
Immobilization of Rh(I)-N-Xantphos and Fe(II)-C-Scorpionate onto Magnetic Nanoparticles: Reusable Catalytic System for Sequential Hydroformylation/Acetalization

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract

1. Introduction
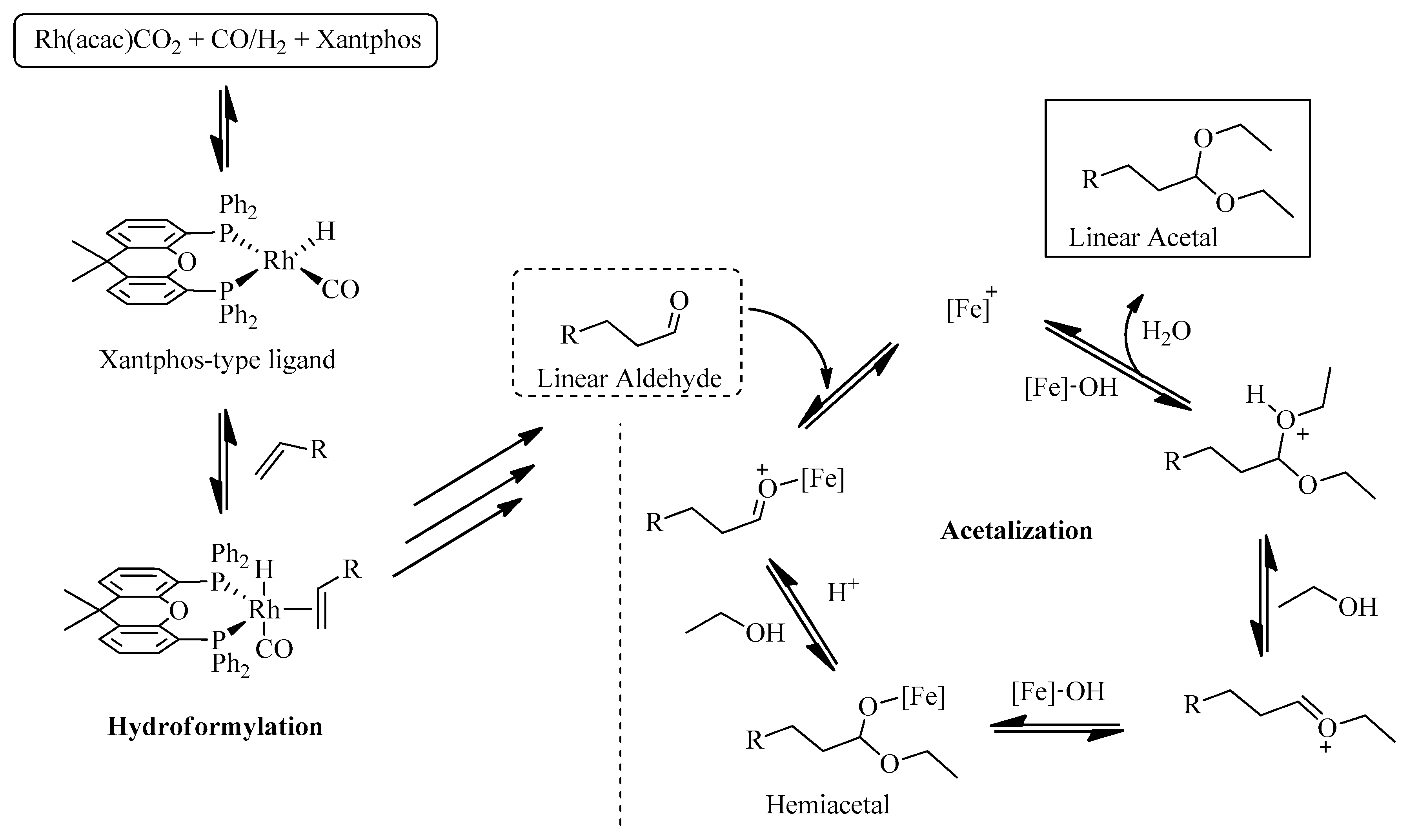
2. Results and Discussion
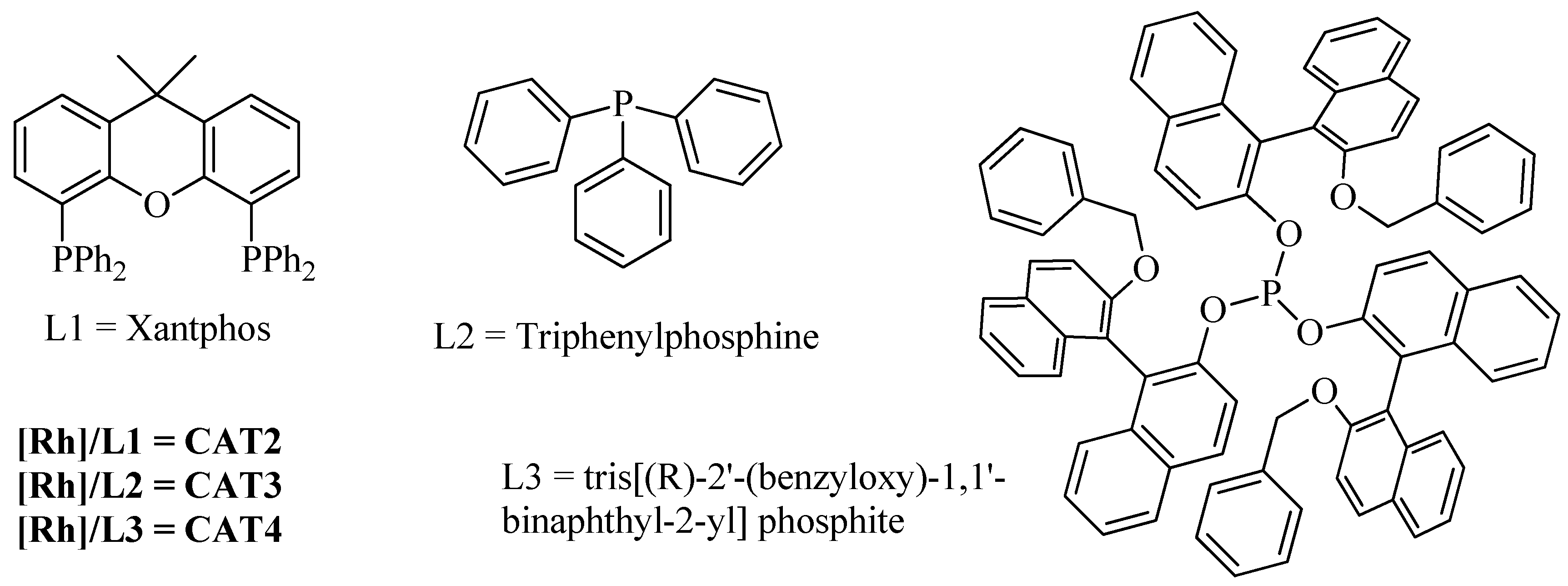
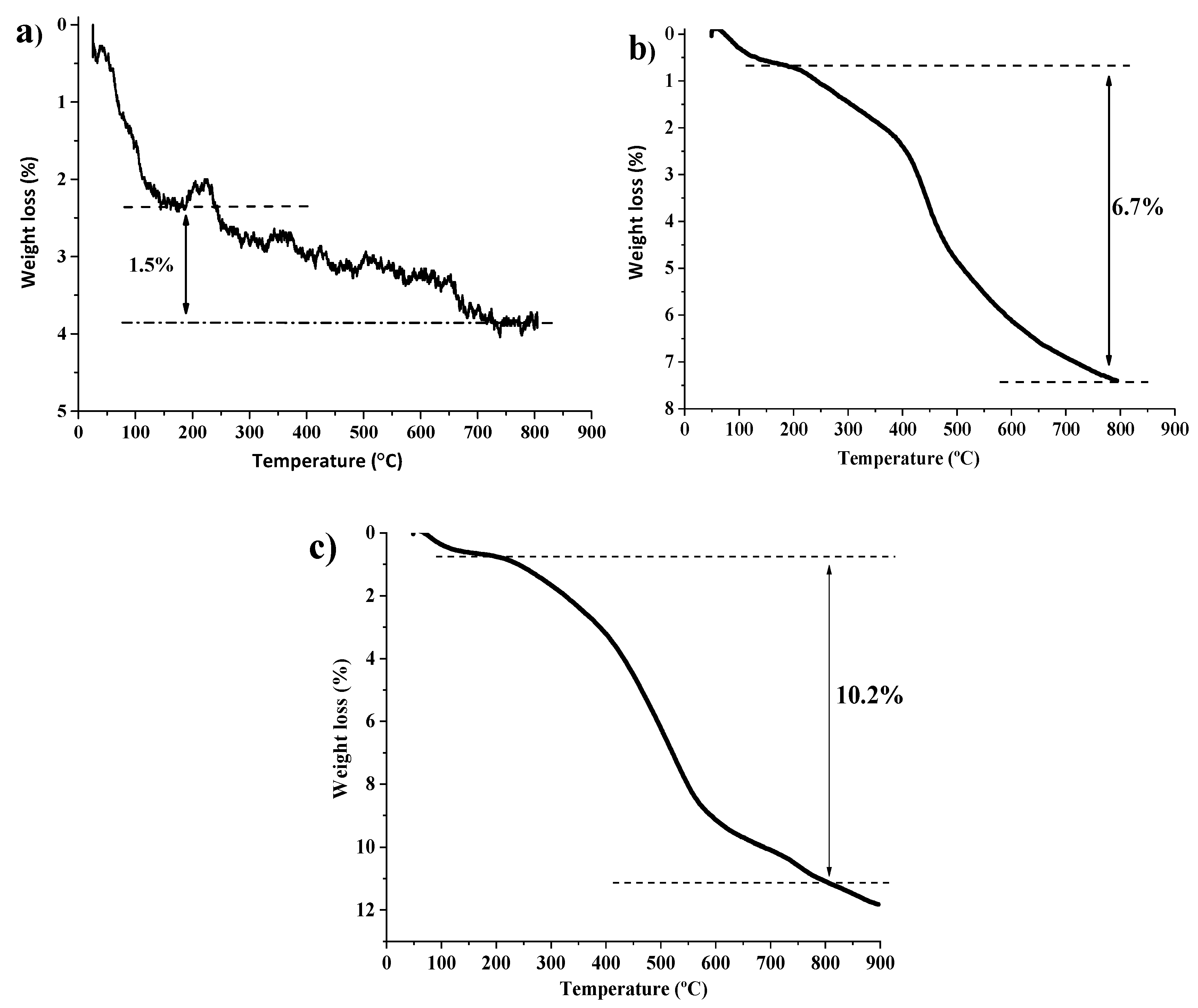
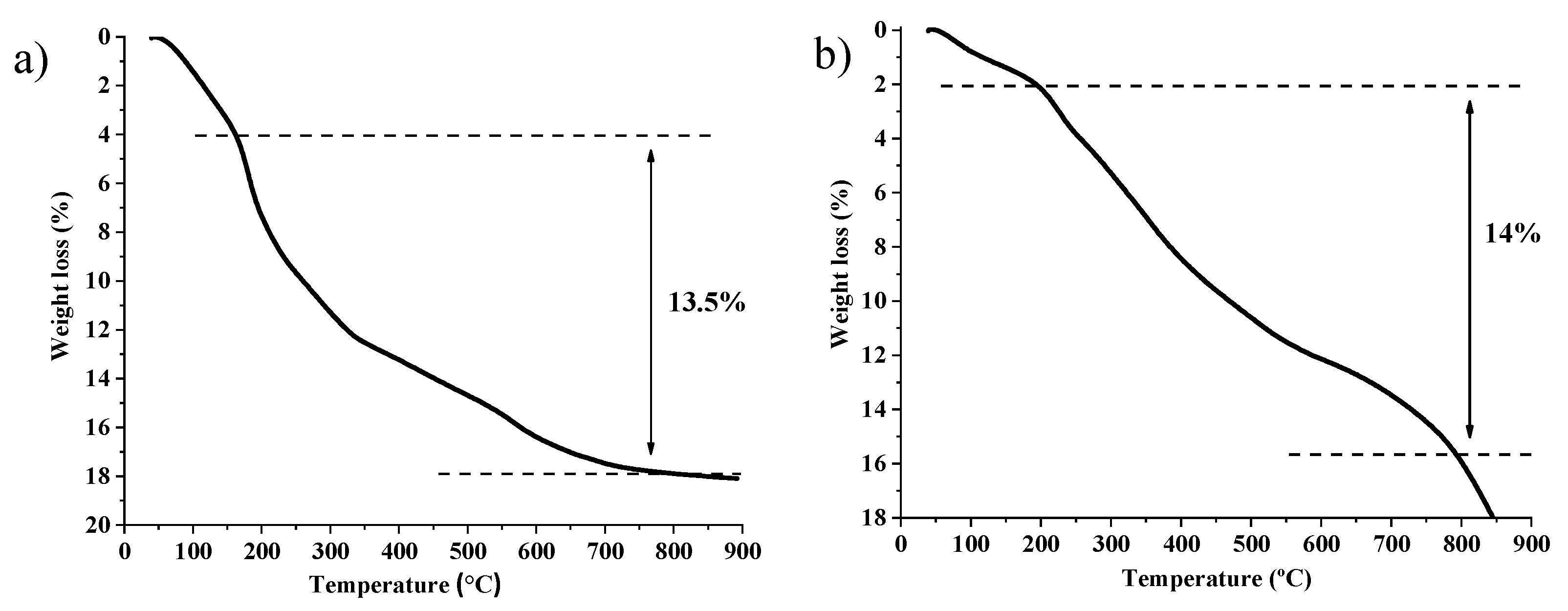
2.1. Synthesis and Characterization of Catalysts
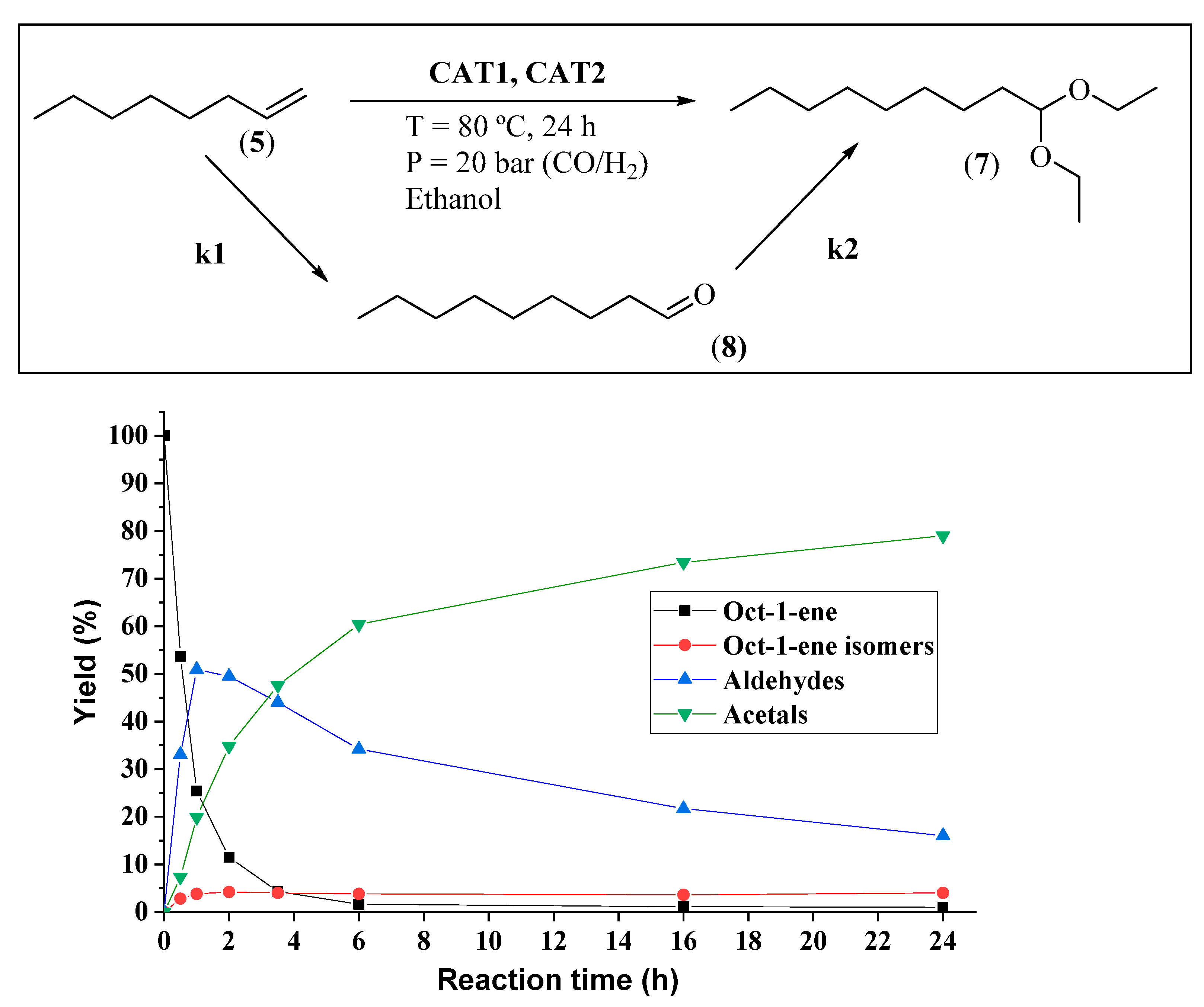
2.2. Homogeneous Hydroformylation/Acetalization of Olefins
2.3. Sequential Heterogeneous Hydroformylation/Acetalization of Olefins
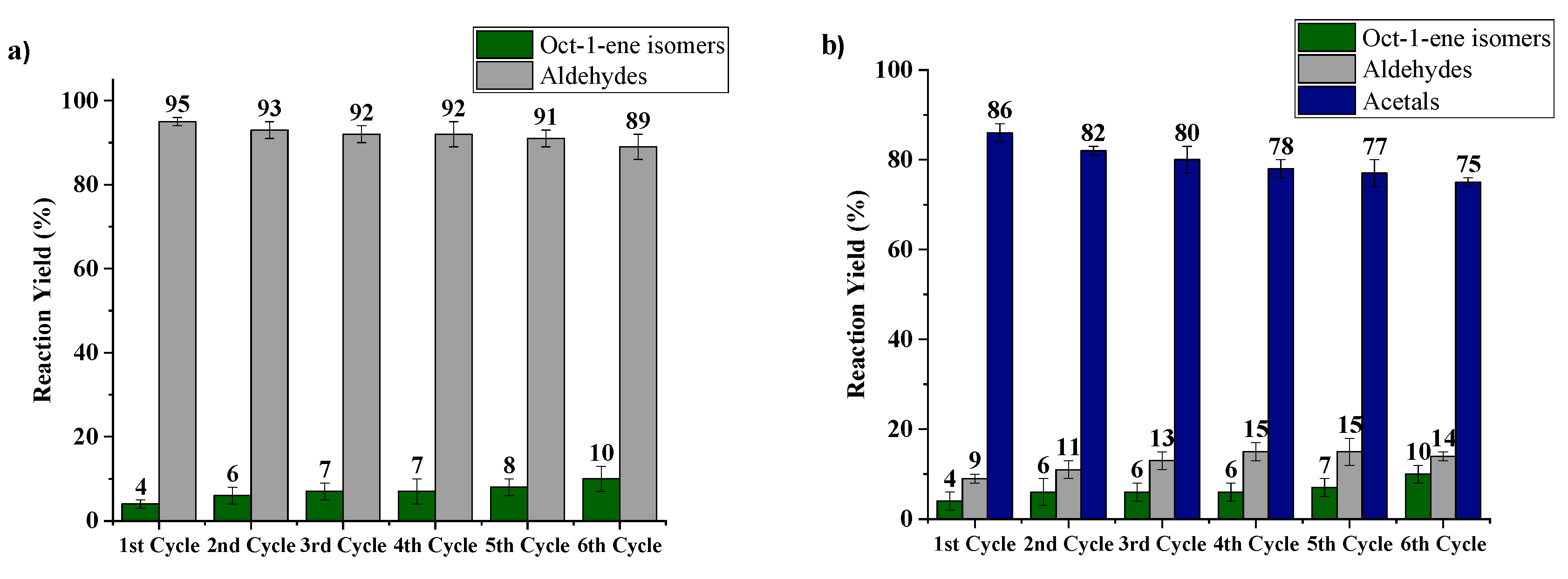
2.4. Reutilization of Heterogeneous Catalysts
3. Experimental
3.1. Chemicals and Materials
3.2. Instrumentation
3.3. Synthesis of New Catalysts
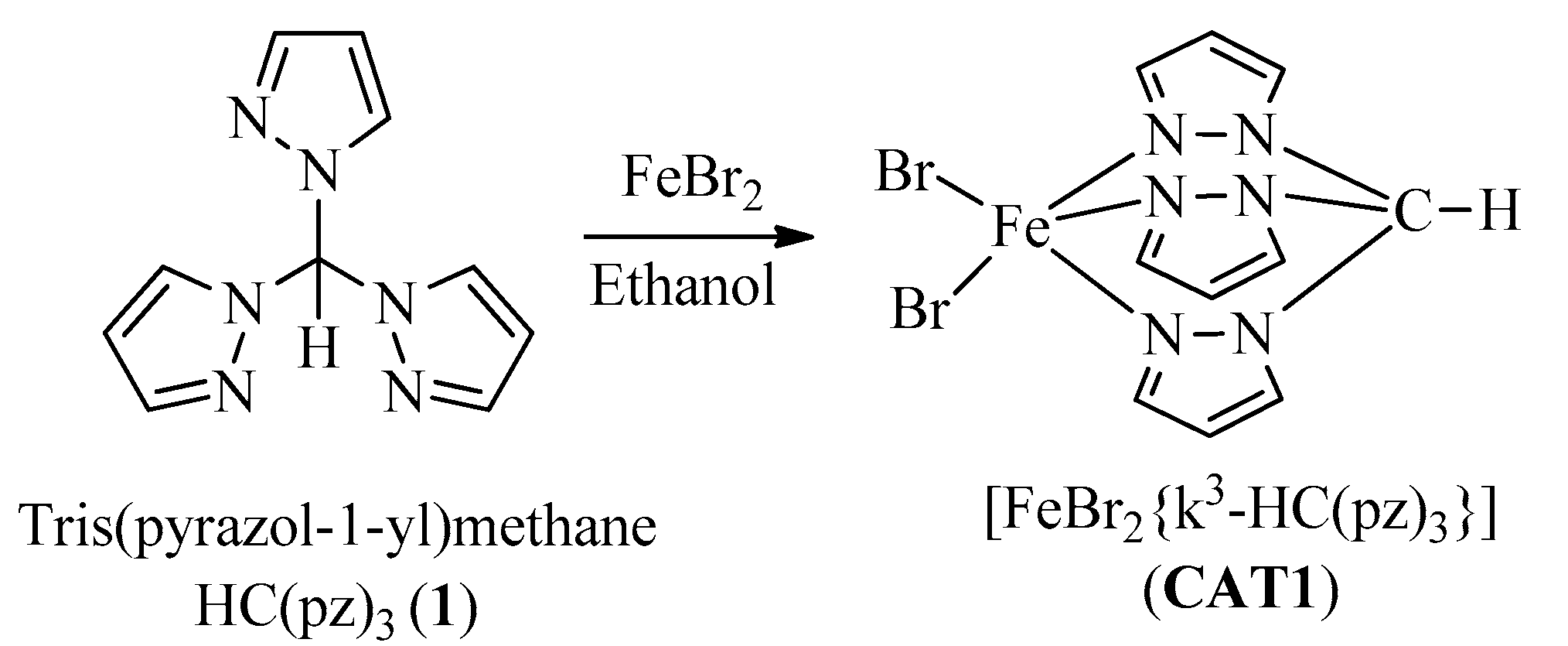
3.3.1. CAT1 Synthesis
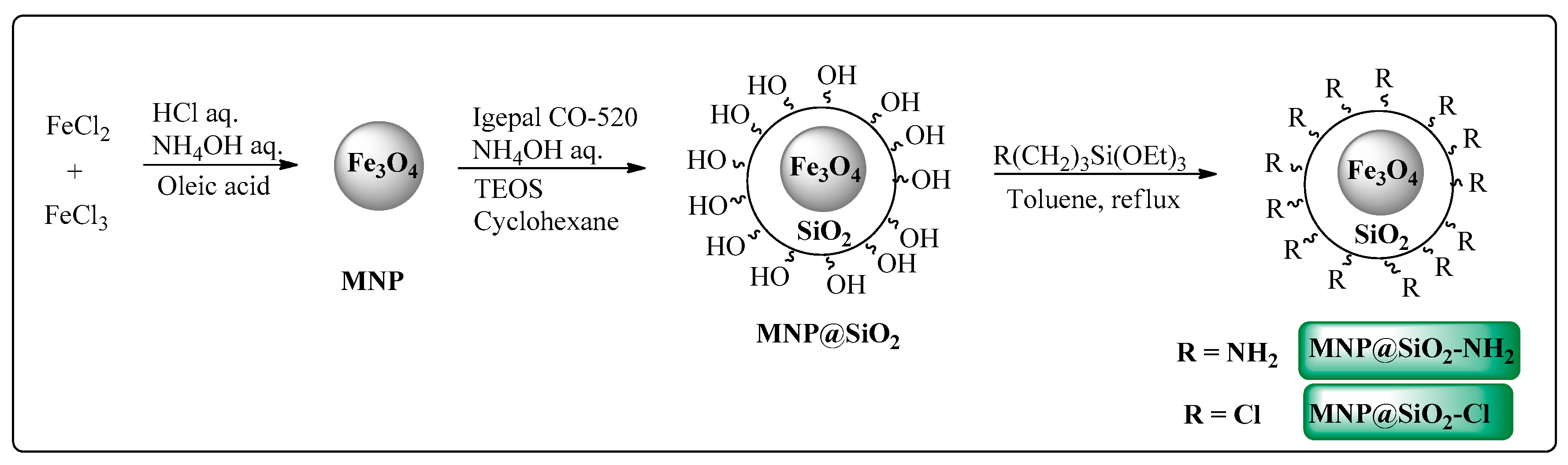
