
Selective Oxidation of Isobutane to Methacrylic Acid and Methacrolein: A Critical Review




Abstract
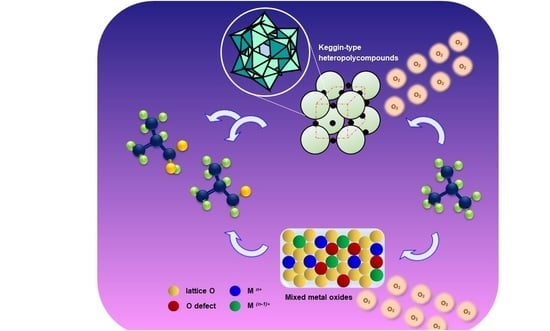
1. Introduction
2. Catalysts for Selective Oxidation of Isobutane to MAA and MAC
2.1. Keggin-Type Heteropolycompounds Catalysts
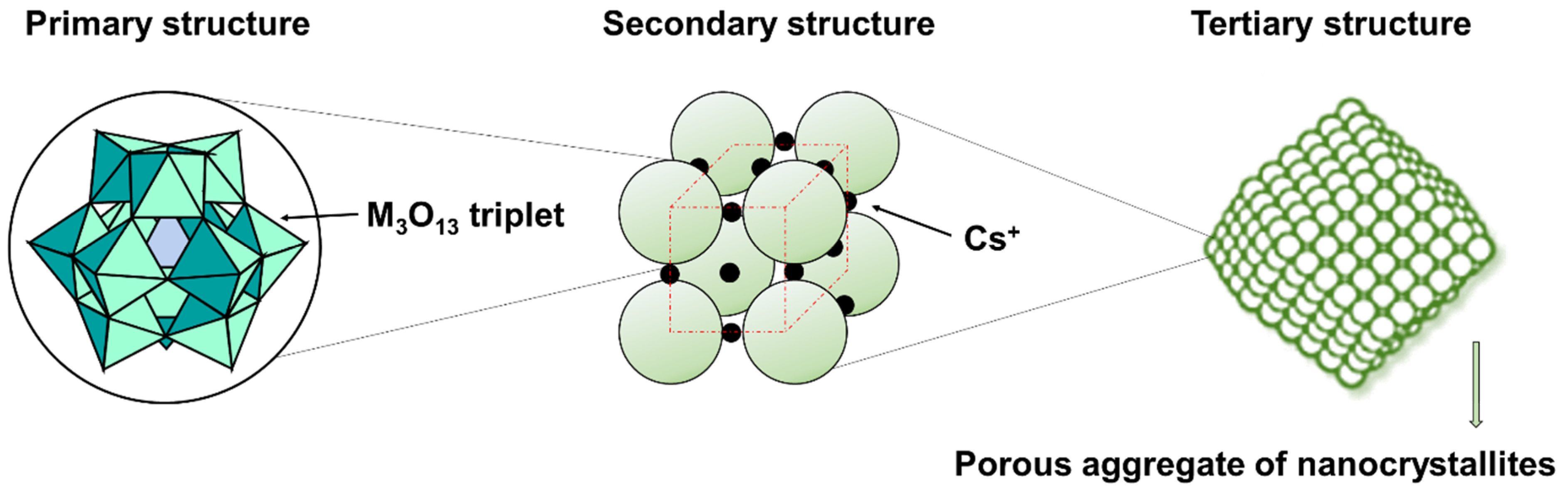
2.1.1. Keggin Structure
2.1.2. Heteropolycompounds for Oxidizing Isobutane to MAA and MAC
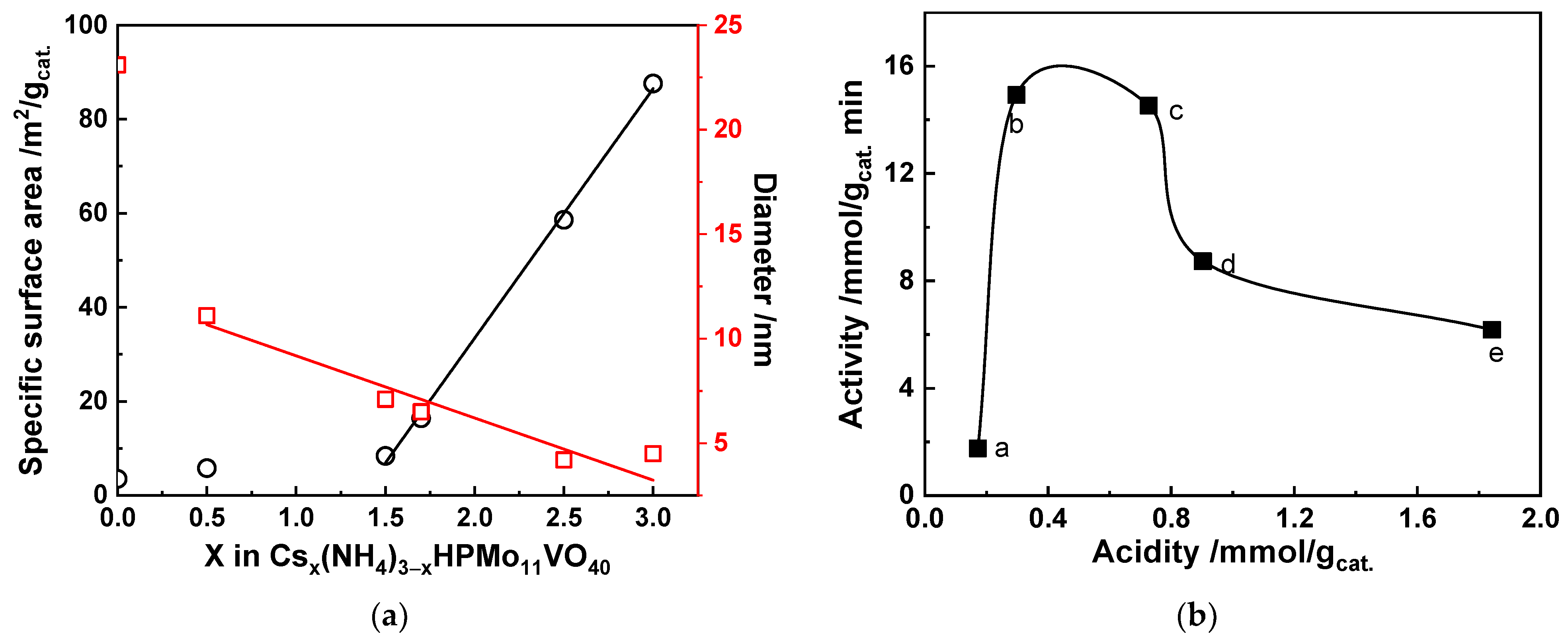
Cesium or/and Ammonium-Based HPCs
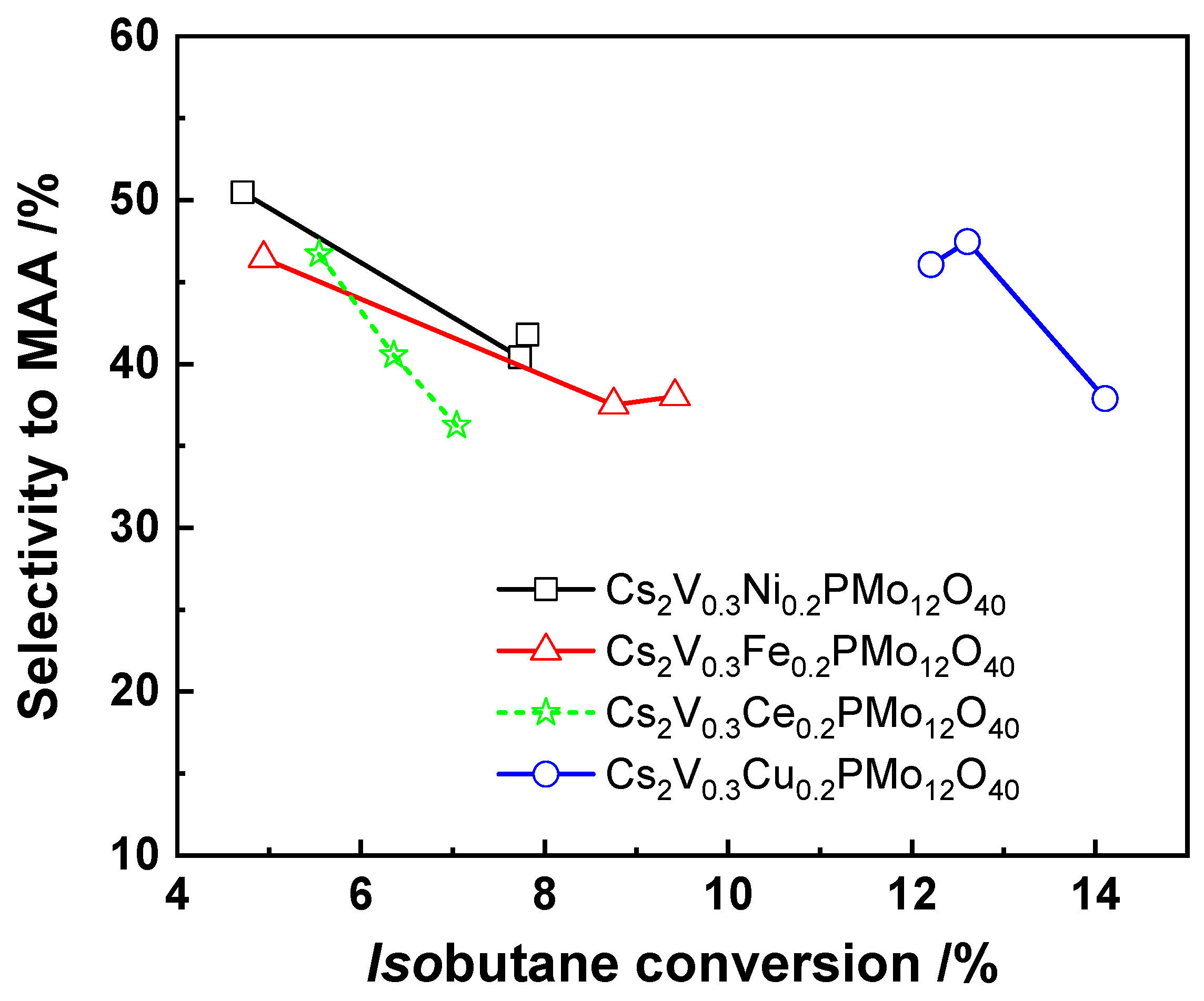
Vanadium-Substituted HPCs
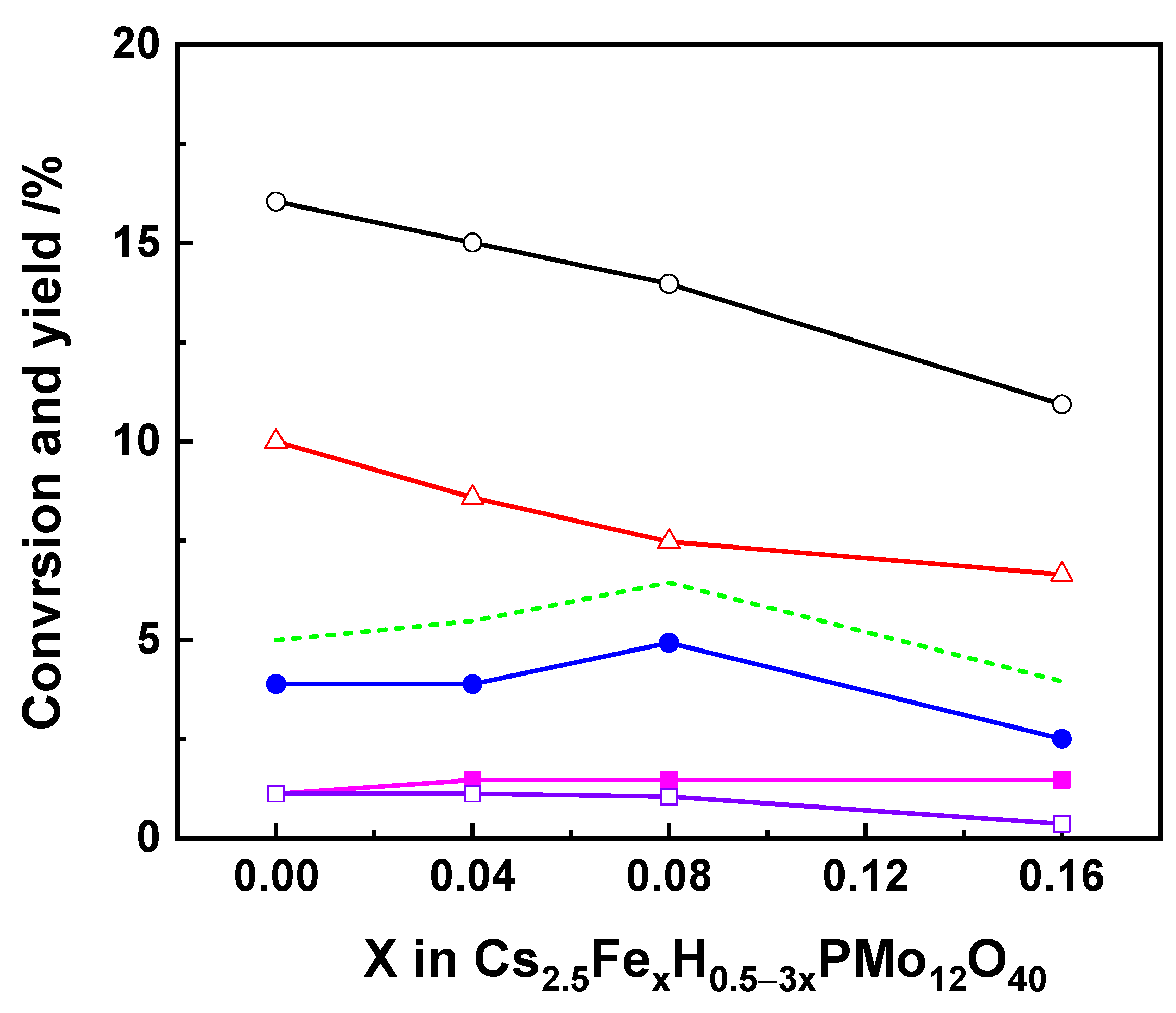
Metal Ions-Substituted HPCs
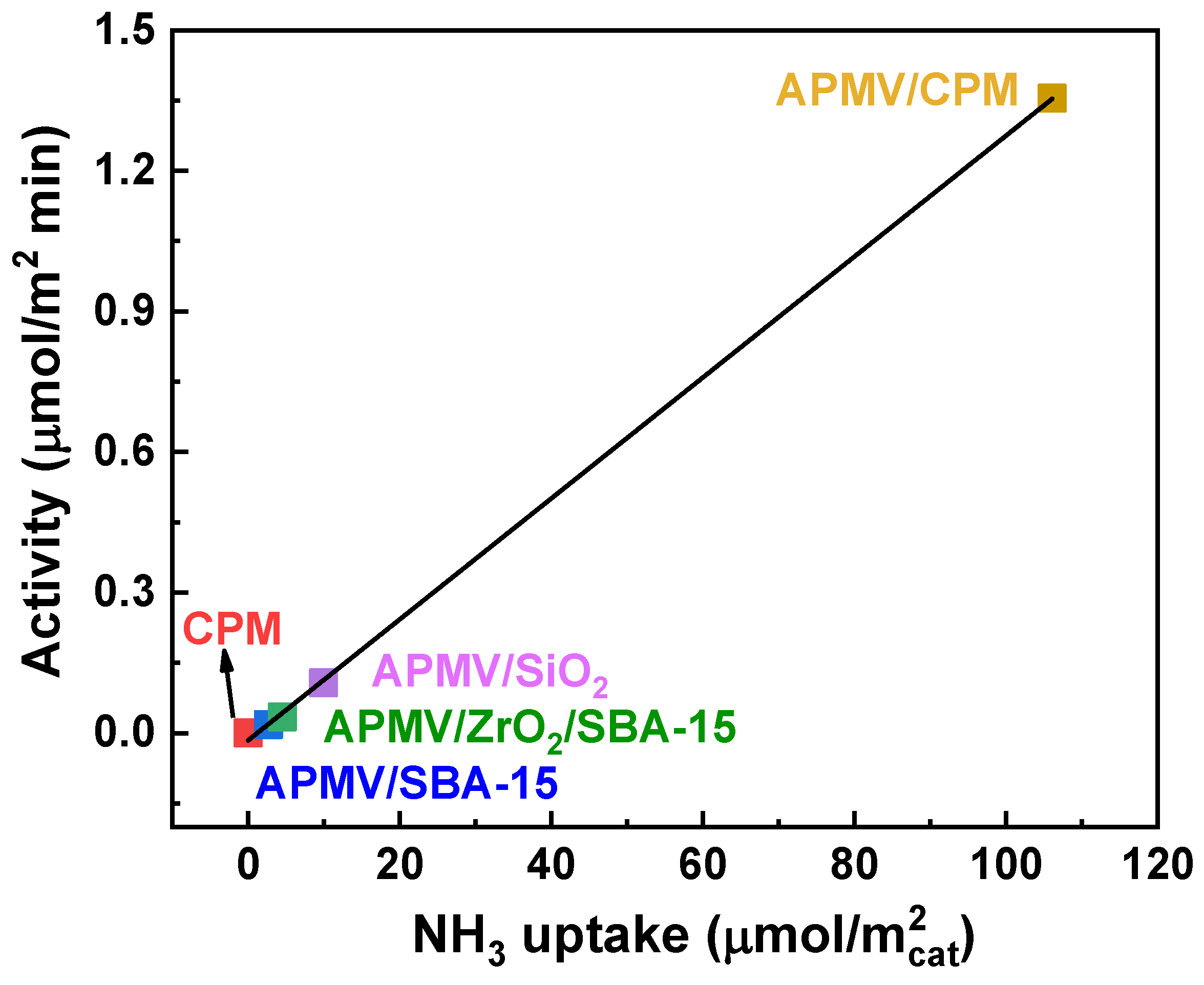
2.1.3. Supported Heteropolycompounds
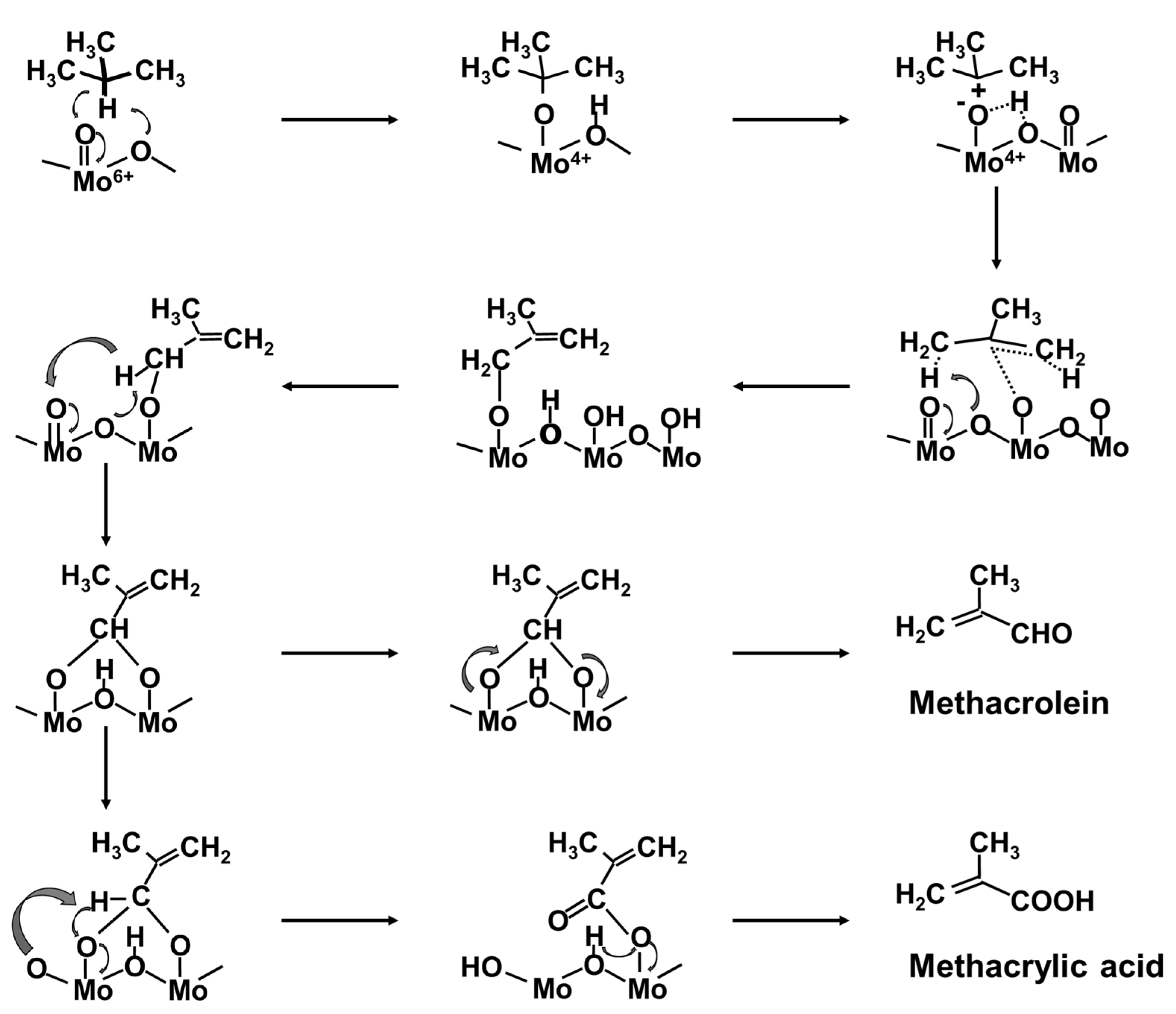
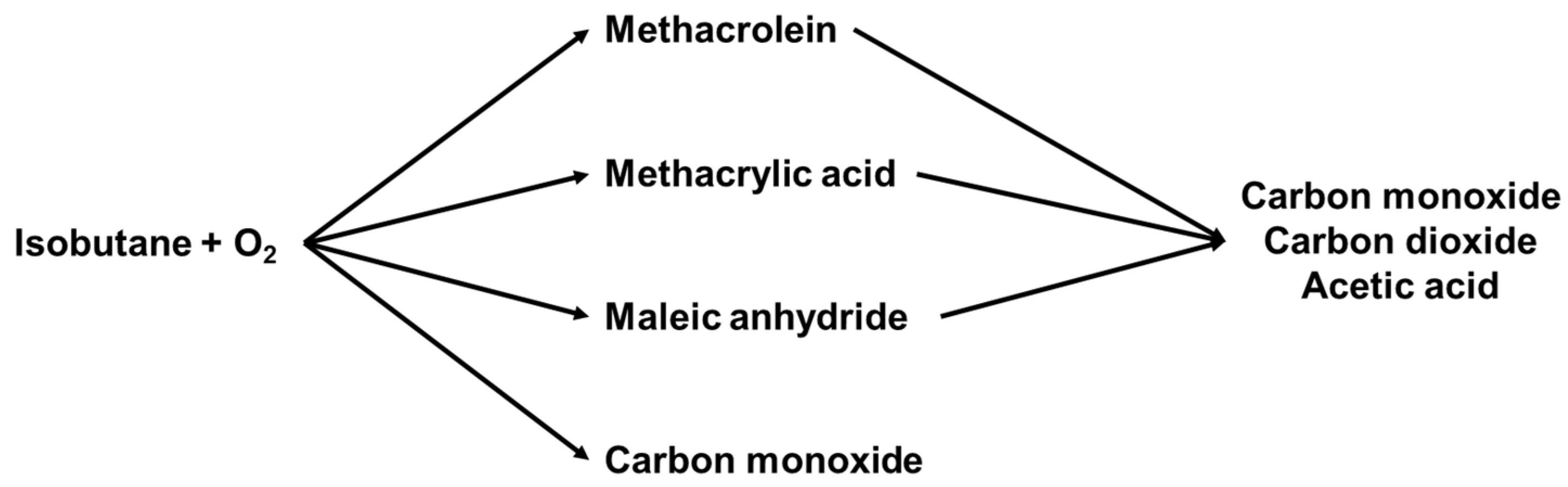
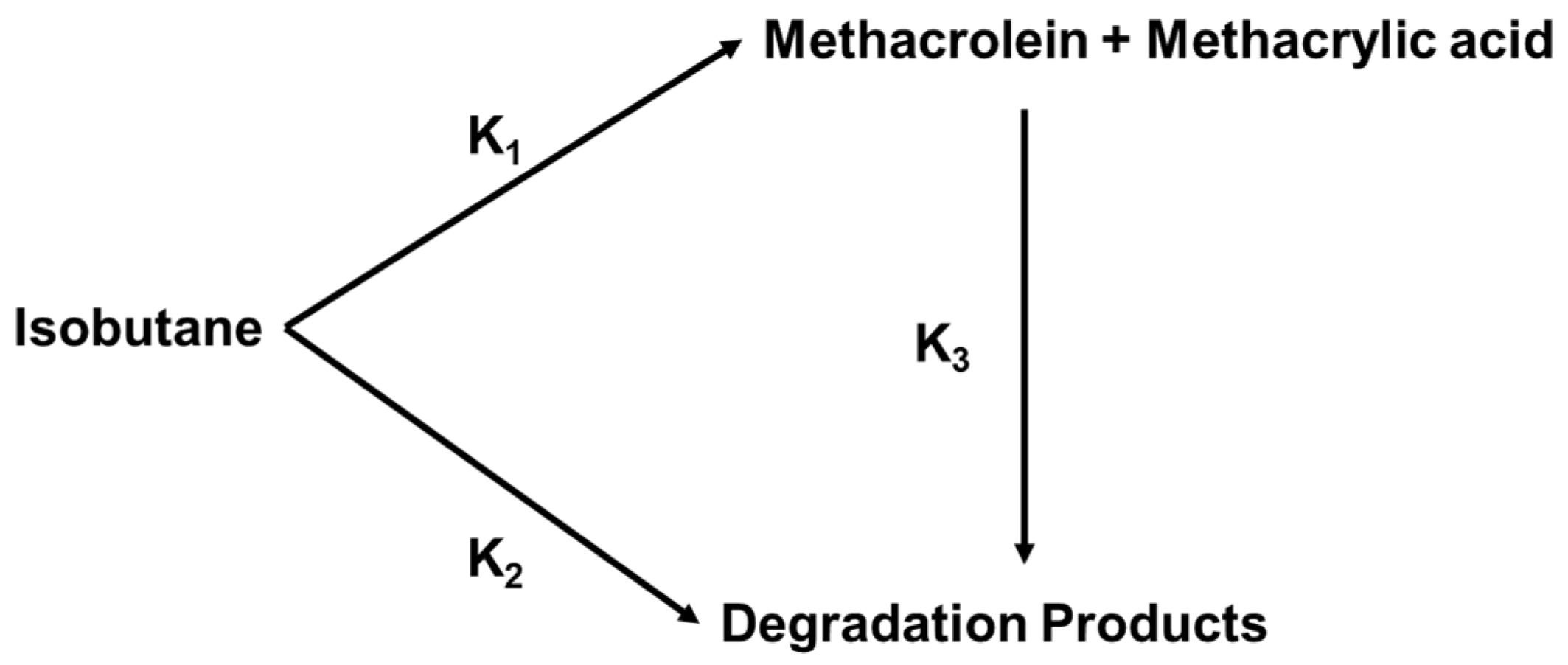
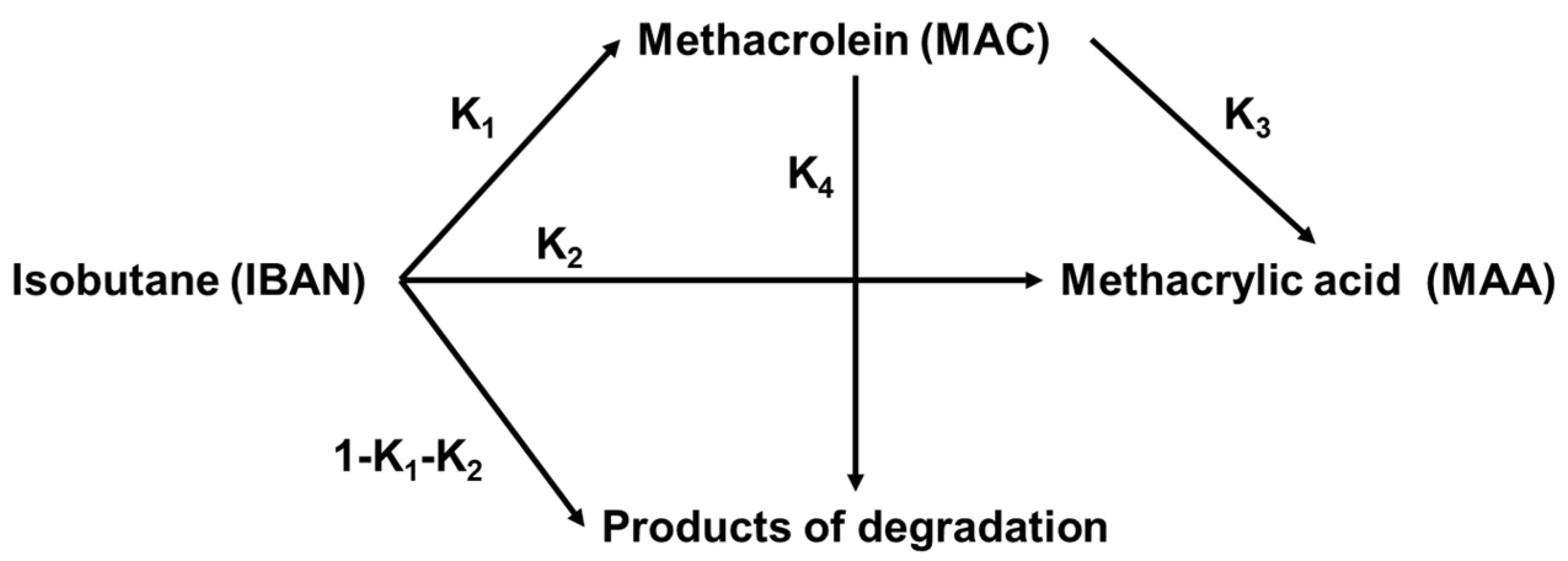
2.1.4. Reaction Mechanism over Heteropolycompounds
2.2. Mixed Metal Oxides
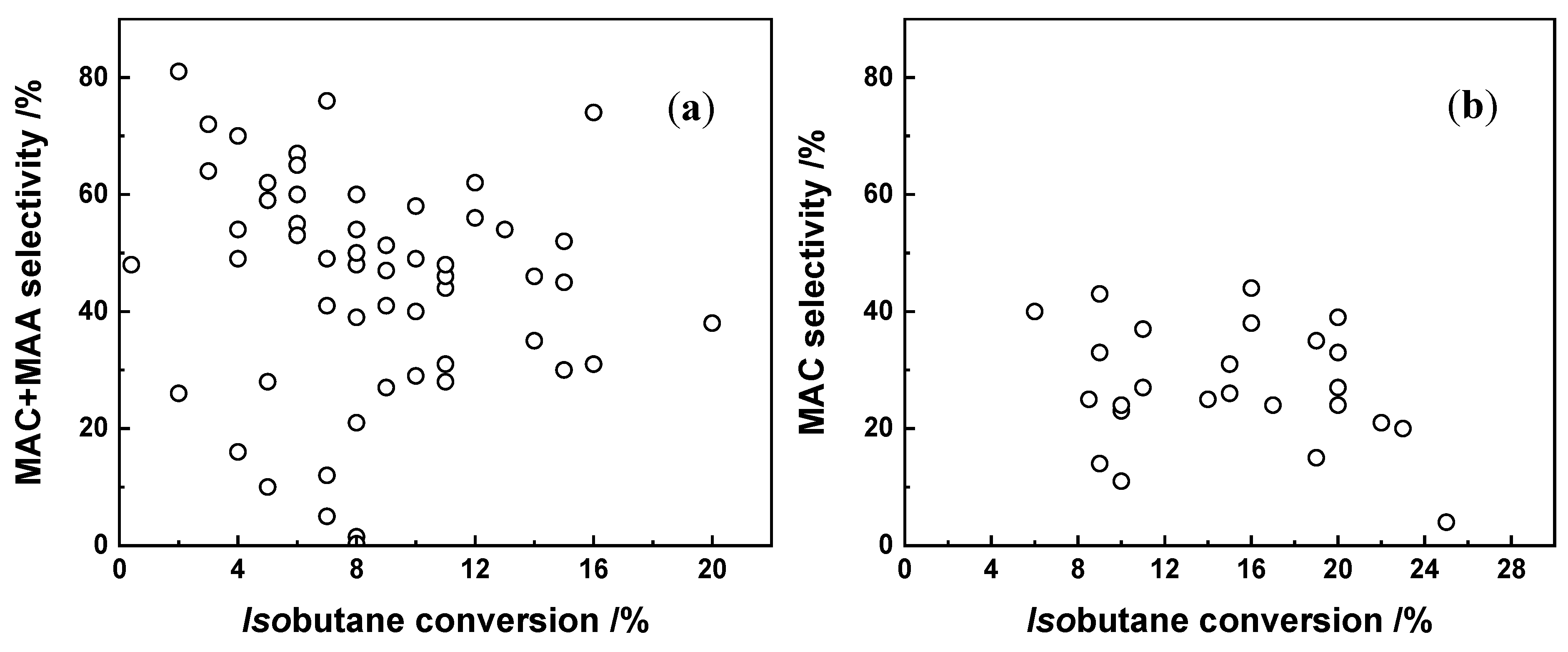
3. Reaction Conditions of Selective Oxidation of Isobutane to MAA and MAC
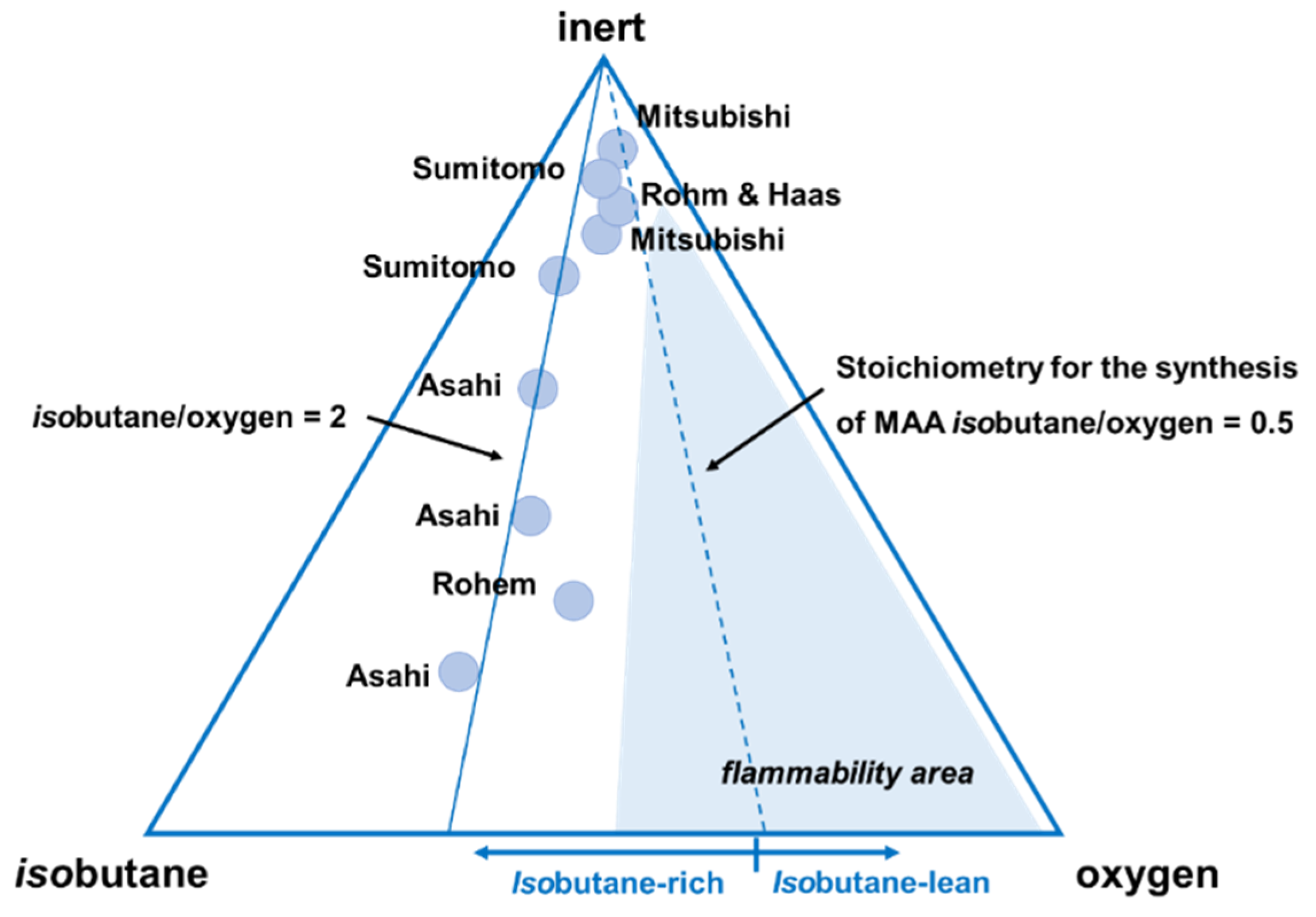
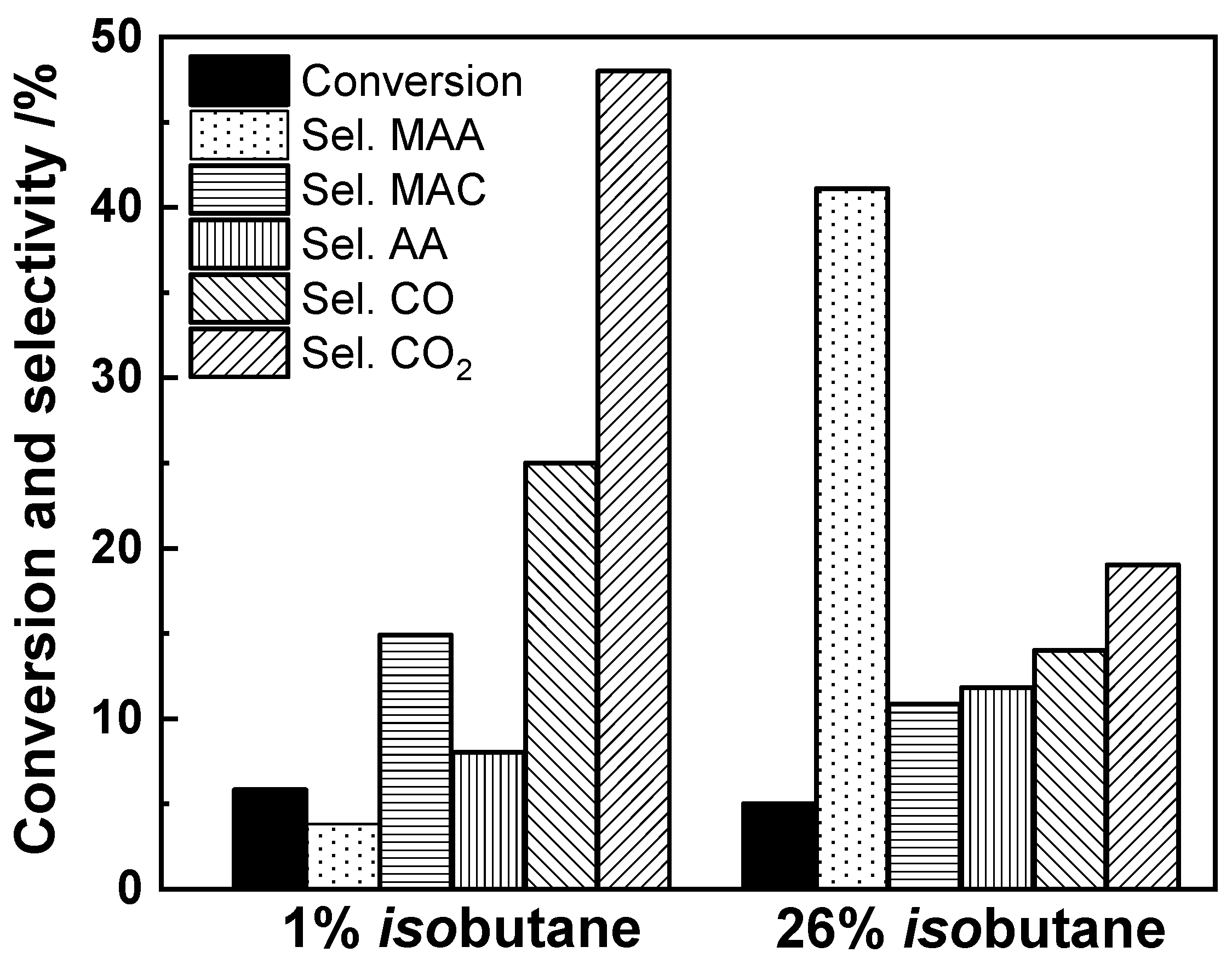
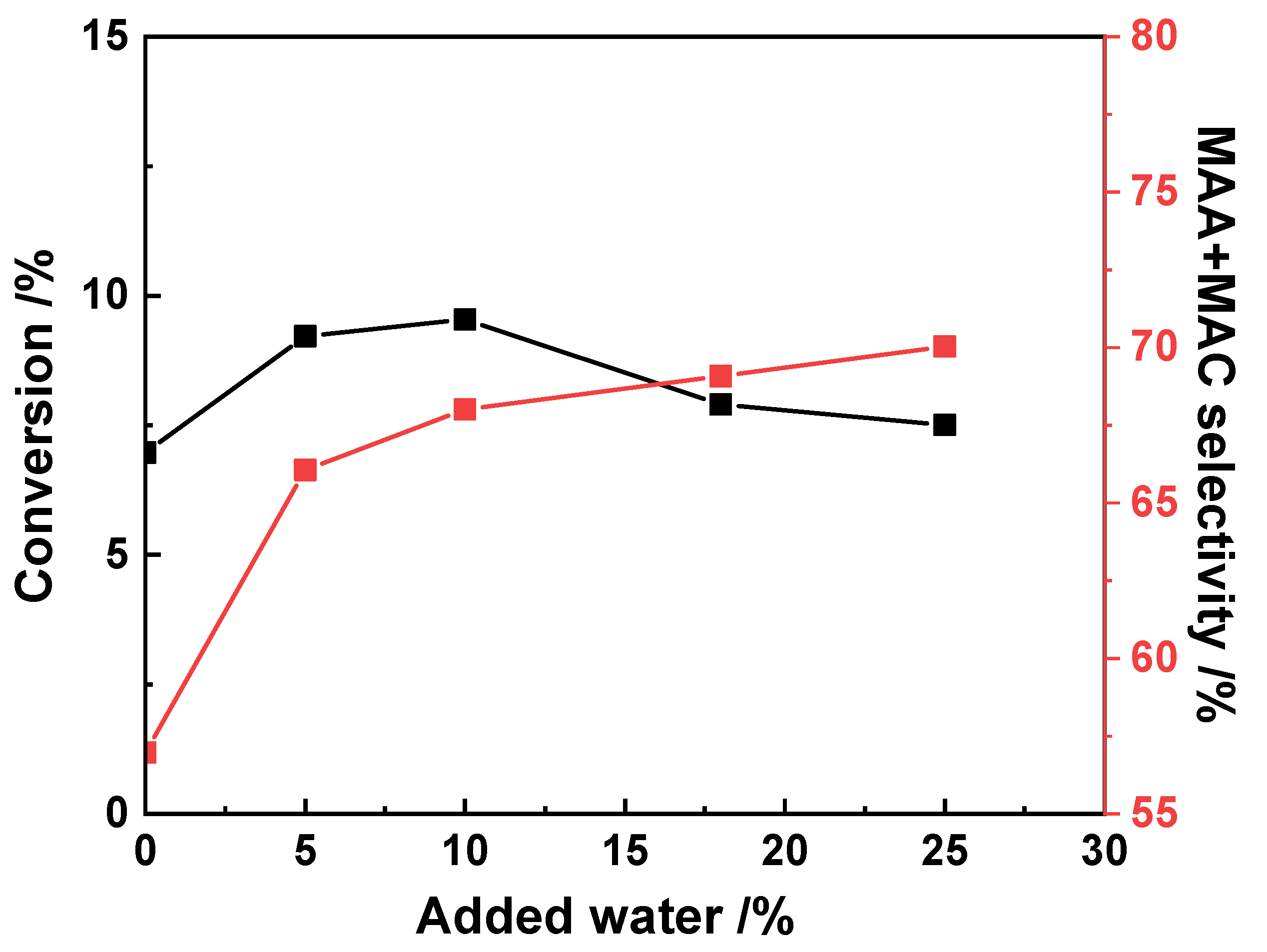
3.1. Effect of the Feed Composition
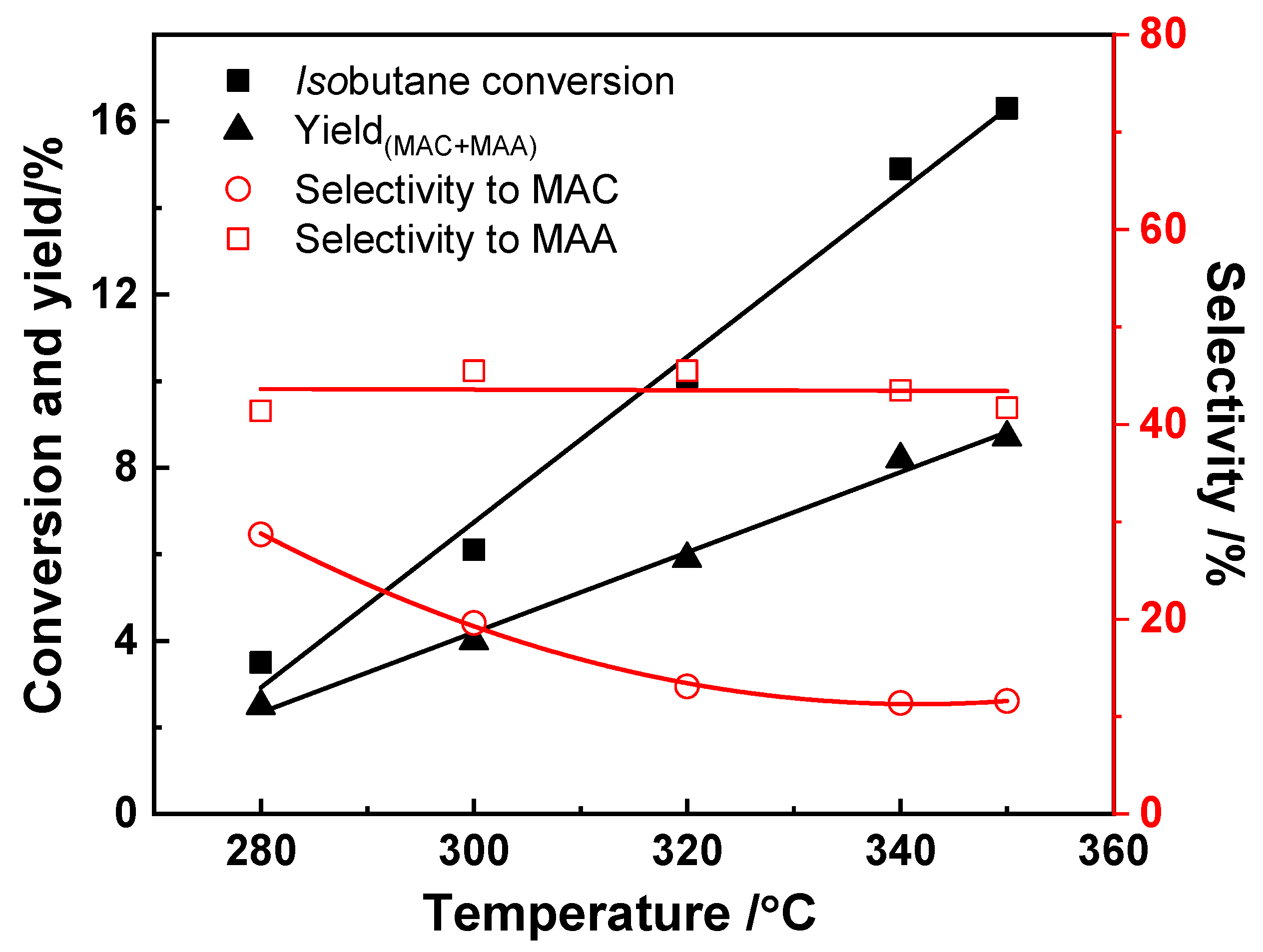
3.2. Effect of Reaction Temperature
3.3. Effect of Contact Time
4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, M.; Zhang, J.; Putaj, P.; Caps, V.; Lefebvre, F.; Pelletier, J.; Basset, J.M. Catalytic oxidation of light alkanes (C1-C4) by heteropoly compounds. Chem. Rev. 2014, 114, 981–1019. [Google Scholar] [CrossRef]
- Cao, H.; Qian, R.; Yu, L. Selenium-catalyzed oxidation of alkenes: Insight into the mechanisms and developing trend. Catal. Sci. Technol. 2020, 10, 3113–3121. [Google Scholar] [CrossRef]
- Chu, W.; Luo, J.; Paul, S.; Liu, Y.; Khodakov, A.; Bordes, E. Synthesis and performance of vanadium-based catalysts for the selective oxidation of light alkanes. Catal. Today 2017, 298, 145–157. [Google Scholar] [CrossRef]
- Song, T.; Ma, Z.; Ren, P.; Yuan, Y.; Xiao, J.; Yang, Y. A Bifunctional iron nanocomposite catalyst for efficient oxidation of alkenes to ketones and 1,2-diketones. ACS Catal. 2020, 10, 4617–4629. [Google Scholar] [CrossRef]
- Venegas, J.M.; Grant, J.T.; McDermott, W.P.; Burt, S.P.; Micka, J.; Carrero, C.A.; Hermans, I. Selective oxidation of n-butane and isobutane catalyzed by boron nitride. ChemCatChem 2017, 9, 2118–2127. [Google Scholar] [CrossRef]
- Al-Mayman, S.I.; Soliman, M.A.; Al-Awadi, A.S.; Al-Zeghayer, Y.S. Reaction kinetics of ethane partial oxidation to acetic acid. Appl. Petrochem. Res. 2018, 8, 29–38. [Google Scholar] [CrossRef]
- Fornés, V.; López, C.; López, H.H.; Martínez, A. Catalytic performance of mesoporous VOx/SBA-15 catalysts for the partial oxidation of methane to formaldehyde. Appl. Catal. A Gen. 2003, 249, 345–354. [Google Scholar] [CrossRef]
- Baerns, M.; Buyevskaya, O. Simple chemical processes based on low molecular-mass alkanes as chemical feedstocks. Catal. Today 1998, 45, 13–22. [Google Scholar] [CrossRef]
- Dury, F.; Gaigneaux, E.M.; Ruiz, P. The active role of CO2 at low temperature in oxidation processes: The case of the oxidative dehydrogenation of propane on NiMoO4 catalysts. Appl. Catal. A Gen. 2003, 242, 187–203. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Z.J.; Wu, T.; Zeng, L.; Gong, J. Nature of the active sites of VOx/Al2O3 catalysts for propane dehydrogenation. ACS Catal. 2016, 6, 5207–5214. [Google Scholar] [CrossRef]
- Mamedov, E.A.; Cortés Corberán, V. Oxidative dehydrogenation of lower alkanes on vanadium oxide-based catalysts. The present state of the art and outlooks. Appl. Catal. A Gen. 1995, 127, 1–40. [Google Scholar] [CrossRef]
