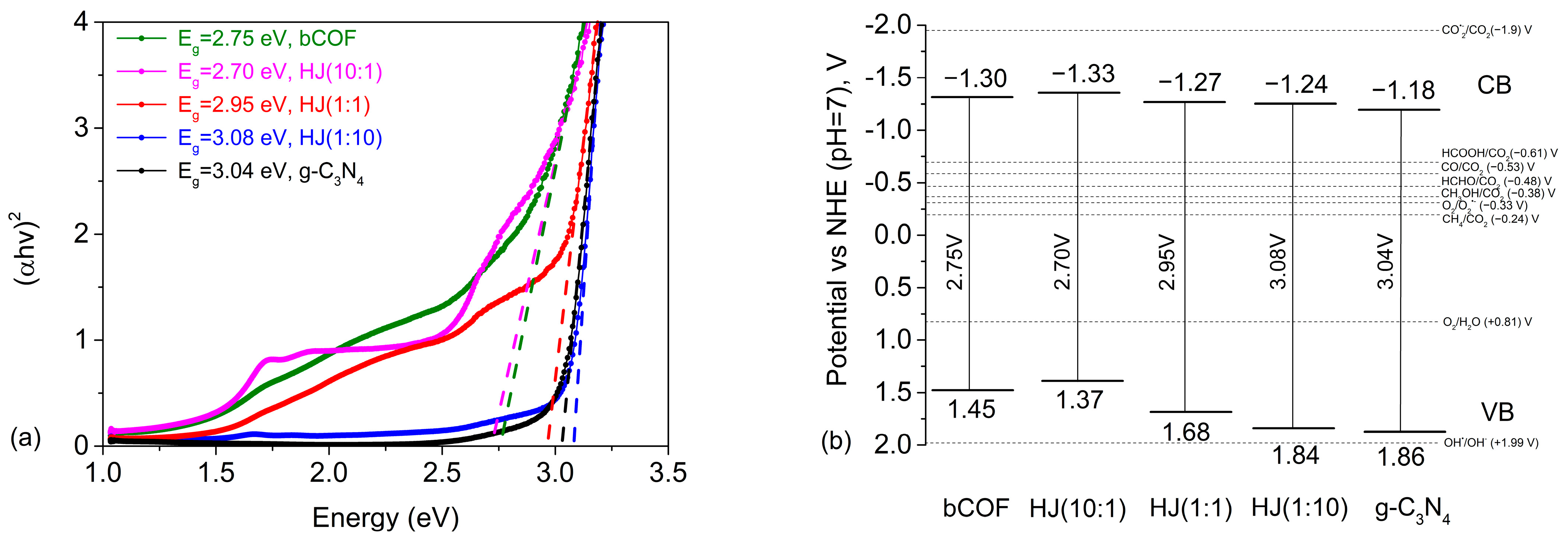
2.3. Photophysical Properties
ΔV = 0.21 and VVB = VCV + Eg/e
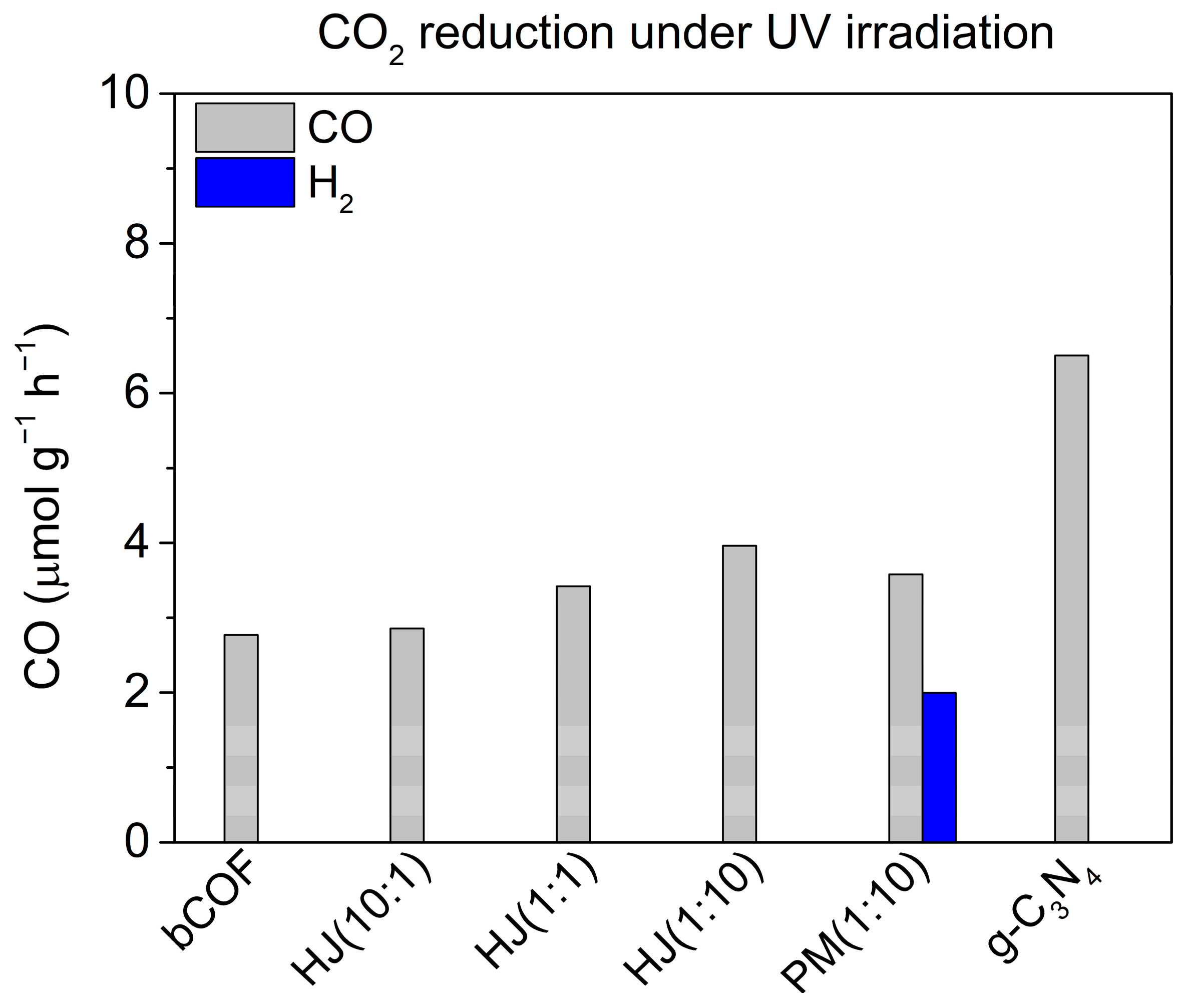
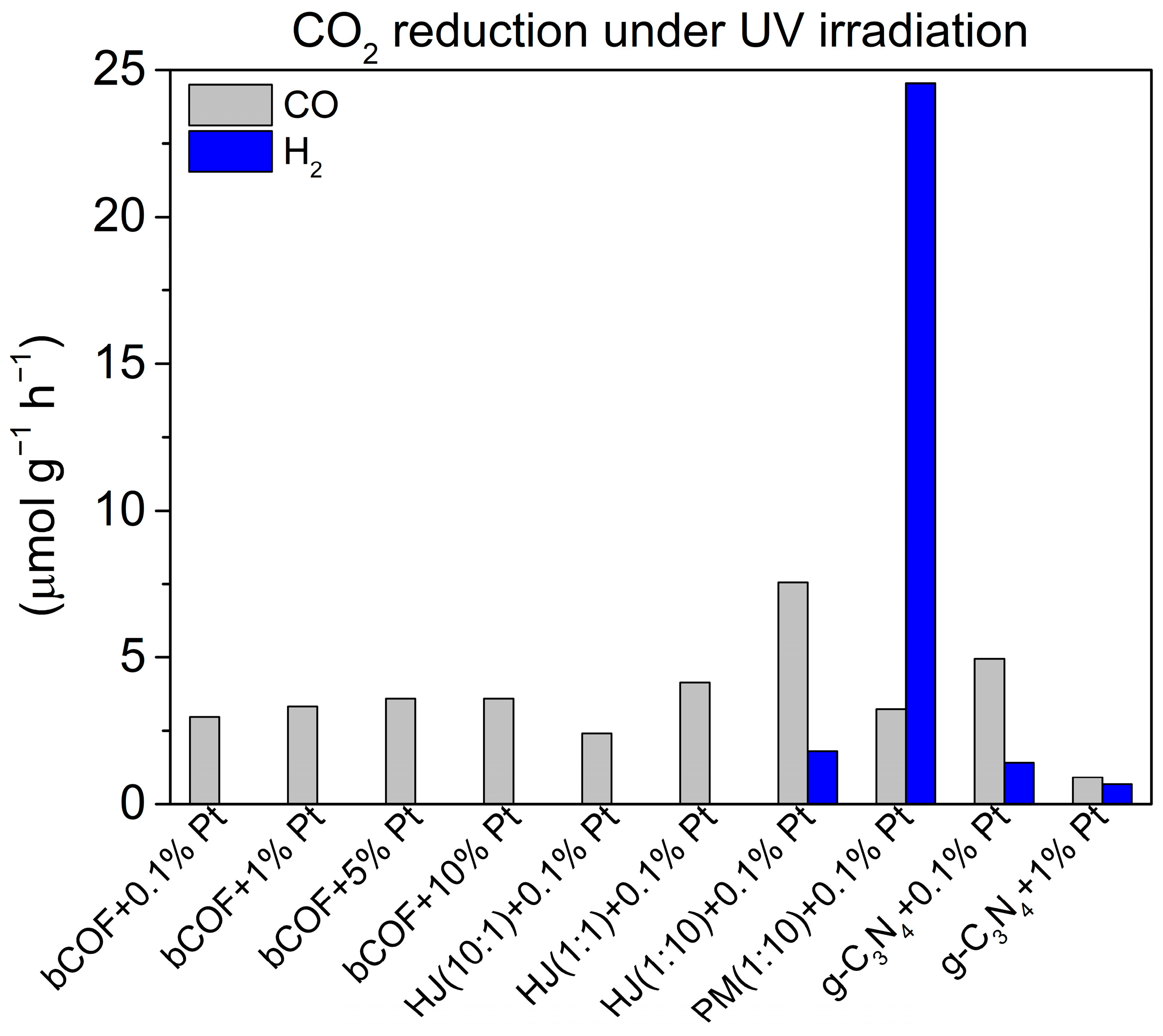
2.4. Photocatalytic Activity
2.4.1. Pristine Materials
2.4.2. Heterojunction Materials
2.4.3. Pristine Materials in Physical Mixture
2.4.4. Effect of Platinum Co-Catalyst
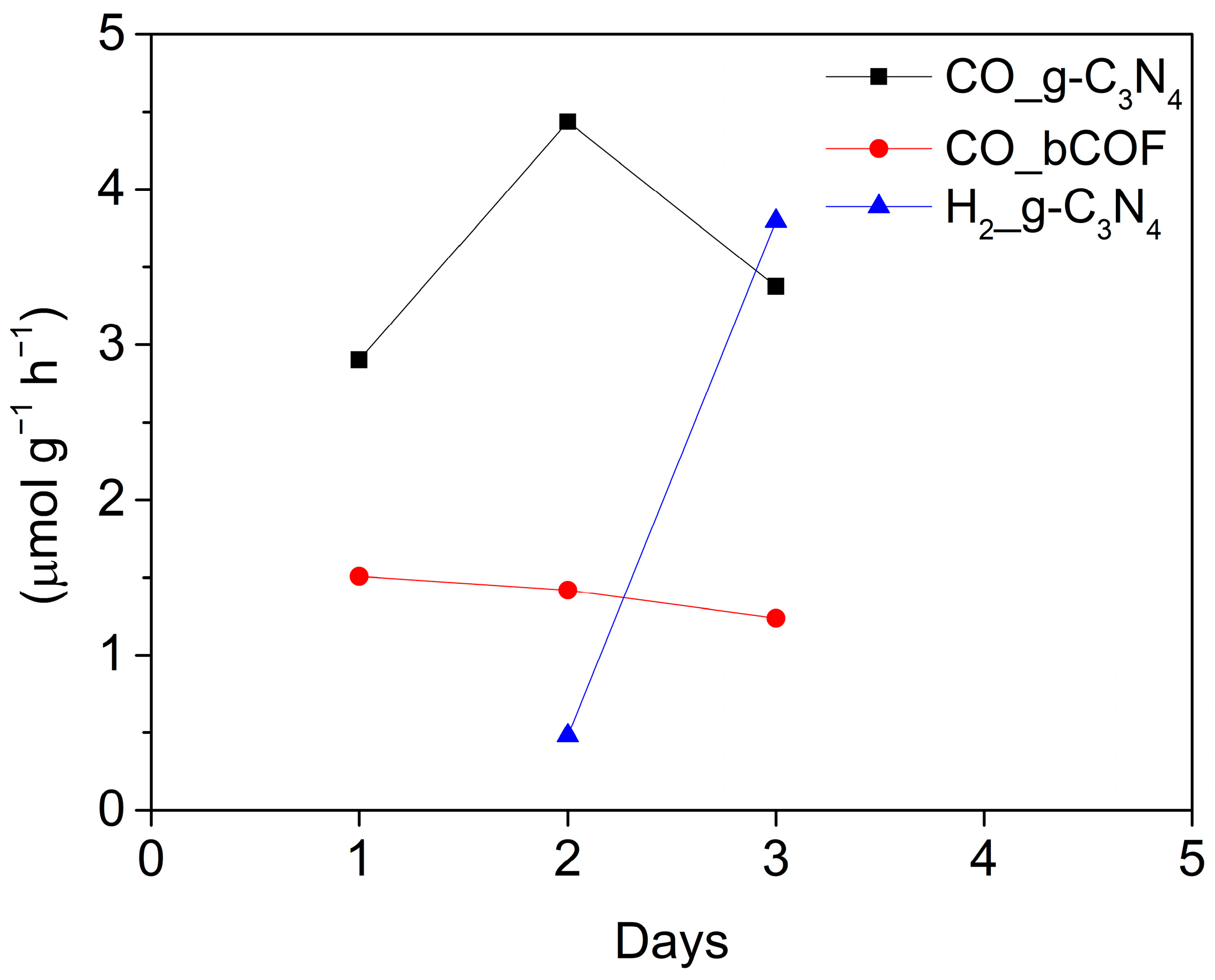
2.4.5. Sustainability and Control Experiments of the Pristine Materials
3. Materials and Methods
3.1. Chemical Reagents
3.2. Photocatalysts
3.3. Synthesis of Heterojunction Materials
3.4. Physical Mixing
3.5. Photocatalytic Reduction
3.6. Characterization Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- He, Y.; Peng, G.; Jiang, Y.; Zhao, M.; Wang, X.; Chen, M.; Lin, S. Environmental Hazard Potential of Nano-Photocatalysts Determined by Nano-Bio Interactions and Exposure Conditions. Small 2020, 16, 1907690. [Google Scholar] [CrossRef]
- Rueda-Marquez, J.J.; Levchuk, I.; Fernández Ibañez, P.; Sillanpää, M. A Critical Review on Application of Photocatalysis for Toxicity Reduction of Real Wastewaters. J. Clean. Prod. 2020, 258, 120694. [Google Scholar] [CrossRef]
- Zhang, J.; Pham, T.H.M.; Ko, Y.; Li, M.; Yang, S.; Koolen, C.D.; Zhong, L.; Luo, W.; Züttel, A. Tandem Effect of Ag@C@Cu Catalysts Enhances Ethanol Selectivity for Electrochemical CO2 Reduction in Flow Reactors. Cell Rep. Phys. Sci. 2022, 3, 100949. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Guo, C.; Wang, Y. Molybdenum Carbide-Based Photocatalysts: Synthesis, Functionalization, and Applications. Langmuir 2022, 38, 12739–12756. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-N.; Soulis, J.; Yang, Y.J.; Biswas, P. Comparison of CO2 Photoreduction Systems: A Review. Aerosol Air Qual. Res. 2014, 14, 533–549. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Gong, J. CO2 Photo-Reduction: Insights into CO2 Activation and Reaction on Surfaces of Photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Zhou, K. Metal–Organic-Framework-Based Catalysts for Photoreduction of CO2. Adv. Mater. 2018, 30, 1705512. [Google Scholar] [CrossRef]