3.3.2. Magnetic Nanoparticles Synthesis and Functionalization
3.3.3. CAT5 Synthesis
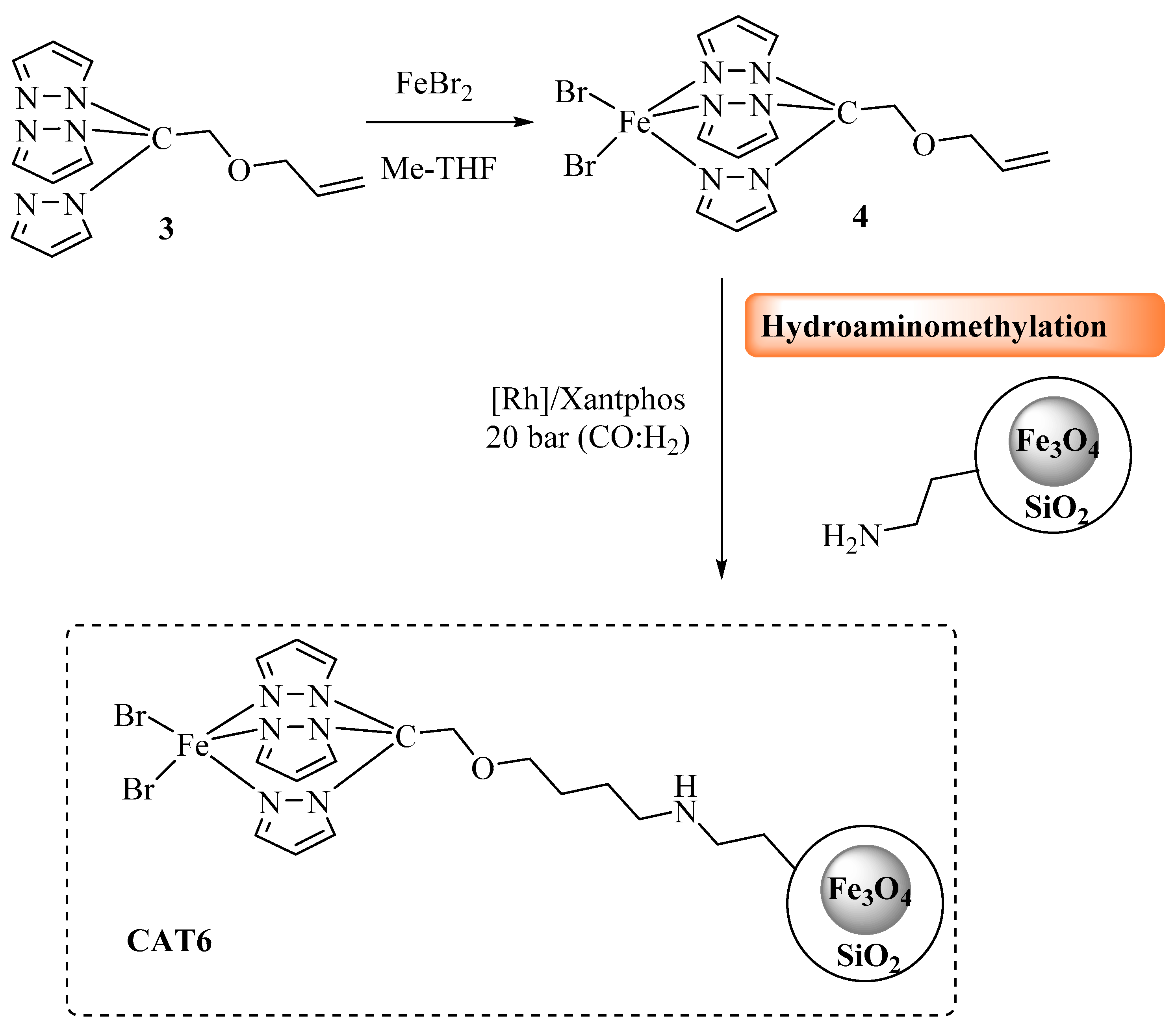
3.3.4. CAT6 Synthesis
3.4. General Methods for the Hydroformylation/Acetalization of Alkenes
3.4.1. Homogeneous Bimetallic System
3.4.2. Heterogeneous One-Pot Bimetallic System
3.4.3. Heterogeneous Sequential Process
3.4.4. Reutilization Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galván, A.; Fañanás, F.J.; Rodríguez, F. Multicomponent and multicatalytic reactions—A synthetic strategy inspired by nature. Eur. J. Inorg. Chem. 2016, 9, 1306–1313. [Google Scholar] [CrossRef]
- Lohr, T.L.; Marks, T.J. Orthogonal tandem catalysis. Nat. Chem. 2015, 7, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Hinzmann, A.; Terhorst, M.; Brauer, W.; Oike, K.; Yavuzer, H.; Asano, Y.; Vorholt, A.J.; Betke, T.; Gröger, H. Approaching bulk chemical nitriles from alkenes: A hydrogen cyanide-free approach through a combination of hydroformylation and biocatalysis. ACS Catal. 2019, 9, 5198–5203. [Google Scholar] [CrossRef]