- Routray, K.; Reddy, K.R.S.K.; Deo, G. Oxidative dehydrogenation of propane on V2O5/Al2O3 and V2O5/TiO2 catalysts: Understanding the effect of support by parameter estimation. Appl. Catal. A Gen. 2004, 265, 103–113. [Google Scholar] [CrossRef]
- Sheng, J.; Yan, B.; Lu, W.D.; Qiu, B.; Gao, X.Q.; Wang, D.; Lu, A.H. Oxidative dehydrogenation of light alkanes to olefins on metal-free catalysts. Chem. Soc. Rev. 2021, 50, 1438–1468. [Google Scholar] [CrossRef]
- Shi, L.; Wang, D.; Lu, A.H. A viewpoint on catalytic origin of boron nitride in oxidative dehydrogenation of light alkanes. Cuihua Xuebao Chin. J. Catal. 2018, 39, 908–913. [Google Scholar] [CrossRef]
- Landi, G.; Lisi, L.; Volta, J.C. Effect of water on the catalytic behaviour of VPO in the selective oxidation of propane to acrylic acid. Chem. Commun. 2003, 3, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Ueda, W.; Vitry, D.; Katou, T. Structural organization of catalytic functions in Mo-based oxides for propane selective oxidation. Catal. Today 2004, 96, 235–240. [Google Scholar] [CrossRef]
- Misono, M. Selective oxidation of butanes. Toward green/sustainable chemistry. Top. Catal. 2002, 21, 89–96. [Google Scholar] [CrossRef]
- Ai, M. Comparison of catalytic properties for partial oxidation between heteropolyacids and phosphates of vanadium and iron. J. Mol. Catal. A Chem. 1996, 114, 3–13. [Google Scholar] [CrossRef]
- Kobayashi, T. Selective oxidation of light alkanes to aldehydes over silica catalysts supporting mononuclear active sites—Acrolein formation from ethane. Catal. Today 2001, 71, 69–76. [Google Scholar] [CrossRef]
- Coindre, V.F.; Kinney, S.M.; Sefton, M.V. Methacrylic acid copolymer coating of polypropylene mesh chamber improves subcutaneous islet engraftment. Biomaterials 2020, 259, 120324. [Google Scholar] [CrossRef]
- Darabi Mahboub, M.J.; Dubois, J.L.; Cavani, F.; Rostamizadeh, M.; Patience, G.S. Catalysis for the synthesis of methacrylic acid and methyl methacrylate. Chem. Soc. Rev. 2018, 47, 7703–7738. [Google Scholar] [CrossRef] [PubMed]
- Mangal, U.; Kim, J.Y.; Seo, J.Y.; Kwon, J.S.; Choi, S. Novel poly (methyl methacrylate) containing and fungal resistance. Materials 2019, 12, 3438. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K. New developments in the production of methyl methacrylate. Appl. Catal. A Gen. 2001, 221, 367–377. [Google Scholar] [CrossRef]
- Smith, A.W.; Jackson, I.T.; Yousefi, J. The use of screw fixation of methyl methacrylate to reconstruct large craniofacial contour defects. Eur. J. Plast. Surg. 1999, 22, 17–21. [Google Scholar] [CrossRef]
- Trimpalis, A.; Giannakakis, G.; Cao, S.; Flytzani-Stephanopoulos, M. NiAu single atom alloys for the selective oxidation of methacrolein with methanol to methyl methacrylate. Catal. Today 2020, 355, 804–814. [Google Scholar] [CrossRef]
- Jing, F.; Katryniok, B.; Dumeignil, F.; Bordes-Richard, E.; Paul, S. Catalytic selective oxidation of isobutane to methacrylic acid on supported (NH4)3HPMo11VO40 catalysts. J. Catal. 2014, 309, 121–135. [Google Scholar] [CrossRef]
- Liu, Y.; He, J.; Chu, W.; Yang, W. Polyoxometalate catalysts with co-substituted VO2+ and transition metals and their catalytic performance for the oxidation of isobutane. Catal. Sci. Technol. 2018, 8, 5774–5781. [Google Scholar] [CrossRef]
- Cavani, F.; Mezzogori, R.; Pigamo, A.; Trifirò, F.; Etienne, E. Main aspects of the selective oxidation of isobutane to methacrylic acid catalyzed by Keggin-type polyoxometalates. Catal. Today 2001, 71, 97–110. [Google Scholar] [CrossRef]