- Chen, J.; Tao, X.; Li, C.; Ma, Y.; Tao, L.; Zheng, D.; Zhu, J.; Li, H.; Li, R.; Yang, Q. Synthesis of Bipyridine-Based Covalent Organic Frameworks for Visible-Light-Driven Photocatalytic Water Oxidation. Appl. Catal. B Environ. 2020, 262, 118271. [Google Scholar] [CrossRef]
- Lu, M.; Liu, J.; Li, Q.; Zhang, M.; Liu, M.; Wang, J.; Yuan, D.; Lan, Y. Rational Design of Crystalline Covalent Organic Frameworks for Efficient CO2 Photoreduction with H2O. Angew. Chem. 2019, 131, 12522–12527. [Google Scholar] [CrossRef]
- Chen, R.; Wang, Y.; Ma, Y.; Mal, A.; Gao, X.Y.; Gao, L.; Qiao, L.; Li, X.B.; Wu, L.Z.; Wang, C. Rational Design of Isostructural 2D Porphyrin-Based Covalent Organic Frameworks for Tunable Photocatalytic Hydrogen Evolution. Nat. Commun. 2021, 12, 1354. [Google Scholar] [CrossRef]
- Cao, S.; Yu, J. Journal of Photochemistry and Photobiology C : Photochemistry Reviews Carbon-Based H2-Production Photocatalytic Materials. J. Photochem. Photobiol. C Photochem. Rev. 2016, 27, 72–99. [Google Scholar] [CrossRef]
- Wen, J.; Xie, J.; Chen, X.; Li, X. A Review on G-C3N4 -Based Photocatalysts. Appl. Surf. Sci. 2017, 391, 72–123. [Google Scholar] [CrossRef]
- Yang, Q.; Luo, M.; Liu, K.; Cao, H.; Yan, H. Covalent Organic Frameworks for Photocatalytic Applications. Appl. Catal. B Environ. 2020, 276, 119174. [Google Scholar] [CrossRef]
- Geng, Q.; Xu, Z.; Wang, J.; Song, C.; Wu, Y.; Wang, Y. Tailoring Covalent Triazine Frameworks Anode for Superior Lithium-Ion Storage via Thioether Engineering. Chem. Eng. J. 2023, 469, 143941. [Google Scholar] [CrossRef]
- Pan, J.; Guo, L.; Zhang, S.; Wang, N.; Jin, S.; Tan, B. Embedding Carbon Nitride into a Covalent Organic Framework with Enhanced Photocatalysis Performance. Chem. Asian J. 2018, 13, 1674–1677. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, Y.; Hu, H.; Chen, L.; Yu, M.; Gao, M.; Wang, S. Metal-Free Catalysts of Graphitic Carbon Nitride–Covalent Organic Frameworks for Efficient Pollutant Destruction in Water. J. Colloid Interface Sci. 2019, 554, 376–387. [Google Scholar] [CrossRef]