- Bondžić, B.P. Rh catalyzed multicomponent tandem and one-pot reactions under hydroformylation conditions. J. Mol. Catal. A-Chem. 2015, 408, 310–334. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Kalck, P.; Urrutigoïty, M. Tandem hydroaminomethylation reaction to synthesize amines from alkenes. Chem. Rev. 2018, 118, 3833–3861. [Google Scholar] [CrossRef] [PubMed]
- Claver, C. Rhodium Catalysis in Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Kollár, L.; Pongrácz, P. Tandem hydroformylation/aldol condensation reactions: Synthesis of unsaturated ketones from olefins. J. Organomet. Chem. 2018, 866, 184–188. [Google Scholar] [CrossRef]
- Pongrácz, P.; Bartal, B.; Kollár, L.; Mika, L.T. Rhodium-catalyzed hydroformylation in γ-valerolactone as a biomass-derived solvent. J. Organomet. Chem. 2017, 847, 140–145. [Google Scholar] [CrossRef]
- Torres, G.M.; Frauenlob, R.; Franke, R.; Börner, A. Production of alcohols via hydroformylation. Catal. Sci. Technol. 2015, 5, 34–54. [Google Scholar] [CrossRef]
- Almeida, A.R.; Dias, R.D.; Monteiro, C.J.P.; Abreu, A.R.; Gois, P.M.P.; Bayon, J.C.; Pereira, M.M. Rhodium-catalysed tandem hydroformylation/arylation reaction with boronic acids. Adv. Synth. Catal. 2014, 356, 1223–1228. [Google Scholar] [CrossRef]
- Liu, H.; Liu, L.; Guo, W.-D.; Lu, Y.; Zhao, X.-L. Phosphine-ligated Ir(III)-complex as a bi-functional catalyst for one-pot tandem hydroformylation-acetalization. J. Catal. 2019, 373, 215–221. [Google Scholar] [CrossRef]
- Wang, P.; Wang, D.L.; Liu, H.; Zhao, X.-L.; Lu, Y.; Liu, Y. Production of alcohols from olefins via one-pot tandem hydroformylation–acetalization–hydrogenolysis over bifunctional catalyst merging RuIII–P complex and RuIII Lewis acid. Organometallics 2017, 36, 2404–2411. [Google Scholar] [CrossRef]