- Kendel, S.; Brown, T. Comprehensive study of isobutane selective oxidation over group i and II phosphomolybdates: Structural and kinetic factors. Catal. Lett. 2011, 141, 1767–1785. [Google Scholar] [CrossRef]
- Sultan, M.; Paul, S.; Fournier, M.; Vanhove, D. Evaluation and design of heteropolycompound catalysts for the selective oxidation of isobutane into methacrylic acid. Appl. Catal. A Gen. 2004, 259, 141–152. [Google Scholar] [CrossRef]
- Védrine, J.C.; Fechete, I. Heterogeneous partial oxidation catalysis on metal oxides. Comptes Rendus Chim. 2016, 19, 1203–1225. [Google Scholar] [CrossRef]
- Krieger, H.; Kirch, L.S. Process for the production of unsaturated acids. U.S. Patent 4260822, 7 April 1981. [Google Scholar]
- Cavani, F.; Mezzogori, R.; Pigamo, A.; Trifirò, F. Combined effects of Sb-doping and of preparation via lacunary precursor for P/Mo-based, Keggin-type polyoxometalates, catalysts for the selective oxidation of isobutane to methacrylic acid. Top. Catal. 2003, 23, 119–124. [Google Scholar] [CrossRef]
- Bordes-Richard, E. Application of concepts in heterogeneous oxidation of hydrocarbons: Mo, V-based oxide catalysts for oxidation of ethane and of n- and i-butanes. Catal. Today 2019, 1–12. [Google Scholar] [CrossRef]
- Misono, M. Unique acid catalysis of heteropoly compounds (heteropolyoxometalates) in the solid state. Chem. Commun. 2001, 1, 1141–1153. [Google Scholar] [CrossRef]
- Mizuno, N.; Misono, M. Heterogeneous catalysis. Chem. Rev. 1998, 98, 199–217. [Google Scholar] [CrossRef]
- Vanhove, D. Catalyst testing at a lab scale in mild oxidation: Can you control the reaction temperature? Appl. Catal. A Gen. 1996, 138, 215–234. [Google Scholar] [CrossRef]
- Pope, M.T.; Müller, A. Polyoxometalate Chemistry: An old field with new dimensions in several disciplines. Angew. Chemie Int. Ed. 2013, 30, 1–59. [Google Scholar] [CrossRef]
- Keggin, J.F. The structure and formula of 12-phosphotungstic acid. Proc. R. Soc. Lond. Ser. A 1934, 144, 75–100. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef]
- Moffat, J.B. A comparison of the catalytic and structural properties of heteropoly compounds: Semi-empirical calculations. J. Mol. Catal. 1984, 26, 385–396. [Google Scholar] [CrossRef]
- Misono, M. Heterogeneous Catalysis by Heteropoly Compounds of Molybdenum and Tungsten. Catal. Rev. 1978, 29, 269–321. [Google Scholar] [CrossRef]
- Barcza, L.; Pope, M.T. Heteroconjugation of inorganic anions in nonaqueous solvents. III. Complexes of polymolybdates and -tungstates with chloral hydrate. J. Phys. Chem. 1975, 79, 92–93. [Google Scholar] [CrossRef]
- Ganapathy, S.; Fournier, M.; Paul, J.F.; Delevoye, L.; Guelton, M.; Amoureux, J.P. Location of protons in anhydrous keggin heteropolyacids H3PMo12O40 and H3PW12O40 by 1H{31P}/31P{1H} REDOR NMR and DFT quantum chemical calculations. J. Am. Chem. Soc. 2002, 124, 7821–7828. [Google Scholar] [CrossRef]
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic chemistry of heteropoly compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar] [CrossRef]
- Li, W.; Ueda, W. Catalytic oxidation of isobutane to methacrylic acid with molecular oxygen over activated pyridinium 12-molybdophosphate. Catal. Lett. 1997, 46, 261–265. [Google Scholar] [CrossRef]
- Min, J.S.; Mizuno, N. Effects of additives on catalytic performance of heteropoly compounds for selective oxidation of light alkanes. Catal. Today 2001, 71, 89–96. [Google Scholar] [CrossRef]
- Mizuno, N.; Tateishi, M.; Iwamoto, M. Oxidation of isobutane catalyzed by CsxH3-xPMo12O40-based heteropoly compounds. J. Catal. 1996, 163, 87–94. [Google Scholar] [CrossRef]