- Wang, X.; Gong, J.; Dong, Y.; An, S.; Zhang, X.; Tian, J. Energy Band Engineering of Hydroxyethyl Group Grafted on the Edge of 3D G-C3N4 Nanotubes for Enhanced Photocatalytic H2 Production. Mater. Today Phys. 2022, 27, 100806. [Google Scholar] [CrossRef]
- Zhong, H.; Hong, Z.; Yang, C.; Li, L.; Xu, Y.; Wang, X.; Wang, R. A Covalent Triazine-Based Framework Consisting of Donor–Acceptor Dyads for Visible-Light-Driven Photocatalytic CO2 Reduction. ChemSusChem 2019, 12, 4493–4499. [Google Scholar] [CrossRef]
- Liu, W.; Li, X.; Wang, C.; Pan, H.; Liu, W.; Wang, K.; Zeng, Q.; Wang, R.; Jiang, J. A Scalable General Synthetic Approach toward Ultrathin Imine-Linked Two-Dimensional Covalent Organic Framework Nanosheets for Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 17431–17440. [Google Scholar] [CrossRef]
- Wang, X.; Fu, Z.; Zheng, L.; Zhao, C.; Wang, X.; Chong, S.Y.; McBride, F.; Raval, R.; Bilton, M.; Liu, L.; et al. Covalent Organic Framework Nanosheets Embedding Single Cobalt Sites for Photocatalytic Reduction of Carbon Dioxide. Chem. Mater. 2020, 32, 9107–9114. [Google Scholar] [CrossRef]
- Ran, L.; Li, Z.; Ran, B.; Cao, J.; Zhao, Y.; Shao, T.; Song, Y.; Leung, M.K.H.; Sun, L.; Hou, J. Engineering Single-Atom Active Sites on Covalent Organic Frameworks for Boosting CO2 Photoreduction. J. Am. Chem. Soc. 2022, 144, 17097–17109. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhu, X.; Huang, L.; Zhang, X.; Zhang, F.; Zhu, W. Azine-Based Covalent Organic Frameworks as Metal-Free Visible Light Photocatalysts for CO2 Reduction with H2O. Appl. Catal. B Environ. 2018, 239, 46–51. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yang, Q.; Yang, W.; Wang, J.; He, F.; Liu, K.; Cao, H.; Yan, H. Defects Engineering Leads to Enhanced Photocatalytic H2 Evolution on Graphitic Carbon Nitride–Covalent Organic Framework Nanosheet Composite. Small 2020, 16, 2001100. [Google Scholar] [CrossRef] [PubMed]