- Li, Y.Q.; Wang, P.; Liu, H.; Lu, Y.; Zhao, X.-L.; Liu, Y. Co-catalysis of a bi-functional ligand containingphosphine and Lewis acidic phosphonium forhydroformylation–acetalization of olefins. Green Chem. 2016, 18, 1798–1806. [Google Scholar] [CrossRef]
- Wang, P.; Liu, H.; Li, Y.-Q.; Zhao, X.-L.; Lu, Y.; Liu, Y. Phosphonium-based aminophosphines as bifunctional ligands for sequential catalysis of one-pot hydroformylation–acetalization of olefins. Catal. Sci. Technol. 2016, 6, 3854–3861. [Google Scholar] [CrossRef]
- Vieira, C.G.; da Silva, J.G.; Penna, C.A.A.; dos Santos, E.N.; Gusevskaya, E.V. Synthesis of fragrance compounds from renewable resources: The aqueous biphasic hydroformylation of acyclic terpenes. Appl. Catal. A 2010, 380, 125–132. [Google Scholar] [CrossRef]
- Freixa, Z.; Pereira, M.M.; Pais, A.A.C.C.; Bayón, J.C. Evidence of a rhodium catalytic species containing a bridging 1,2-diphosphine in styrene hydroformylation. J. Chem. Soc. Dalton Trans. 1999, 18, 3245–3251. [Google Scholar] [CrossRef]