- Cavani, F.; Etienne, E.; Mezzogori, R.; Pigamo, A.; Trifirò, F. Improvement of catalytic performance in isobutane oxidation to methacrylic acid of Keggin-type phosphomolybdates by preparation via lacunary precursors: Nature of the active sites. Catal. Lett. 2001, 75, 99–105. [Google Scholar] [CrossRef]
- Cavani, F.; Mezzogori, R.; Pigamo, A.; Trifirò, F. Improved catalytic performance of Keggin-type polyoxometalates in the oxidation of isobutane to methacrylic acid under hydrocarbon-lean conditions using antimony-doped catalysts. Chem. Eng. J. 2001, 82, 33–42. [Google Scholar] [CrossRef]
- Jing, F.; Katryniok, B.; Dumeignil, F.; Bordes-Richard, E.; Paul, S. Catalytic selective oxidation of isobutane over Csx(NH4)3-xHPMo11VO40 mixed salts. Catal. Sci. Technol. 2014, 4, 2938–2945. [Google Scholar] [CrossRef]
- Mizuno, N.; Yahiro, H. Oxidation of isobutane catalyzed by partially salified cesium molybdovanadophosphoric acids. J. Phys. Chem. B 1998, 102, 437–443. [Google Scholar] [CrossRef]
- He, J.; Liu, Y.; Chu, W.; Yang, W. Effect of V-containing precursors on the structure and catalytic performance of Cs-substituted phosphomolybdates for isobutane oxidation. Appl. Catal. A Gen. 2018, 556, 104–112. [Google Scholar] [CrossRef]
- Min, J.S.; Mizuno, N. Iron as an effective additive for enhancement of catalytic performance of cesium hydrogen salt of molybdophosphoric acid for selective oxidation of isobutane, propane, and ethane under oxygen-rich and -poor conditions and the catalyst design. Catal. Today 2001, 66, 47–52. [Google Scholar] [CrossRef]
- Mizuno, N. Catalytic performance of Cs2.5Fe0.08H1.26PVMo11O40 for direct oxidation of lower alkanes. J. Mol. Catal. A Chem. 1996, 114, 309–317. [Google Scholar] [CrossRef]
- Knapp, C.; Ui, T.; Nagai, K.; Mizuno, N. Stability of iron in the Keggin anion of heteropoly acid catalysts for selective oxidation of isobutane. Catal. Today 2001, 71, 111–119. [Google Scholar] [CrossRef]
- Etienne, E.; Cavani, F.; Mezzogori, R.; Trifirò, F.; Calestani, G.; Gengembre, L.; Guelton, M. Chemical-physical characterization of Fe-doped, Keggin-type P/Mo polyoxometalates, catalysts for the selective oxidation of isobutane to methacrylic acid. Appl. Catal. A Gen. 2003, 256, 275–290. [Google Scholar] [CrossRef]
- Langpape, M.; Millet, J.M.M.; Ozkan, U.S.; Boudeulle, M. Study of cesium or cesium-transition metal-substituted Keggin-type phosphomolybdic acid as isobutane oxidation catalysts I. Structural characterization. J. Catal. 1999, 181, 80–90. [Google Scholar] [CrossRef]
- Wu, S.J.; Kan, Q.B.; Ding, W.L.; Shang, F.P.; Liu, H.; Guan, J.Q. Partial oxidation of isobutane to methacrolein over Te(1.5+0.5x)PMo12−xVxOn heteropolycompounds with tellurium as counter cations. React. Kinet. Mech. Catal. 2012, 106, 157–164. [Google Scholar] [CrossRef]
- Huynh, Q.; Selmi, A.; Corbel, G.; Lacorre, P.; Millet, J.M.M. Atypical synergetic effect between Te- and V-substituted phosphomolybdic cesium salt and LAMOX-type phases for the oxidation of isobutane into methacrylic acid. J. Catal. 2009, 266, 64–70. [Google Scholar] [CrossRef]
- Langpape, M.; Millet, J.M.M. Effect of iron counter-ions on the redox properties of the Keggin-type molybdophosphoric heteropolyacid. Part I. An experimental study on isobutane oxidation catalysts. Appl. Catal. A Gen. 2000, 200, 89–101. [Google Scholar] [CrossRef]
- Jalowiecki-Duhamel, L.; Monnier, A.; Barbaux, Y.; Hecquet, G. Oxidation of isobutane on a heteropolycompound hydrogen reservoir. Catal. Today 1996, 32, 237–241. [Google Scholar] [CrossRef]
- Liu, S.; Chen, L.; Wang, G.; Liu, J.; Gao, Y.; Li, C.; Shan, H. Effects of Cs-substitution and partial reduction on catalytic performance of Keggin-type phosphomolybdic polyoxometalates for selective oxidation of isobutane. J. Energy Chem. 2016, 25, 85–92. [Google Scholar] [CrossRef]