- Papailias, I.; Todorova, N.; Giannakopoulou, T.; Ioannidis, N.; Boukos, N.; Athanasekou, C.P.; Dimotikali, D.; Trapalis, C. Chemical vs Thermal Exfoliation of G-C3N4 for NOx Removal under Visible Light Irradiation. Appl. Catal. B Environ. 2018, 239, 16–26. [Google Scholar] [CrossRef]
- Song, X.; Wu, Y.; Zhang, X.; Li, X.; Zhu, Z.; Ma, C.; Yan, Y.; Huo, P.; Yang, G. Boosting Charge Carriers Separation and Migration Efficiency via Fabricating All Organic van Der Waals Heterojunction for Efficient Photoreduction of CO2. Chem. Eng. J. 2021, 408, 127292. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Dallas, P.; Sanakis, Y.; Stamopoulos, D.; Trapalis, C.; Niarchos, D. Synthesis and Characterization of a π-Conjugate, Covalent Layered Network Derived from Condensation Polymerization of the 4,4′-Bipyridine-Cyanuric Chloride Couple. Eur. Polym. J. 2006, 42, 2940–2948. [Google Scholar] [CrossRef]
- Lei, K.; Wang, D.; Ye, L.; Kou, M.; Deng, Y.; Ma, Z.; Wang, L.; Kong, Y. A Metal-Free Donor–Acceptor Covalent Organic Framework Photocatalyst for Visible-Light-Driven Reduction of CO2 with H2O. ChemSusChem 2020, 13, 1725–1729. [Google Scholar] [CrossRef]
- Liu, N.; Li, T.; Zhao, Z.; Liu, J.; Luo, X.; Yuan, X.; Luo, K.; He, J.; Yu, D.; Zhao, Y. From Triazine to Heptazine: Origin of Graphitic Carbon Nitride as a Photocatalyst. ACS Omega 2020, 5, 12557–12567. [Google Scholar] [CrossRef]
- Ye, H.; Gong, N.; Cao, Y.; Fan, X.; Song, X.; Li, H.; Wang, C.; Mei, Y.; Zhu, Y. Insights into the Role of Protonation in Covalent Triazine Framework-Based Photocatalytic Hydrogen Evolution. Chem. Mater. 2022, 34, 1481–1490. [Google Scholar] [CrossRef]
- Luo, M.; Yang, Q.; Liu, K.; Cao, H.; Yan, H. Boosting Photocatalytic H2 Evolution on G-C3N4 by Modifying Covalent Organic Frameworks (COFs). Chem. Commun. 2019, 55, 5829–5832. [Google Scholar] [CrossRef] [PubMed]
- Bika, P.; Giannakopoulou, T.; Osokin, V.; Li, M.; Todorova, N.; Kaidatzis, A.; Taylor, R.A.; Trapalis, C.; Dallas, P. An Insight Study into the Parameters Altering the Emission of a Covalent Triazine Framework. J. Mater. Chem. C 2021, 9, 13770–13781. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, K.; Wu, L.; Liu, K.; Huang, R.; Long, Z.; Tong, M.; Chen, G. Facile Synthesis of Crystalline Viologen-Based Porous Ionic Polymers with Hydrogen-Bonded Water for Efficient Catalytic CO2 Fixation under Ambient Conditions. RSC Adv. 2020, 10, 3606–3614. [Google Scholar] [CrossRef]
- Sun, B.W.; Yu, H.Y.; Yang, Y.J.; Li, H.J.; Zhai, C.Y.; Qian, D.J.; Chen, M. New Complete Assignment of X-Ray Powder Diffraction Patterns in Graphitic Carbon Nitride Using Discrete Fourier Transform and Direct Experimental Evidence. Phys. Chem. Chem. Phys. 2017, 19, 26072–26084. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, L.; Zong, R.; Zhu, Y. Photocatalytic Activity Enhanced via G-C3N4 Nanoplates to Nanorods. J. Phys. Chem. C 2013, 117, 9952–9961. [Google Scholar] [CrossRef]
- Xing, Y.; Yin, L.; Zhao, Y.; Du, Z.; Tan, H.Q.; Qin, X.; Ho, W.; Qiu, T.; Li, Y.G. Construction of the 1D Covalent Organic Framework/2D g-C3N4Heterojunction with High Apparent Quantum Efficiency at 500 Nm. ACS Appl. Mater. Interfaces 2020, 12, 51555–51562. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.N.; Nguyen, D.T.; Becerra, J.; Sakar, M.; Ahad, J.M.E.; Jautzy, J.J.; Mindorff, L.M.; Béland, F.; Do, T.O. Manifestation of an Enhanced Photoreduction of CO2 to CO over the in Situ Synthesized RGO-Covalent Organic Framework under Visible Light Irradiation. ACS Appl. Energy Mater. 2021, 4, 6005–6014. [Google Scholar] [CrossRef]
- Vyas, V.S.; Haase, F.; Stegbauer, L.; Savasci, G.; Podjaski, F.; Ochsenfeld, C.; Lotsch, B.V. A Tunable Azine Covalent Organic Framework Platform for Visible Light-Induced Hydrogen Generation. Nat. Commun. 2015, 6, 8508. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Tian, Y.; Xu, Z.; Wang, W.; Qiu, M.; Wang, H.; Li, X.; Zhu, G.; Wang, Y. Covalent Organic Framework/g-C3N4 van Der Waals Heterojunction toward H2 Production. Inorg. Chem. 2023, 62, 3271–3277. [Google Scholar] [CrossRef]