- Mizuno, N.; Tateishi, M.; Iwamoto, M. Enhancement of catalytic activity of Cs2.5Ni0.08H0.34PMo12O40 by V5+-substitution for oxidation of isobutane into methacrylic a. Appl. Catal. A Gen. 1994, 118. [Google Scholar] [CrossRef]
- Mizuno, N.; Han, W.; Kudo, T. Selective oxidation of ethane, propane, and isobutane catalyzed by copper-containing Cs2.5H1.5PVMo11O40 under oxygen-poor conditions. J. Catal. 1998, 178, 391–394. [Google Scholar] [CrossRef]
- Huynh, Q.; Schuurman, Y.; Delichere, P.; Loridant, S.; Millet, J.M.M. Study of Te and V as counter-cations in Keggin type phosphomolybdic polyoxometalate catalysts for isobutane oxidation. J. Catal. 2009, 261, 166–176. [Google Scholar] [CrossRef]
- Ding, W.; Liu, L.; Shang, F.; Hu, J.; Kan, Q.; Guan, J. Preparation of Te(12-X)/4PMo12-xVxOn mixed oxides from heteropolycompound precursors for selective oxidation of isobutane. Catal. Commun. 2012, 18, 81–84. [Google Scholar] [CrossRef]
- Schindler, G.P.; Ui, T.; Nagai, K. Kinetics of isobutane selective oxidation over Mo-V-P-As-Cs-Cu-O heteropoly acid catalyst. Appl. Catal. A Gen. 2001, 206, 183–195. [Google Scholar] [CrossRef]
- Jing, F.; Katryniok, B.; Bordes-richard, E.; Paul, S. Improvement of the catalytic performance of supported (NH4)3HPMo11VO40 catalysts in isobutane selective oxidation. Catal. Today 2013, 203, 32–39. [Google Scholar] [CrossRef]
- Cai, X.; Ma, Y.; Zhou, Q.; Zhang, Z.; Chu, W.; Yang, W. Synergistic effects of phases in the selective oxidation of isobutane over supported (NH4)3HPMo11VO40 catalysts. React. Kinet. Mech. Catal. 2021. [Google Scholar] [CrossRef]
- Ballarini, N.; Candiracci, F.; Cavani, F.; Degrand, H.; Dubois, J.L.; Lucarelli, G.; Margotti, M.; Patinet, A.; Pigamo, A.; Trifirò, F. The dispersion of Keggin-type P/Mo polyoxometalates inside silica gel, and the preparation of catalysts for the oxidation of isobutane to methacrolein and methacrylic acid. Appl. Catal. A Gen. 2007, 325, 263–269. [Google Scholar] [CrossRef]
- Paul, S.; Chu, W.; Sultan, M.; Bordes-Richard, E. Keggin-type H4PVMo11O40-based catalysts for the isobutane selective oxidation. Sci. China Chem. 2010, 53, 2039–2046. [Google Scholar] [CrossRef]
- Busca, G.; Cavani, F.; Etienne, E.; Finocchio, E.; Galli, A.; Selleri, G.; Trifirò, F. Reactivity of Keggin-type heteropolycompounds in the oxidation of isobutane to methacrolein and methacrylic acid: Reaction mechanism. J. Mol. Catal. A Chem. 1996, 114, 343–359. [Google Scholar] [CrossRef]
- Centi, G.; Cavani, F.; Trifirò, F. Selective Oxidation by Heterogeneous Catalysis; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Paul, S.; Le Courtois, V.; Vanhove, D. Kinetic investigation of isobutane selective oxidation over a heteropolyanion catalyst. Ind. Eng. Chem. Res. 1997, 36, 3391–3399. [Google Scholar] [CrossRef]
- Guan, J.; Xu, H.; Song, K.; Liu, B.; Shang, F.; Yu, X.; Kan, Q. Selective oxidation and oxidative dehydrogenation of isobutane over hydrothermally synthesized Mo-V-O mixed oxide catalysts. Catal. Lett. 2008, 126, 293–300. [Google Scholar] [CrossRef]
- Guan, J.; Song, K.; Xu, H.; Wang, Z.; Ma, Y.; Shang, F.; Kan, Q. Oxidation of isobutane and isobutene to methacrolein over hydrothermally synthesized Mo-V-Te-O mixed oxide catalysts. Catal. Commun. 2009, 10, 528–532. [Google Scholar] [CrossRef]
- Guan, J.; Jing, S.; Wu, S.; Xu, H.; Wang, Z.; Kan, Q. Selective oxidation of isobutane over Mo-V-Te mixed oxide catalysts with different tellurium contents. React. Kinet. Catal. Lett. 2007, 90, 27–33. [Google Scholar] [CrossRef]