- Pan, Z.; Niu, P.; Liu, M.; Zhang, G.; Zhu, Z.; Wang, X. Molecular Junctions on Polymeric Carbon Nitrides with Enhanced Photocatalytic Performance. ChemSusChem 2020, 13, 888–892. [Google Scholar] [CrossRef]
- Shahbazi, H.; Shafei, A.; Sheibani, S. The Effect of Carbon Nanotubes Functionalization on the Band-Gap Energy of TiO2-CNT Nanocomposite. AIP Conf. Proc. 2018, 1920, 020040. [Google Scholar] [CrossRef]
- Oh, Y.; Le, V.D.; Maiti, U.N.; Hwang, J.O.; Park, W.J.; Lim, J.; Lee, K.E.; Bae, Y.S.; Kim, Y.H.; Kim, S.O. Selective and Regenerative Carbon Dioxide Capture by Highly Polarizing Porous Carbon Nitride. ACS Nano 2015, 9, 9148–9157. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhi, Y.; Feng, X.; Ding, X.; Zou, Y.; Liu, X.; Mu, Y. An Azine-Linked Covalent Organic Framework: Synthesis, Characterization and Efficient Gas Storage. Chem. Eur. J. 2015, 21, 12079–12084. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jia, G.; Hu, Y.; Fan, G.; Tsang, Y.H.; Li, Z.; Zou, Z. Two-Dimensional Nanomaterials for Photocatalytic CO2 Reduction to Solar Fuels. Sustain. Energy Fuels 2017, 1, 1875–1898. [Google Scholar] [CrossRef]
- Zhong, W.; Sa, R.; Li, L.; He, Y.; Li, L.; Bi, J.; Zhuang, Z.; Yu, Y.; Zou, Z. A Covalent Organic Framework Bearing Single Ni Sites as a Synergistic Photocatalyst for Selective Photoreduction of CO2 to CO. J. Am. Chem. Soc. 2019, 141, 7615–7621. [Google Scholar] [CrossRef] [PubMed]
- Todorova, N.; Papailias, I.; Giannakopoulou, T.; Ioannidis, N.; Boukos, N.; Dallas, P.; Edelmannová, M.; Reli, M.; Kočí, K.; Trapalis, C. Photocatalytic H2 evolution, CO2 reduction, and Noxoxidation by Highly Exfoliated g-C3N4. Catalysts 2020, 10, 1147. [Google Scholar] [CrossRef]
- López-Magano, A.; Platero-Prats, A.E.; Cabrera, S.; Mas-Ballesté, R.; Alemán, J. Incorporation of Photocatalytic Pt(II) Complexes into Imine-Based Layered Covalent Organic Frameworks (COFs) through Monomer Truncation Strategy. Appl. Catal. B Environ. 2020, 272, 119027. [Google Scholar] [CrossRef]