- Guan, J.; Xu, C.; Liu, B.; Yang, Y.; Ma, Y.; Kan, Q. Partial oxidation of isobutane over hydrothermally synthesized Mo-V-Te-O mixed oxide catalysts. Catal. Lett. 2008, 126, 301–307. [Google Scholar] [CrossRef]
- Guan, J.; Xu, C.; Wang, Z.; Yang, Y.; Liu, B.; Shang, F.; Shao, Y.; Kan, Q. Selective oxidation of isobutane and isobutene to methacrolein over Te-Mo mixed oxide catalysts. Catal. Lett. 2008, 124, 428–433. [Google Scholar] [CrossRef]
- Guan, J.; Wang, H.; Yang, Y.; Liu, B.; Yu, X.; Ma, Y.; Kan, Q. Effect of pH on the catalytic properties of Mo-V-Te-P-O catalysts for selective oxidation of isobutane. Catal. Lett. 2009, 131, 512–516. [Google Scholar] [CrossRef]
- Guan, J.; Jia, M.; Jing, S.; Wang, Z.; Xing, L.; Xu, H.; Kan, Q. Selective oxidation of isobutane to methacrolein over Mo-V-Te-Sb mixed oxide catalysts with different antimony contents. Catal. Lett. 2006, 108, 125–129. [Google Scholar] [CrossRef]
- Stuyven, B.; Emmerich, J.; Eloy, P.; Van Humbeeck, J.; Kirschhock, C.E.A.; Jacobs, P.A.; Martens, J.A.; Breynaert, E. Molybdenum-vanadium-antimony mixed oxide catalyst for isobutane partial oxidation synthesized using magneto hydrodynamic forces. Appl. Catal. A Gen. 2014, 474, 18–25. [Google Scholar] [CrossRef][Green Version]
- Shishido, T.; Inoue, A.; Konishi, T.; Matsuura, I.; Takehira, K. Oxidation of isobutane over Mo-V-Sb mixed oxide catalyst. Catal. Lett. 2000, 68, 215–221. [Google Scholar] [CrossRef]
- Guan, J.; Wu, S.; Wang, H.; Jing, S.; Wang, G.; Zhen, K.; Kan, Q. Synthesis and characterization of MoVTeCeO catalysts and their catalytic performance for selective oxidation of isobutane and isobutylene. J. Catal. 2007, 251, 354–362. [Google Scholar] [CrossRef]
- Guan, J.; Wu, S.; Jia, M.; Huang, J.; Jing, S.; Xu, H.; Wang, Z.; Zhu, W.; Xing, H.; Wang, H.; et al. Effect of antimony doping on the catalytic behavior of Mo-V-Te-P mixed oxide catalysts in oxidation of isobutane. Catal. Commun. 2007, 8, 1219–1223. [Google Scholar] [CrossRef]
- Sun, X.D.; Yi, X.D.; Hua, W.Q.; Jin, H.; Weng, W.Z.; Wan, H.L. Selective oxidation of isobutane to methacrolein over MoVTe mixed oxide supported on SBA-3 and SiO2. Fuel Process. Technol. 2011, 92, 1662–1669. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Wang, F.; Zhong, S.; Jiang, S.; Wang, S. Effect of Bi promoter on the performances of selective oxidation of isobutane to methacrolein over MoVO/AlPO4 catalysts. J. Nat. Gas Chem. 2012, 21, 165–169. [Google Scholar] [CrossRef]
- Cavani, F.; Teles, J.H. Sustainability in catalytic oxidation: An alternative approach or a structural evolution? ChemSusChem 2009, 2, 508–534. [Google Scholar] [CrossRef]
- Ballarini, N.; Cavani, F.; Degrand, H.; Etienne, E.; Pigamo, A.; Trifirò, F.; Dubois, J.L. The oxidation of isobutane to methacrylic acid: An alternative technology for MMA production. In Methods and Reagents for Green Chemistry: An Introduction; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 265–279. [Google Scholar] [CrossRef]
- Sultan, M.; Paul, S.; Vanhove, D. Kinetic effects of chemical modifications of PMo12 catalysts for the selective oxidation of isobutane. Stud. Surf. Sci. Catal. 1999, 122, 283–290. [Google Scholar] [CrossRef]
- Dai, X.P.; Li, R.J.; Yu, C.C.; Hao, Z.P. Unsteady-state direct partial oxidation of methane to synthesis gas in a fixed-bed reactor using AFeO3 (A = La, Nd, Eu) perovskite-type oxides as oxygen storage. J. Phys. Chem. B 2006, 110, 22525–22531. [Google Scholar] [CrossRef]
- Emig, G.; Uihlein, K.; Häcker, C.J. Separation of catalyst oxidation and reduction—an alternative to the conventional oxidation of n-butane to maleic anhydride? Stud. Surf. Sci. Catal. 1994, 82, 243–251. [Google Scholar] [CrossRef]