- Almansaf, Z.; Hu, J.; Zanca, F.; Shahsavari, H.R.; Kampmeyer, B.; Tsuji, M.; Maity, K.; Lomonte, V.; Ha, Y.; Mastrorilli, P.; et al. Pt(II)-Decorated Covalent Organic Framework for Photocatalytic Difluoroalkylation and Oxidative Cyclization Reactions. ACS Appl. Mater. Interfaces 2021, 13, 6349–6358. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; He, H.; Sun, L.; Wang, H.; Fang, X.; Zhao, Y.; Zheng, D.; Qi, Y.; Li, Z.; et al. In Situ Photodeposition of Platinum Clusters on a Covalent Organic Framework for Photocatalytic Hydrogen Production. Nat. Commun. 2022, 13, 1355. [Google Scholar] [CrossRef]
- Bika, P.; Ioannidis, N.; Gatou, M.A.; Sanakis, Y.; Dallas, P. Copper Coordination and the Induced Morphological Changes in Covalent Organic Frameworks. Langmuir 2022, 38, 3082–3089. [Google Scholar] [CrossRef]
- Bratsos, I.; Tampaxis, C.; Spanopoulos, I.; Demitri, N.; Charalambopoulou, G.; Vourloumis, D.; Steriotis, T.A.; Trikalitis, P.N. Heterometallic In(III)-Pd(II) Porous Metal-Organic Framework with Square-Octahedron Topology Displaying High CO2 Uptake and Selectivity toward CH4 and N2. Inorg. Chem. 2018, 57, 7244–7251. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zou, R.; Zhao, Y. Covalent Organic Frameworks for CO2 Capture. Adv. Mater. 2016, 28, 2855–2873. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.Q.; Liu, C.Y.; Wang, X.M.; Zhang, X.M. Novel Covalent Triazine Framework for High-Performance CO2 Capture and Alkyne Carboxylation Reaction. ACS Appl. Mater. Interfaces 2018, 10, 27972–27978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, R.; Ma, Y.; Chen, R.; Sun, X.; Sun, X. N-Rich Covalent Organic Frameworks with Different Pore Size for High-Pressure CO2 Adsorption. Microporous Mesoporous Mater. 2019, 285, 70–79. [Google Scholar] [CrossRef]