- Song, N.; Rhodes, C.; Bartley, J.K.; Taylor, S.H.; Chadwick, D.; Hutchings, G.J. Oxidation of isobutene to methacrolein using bismuth molybdate catalysts: Comparison of operation in periodic and continuous feed mode. J. Catal. 2005, 236, 282–291. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, J.; Dong, Q.; Liu, J. Selective oxidation of propane by lattice oxygen of vanadium-phosphorous oxide in a pulse reactor. J. Nat. Gas Chem. 2005, 14, 88–94. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | Conversion a, % | Selectivity, % | |||
|---|---|---|---|---|---|
| MAA | MAC | CO | CO2 | ||
| 0 | 10 | 27 | 12 | 30 | 26 |
| 1 | 15 | 36 | 9 | 25 | 24 |
| 2 | 13 | 28 | 8 | 25 | 33 |
| 3 | 12 | 10 | 8 | 35 | 38 |
| Catalyst | Reaction T, °C | Conversion, % | Selectivity, % | Yield, % (MAA + MAC) | Ref. | ||
|---|---|---|---|---|---|---|---|
| MAA | MAC | COx | |||||
| H3PMo12O40 | 340 | 5 | 3 | 7 | 82 | 0.5 | [61] |
| H3PMo12O40 | 340 | 4 | 4 | 12 | - | 0.6 | [30] |
| H3PMo12O40 | 300 | 12 | 58 | 4 | 22 | 7.4 | [46] |
| Cs2HPMo12O40 | 340 | 11 | 34 | 10 | 50 | 4.8 | [47] |
| Cs2HPMo12O40 | 350 | 9 | 44 | 7.3 | 33 | 4.6 | [53] |
| Cs2.5H0.5PMo12O40 | 340 | 16 | 24 | 7 | 62 | 5.1 | [48] |
| (NH4)3PMo12O40 | 340 | 4 | 33 | 21 | - | 2.2 | [30] |
| (NH4)3PMo12O40 | 350 | 7 | 42 | 7 | - | 3.4 | [28] |
| (NH4)3PMo12O40 | 350 | 8 | 41 | 7 | - | 3.8 | [33] |
| (NH4)3PMo12O40 | 380 | 8 | 41 | 13 | 37 | 4.3 | [49] |
| (NH4)3PMo12O40 | 350 | 6 | 43 | 12 | 36 | 3.3 | [50] |
| Cs0.5(NH4)2.5HPMo11VO40 | 340 | 4 | 41 | 29 | 17 | 2.9 | [51] |
| Cs1.15(NH4)1.85HPMo11VO40 | 340 | 6 | 45 | 15 | - | 3.6 | [30] |
| Cs1.5(NH4)1.5HPMo11VO40 | 340 | 6 | 45 | 22 | 19 | 3.9 | [51] |
| Cs1.7(NH4)1.3HPMo11VO40 | 340 | 10 | 44 | 14 | 26 | 5.5 | [51] |
| Cs1.75(NH4)1.25HPMo11VO40 | 340 | 10 | 32 | 8 | - | 4 | [30] |
| Cs2.4(NH4)0.6HPMo11VO40 | 340 | 2 | 10 | 16 | - | 0.5 | [30] |
| Cs2.5(NH4)0.5HPMo11VO40 | 340 | 10 | 19 | 10 | 53 | 2.9 | [51] |
| H4PMo11VO40 | 340 | 3 | 25 | 39 | - | 1.9 | [30] |
| H5PMo10V2O40 | 340 | 5 | 34 | 28 | 36 | 3 | [52] |
| Cs1.6H2.4P1.7Mo11V1.1O40 | 350 | 11 | 38 | 8 | 15 | 4.8 | [62] |
| (NH4)3HPMo11VO40 | 340 | 2 | 49 | 32 | - | 1.6 | [30] |
| (NH4)4PMo11VO40 | 350 | 3 | 52 | 20 | - | 2.2 | [28] |
| Cs2V0.3PMo11VO40 | 350 | 6 | 55 | 10 | 23 | 4 | [53] |
| Cs2V0.3Cu0.2PMo12O40 | 350 | 13 | 48 | 6 | 31 | 7 | [27] |
| Cs2.5Fe0.08H0.26PMo12O40 | 340 | 15 | 30 | 40 | 4.5 | [54] | |
| Cs2.5Fe0.08H1.26PMo12O40 | 340 | 14 | 35 | 11 | 53 | 6.3 | [55] |
| Cs2V0.2Fe0.2PMo12O40 | 350 | 9 | 38 | 9 | 39 | 4.2 | [27] |
| Cs2Fe0.2H0.4PMo12O40 | 340 | 7 | 24 | 17 | 50 | 2.9 | [58] |
| Cs2Fe0.2H0.4PMo12O40 | 340 | 7 | 24 | 17 | 46 | 2.9 | [61] |
| Cs2(NH4)xFe0.2PMo12O40 | 360 | 20 | 35 | 3 | 27 | 7.6 | [63] |
| K1Fe0.5(NH4)1.5PMo12O40 | 400 | 4 | 30 | 19 | 17 | 2 | [57] |
| Cs1.5Fe0.5(NH4)2PMo12O40 | 350 | 8 | 21 | 6 | - | 2.4 | [56] |
| Cs2Ni0.08H1.34PVMo11O40 | 320 | 15 | 36 | 9 | - | 6.8 | [64] |
| Cs2V0.3Ni0.2PMo12O40 | 350 | 8 | 41 | 9 | 34 | 4 | [27] |
| Cs2.5Cu0.08H1.34PVMo11O40 | 340 | 14 | - | 35 | 36 | 4.9 | [65] |
| Cs2Cu0.2H0.6PMo12O40 | 340 | 8 | 6 | 15 | 72 | 1.7 | [58] |
| Cs2V0.3Cu0.2PMo12O40 | 350 | 13 | 48 | 6 | 31 | 7 | [27] |
| Cs2V0.2Ce0.2PMo12O40 | 350 | 6 | 41 | 12 | 34 | 3.2 | [27] |
| Cs2Te0.2HxPMo12O40 | 330 | 7 | 60 | 16 | 18 | 5.3 | [66] |
| Cs2Te0.3(VO)0.1HxPMo12O40 | 360 | 16 | 71 | 3 | 19 | 11.8 | [60] |
| Te2PMo11On | 350 | 10 | 27 | 22 | 42 | 4.9 | [59] |
| Te2.25PMo9V3On | 390 | 12 | 22 | 34 | 36 | 6.7 | [67] |
| (NH4)3PMo12O40/Sb0.23Ox | 350 | 8 | 50 | 10 | 30 | 4.8 | [50] |
| Mo12V0.5P1.5As0.4Cu0.3Cs1.4Ox | 370 | 0.4 | 18 | 30 | - | 0.2 | [68] |
| Catalyst | Acidity, NH3 Uptake, mol/m2 cat (mmol/gcat) | SBET, (m2/g) | Conversion a (%) | Selectivity (mol.%) | ||||
|---|---|---|---|---|---|---|---|---|
| 130–300 °C, Weak | 300–450 °C, Medium | 450–560 °C, Strong | 560–700 °C, Very Strong | Total Acidity | IBAN | MAC + MAA | ||
| APMV/CPM | 2.9 (0.05) | 12.4 (0.21) | 52.4 (0.89) | 38.2 (0.65) | 105.9 (1.80) | 17 | 15.3 | 52.0 |
| APMV/SiO2 | 1.1 (0.17) | 0.9 (0.14) | 7.76 (1.23) | - | 9.7 (1.54) | 159 | 11.3 | 27.4 |
| APMV/ZrO2/SBA-15 | 2.1 (0.6) | 2.2 (0.62) | - | - | 4.3 (1.22) | 287 | 6.6 | 11.2 |
| APMV/SBA-15 | 0.4 (0.16) | 1.0 (0.35) | 0.9 (0.32) | 0.1 (0.03) | 2.4 (0.87) | 368 | 5.0 | 27.7 |
| Catalysts | Reaction T, °C | Conversion, % | Selectivity % | Yield, % (MAA + MAC) | Ref. | ||
|---|---|---|---|---|---|---|---|
| MAA | MAC | COx | |||||
| (NH4)3HPMo11VO40/SBA-15 | 340 | 5 | 10 | 18 | 42 | 30 | [69] |
| (NH4)3HPMo11VO40/ZrO2/SBA-15 | 340 | 7 | 2 | 10 | 64 | 0.7 | [69] |
| (NH4)3HPMo11VO40/SiO2 | 340 | 11 | 13 | 15 | 43 | 3 | [69] |
| H3PMo12O40/SiO2 | 350 | 7 | 40 | 9 | 39 | 3.4 | [71] |
| (NH4)3HPMo11VO40/Molecular sieve | 340 | 8 | 0.6 | 0.9 | 80 | 0.1 | [70] |
| (NH4)3HPMo11VO40/CeO2 | 340 | 8 | 0.3 | 0 | 99 | 0.02 | [70] |
| (NH4)3HPMo11VO40/WO3/ZrO2 | 340 | 7 | 0.9 | 4 | 85 | 0.3 | [70] |
| (NH4)3HPMo11VO40/Cs2.5H0.5PMo12O40 | 340 | 9 | 35 | 6 | 38 | 4.6 | [70] |
| (NH4)3HPMo11VO40/Cs3PMo12O40 | 340 | 11 | 43 | 5 | 42 | 5.4 | [70] |
| (NH4)3HPMo11VO40/Cs3HPMo11VO40 | 340 | 10 | 35 | 5 | 47 | 4 | [70] |
| (NH4)3HPMo11VO40/Cs4PMo11VO40 | 340 | 8 | 32 | 7 | 37 | 3.1 | [70] |
| (NH4)3HPMo11VO40/Cs3PMo12O40 | 340 | 15 | 42 | 10 | 31 | 8 | [69] |
| H4PVMo11O40/Cs3HPVMo11O40 | 340 | 5 | 42 | 17 | 32 | 3 | [72] |
| H4PVMo11O40/Cs3PMo12O40 | 340 | 11 | 24 | 7 | 54 | 3.4 | [72] |
| Catalysts | Reaction T., °C | Conversion, % | Selectivity % | Yields, % (MAA + MAC) | Ref. | ||
|---|---|---|---|---|---|---|---|
| MAA | MAC | COx | |||||
| MoV0.3 | 420 | 6.4 | 3.1 | 40.4 | 35 | 2.8 | [76] |
| MoV0.3Te0.25 | 420 | 15.6 | 3.5 | 44.2 | 25 | 7.4 | [77] |
| MoV0.3Te0.23 | 440 | 21.3 | 16 | 17 | 33 | 7.1 | [78] |
| MoV0.3Te0.25 | 420 | 20.1 | 11.7 | 12.2 | 59 | 4.8 | [79] |
| TeMoO | 380 | 20 | 3.8 | 33.5 | 26 | 7.5 | [80] |
| MoVTePO | 380 | 12.7 | 10.9 | 37 | 37 | 6.2 | [81] |
| MoV0.3Te0.23Sb0.5 | 470 | 20 | 5 | 39 | 26 | 8.8 | [82] |
| Mo12V3Sb85Ox | 470 | 4.5 | - | 30 | 69 | 1.4 | [83] |
| MoV1Sb10Ox | 440 | 8.1 | - | 17.6 | 59 | 1.4 | [84] |
| MoV0.3Te0.23Ce0.2 | 420 | 20.2 | 20 | 33 | 28 | 10.7 | [85] |
| MoV0.3Te0.23P0.3 | 440 | 9.4 | 15 | 33 | 33 | 4.5 | [86] |
| MoV0.8Te0.23Ox/SiO2 | 440 | 9.2 | - | 33.7 | 54 | 3.1 | [87] |
| MoV0.8Te0.23Ox/SBA-3 | 440 | 13.4 | - | 28 | 56 | 3.7 | [87] |
| MoV0.3Bi0.3O/AlPO4 | 540 | 9.5 | - | 45.1 | 43 | 4.3 | [88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Paul, S.; Dumeignil, F.; Katryniok, B. Selective Oxidation of Isobutane to Methacrylic Acid and Methacrolein: A Critical Review. Catalysts 2021, 11, 769. https://doi.org/10.3390/catal11070769
Zhang L, Paul S, Dumeignil F, Katryniok B. Selective Oxidation of Isobutane to Methacrylic Acid and Methacrolein: A Critical Review. Catalysts. 2021; 11(7):769. https://doi.org/10.3390/catal11070769
Chicago/Turabian StyleZhang, Li, Sébastien Paul, Franck Dumeignil, and Benjamin Katryniok. 2021. "Selective Oxidation of Isobutane to Methacrylic Acid and Methacrolein: A Critical Review" Catalysts 11, no. 7: 769. https://doi.org/10.3390/catal11070769
APA StyleZhang, L., Paul, S., Dumeignil, F., & Katryniok, B. (2021). Selective Oxidation of Isobutane to Methacrylic Acid and Methacrolein: A Critical Review. Catalysts, 11(7), 769. https://doi.org/10.3390/catal11070769