- Hug, S.; Stegbauer, L.; Oh, H.; Hirscher, M.; Lotsch, B.V. Nitrogen-Rich Covalent Triazine Frameworks as High-Performance Platforms for Selective Carbon Capture and Storage. Chem. Mater. 2015, 27, 8001–8010. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kanazawa, S.; Kofuji, Y.; Sakamoto, H.; Ichikawa, S.; Tanaka, S.; Hirai, T. Sunlight-Driven Hydrogen Peroxide Production from Water and Molecular Oxygen by Metal-Free Photocatalysts. Angew. Chem.-Int. Ed. 2014, 53, 13454–13459. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kanazawa, S.; Sugano, Y.; Tsukamoto, D.; Sakamoto, H.; Ichikawa, S.; Hirai, T. Highly Selective Production of Hydrogen Peroxide on Graphitic Carbon Nitride (g-C3N4) Photocatalyst Activated by Visible Light. ACS Catal. 2014, 4, 774–780. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, L.; Xu, D.; Cheng, B.; Yu, J. Adsorption Investigation of CO2 on G-C3N4 Surface by DFT Calculation. J. CO2 Util. 2017, 21, 327–335. [Google Scholar] [CrossRef]
- Giannakopoulou, T.; Papailias, I.; Todorova, N.; Boukos, N.; Liu, Y.; Yu, J.; Trapalis, C. Tailoring the Energy Band Gap and Edges’ Potentials of g-C3N4/TiO2 composite Photocatalysts for NOxremoval. Chem. Eng. J. 2017, 310, 571–580. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bika, P.; Papailias, I.; Giannakopoulou, T.; Tampaxis, C.; Steriotis, T.A.; Trapalis, C.; Dallas, P. Prominent COF, g-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction. Catalysts 2023, 13, 1331. https://doi.org/10.3390/catal13101331
Bika P, Papailias I, Giannakopoulou T, Tampaxis C, Steriotis TA, Trapalis C, Dallas P. Prominent COF, g-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction. Catalysts. 2023; 13(10):1331. https://doi.org/10.3390/catal13101331
Chicago/Turabian StyleBika, Panagiota, Ilias Papailias, Tatiana Giannakopoulou, Christos Tampaxis, Theodore A. Steriotis, Christos Trapalis, and Panagiotis Dallas. 2023. "Prominent COF, g-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction" Catalysts 13, no. 10: 1331. https://doi.org/10.3390/catal13101331
APA StyleBika, P., Papailias, I., Giannakopoulou, T., Tampaxis, C., Steriotis, T. A., Trapalis, C., & Dallas, P. (2023). Prominent COF, g-C3N4, and Their Heterojunction Materials for Selective Photocatalytic CO2 Reduction. Catalysts, 13(10), 1331. https://doi.org/10.3390/catal13101331