- de Freitas, M.C.; Vieira, C.G.; dos Santos, E.N.; Gusevskaya, E.V. Synthesis of fragrance compounds from biorenewables: Tandem hydroformylation–acetalization of bicyclic monoterpenes. ChemCatChem 2013, 5, 1884–1890. [Google Scholar] [CrossRef]
- Vieira, C.G.; dos Santos, E.N.; Gusevskaya, E.V. Synthesis of fragrance compounds from acyclic monoterpenes: Rhodium catalyzed hydroformylation and tandem hydroformylation/acetalization of linalool and citronellene. Appl. Catal. A 2013, 466, 208–215. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, P.K. Recent developments in the synthesis of supported catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef] [PubMed]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes and Applications in Organic Synthesis; VCH: Weinheim, Germany, 2016. [Google Scholar]
- Rodrigues, F.M.S.; Carrilho, R.M.B.; Pereira, M.M. Reusable catalysts for hydroformylation based reactions. Eur. J. Inorg. Chem. 2021. [Google Scholar] [CrossRef]
- Hanf, S.; Rupflin, L.A.; Gläser, R.; Schunk, S.A. Current state of the art of the solid Rh-based catalyzed hydroformylation of short-chain olefins. Catalysts 2020, 10, 510. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Yan, L.; Ding, Y. A mini review on strategies for heterogenization of rhodium-based hydroformylation catalysts. Front. Chem. Sci. Eng. 2018, 12, 113–123. [Google Scholar] [CrossRef]
- Molnár, Á.; Papp, A. Catalyst recycling - A survey of recent progress and current status. Coord. Chem. Rev. 2017, 349, 1–65. [Google Scholar] [CrossRef]
- Neves, A.C.B.; Calvete, M.J.F.; Pinho e Melo, T.M.V.D.; Pereira, M.M. Immobilized catalysts for hydroformylation reactions: A versatile tool for aldehyde synthesis. Eur. J. Org. Chem. 2012, 32, 6309–6320. [Google Scholar] [CrossRef]
- Sandee, A.J.; Reek, J.N.H.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. A silica-supported, switchable, and recyclable hydroformylation−hydrogenation catalyst. J. Am. Chem. Soc. 2001, 123, 8468–8476. [Google Scholar] [CrossRef]
- Ricken, S.; Osinski, P.W.; Eilbracht, P.; Haag, R. A new approach to dendritic supported NIXANTPHOS-based hydroformylation catalysts. J. Mol. Catal. A Chem. 2006, 257, 78–88. [Google Scholar] [CrossRef]
- Joumaa, A.; Gayet, F.; Garcia-Suarez, E.J.; Himmelstrup, J.; Riisager, A.; Poli, R.; Manoury, E. Synthesis of Nixantphos core-functionalized amphiphilic nanoreactors and application to rhodium-catalyzed aqueous biphasic 1-octene hydroformylation. Polymers 2020, 12, 1107. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Luque, R.; Fihri, A.; Zhu, H.; Bouhrara, M.; Bassett, J.-M. Magnetically recoverable nanocatalysts. Chem. Rev. 2011, 111, 3036–3075. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.N.; Bououdina, M.; Jimoh, A.A.; Aziz, M.A.; Helal, A.; Hakeem, A.S.; Yamania, Z.H.; Kimc, T.-J. The rhodium complex of bis(diphenylphosphinomethyl) dopamine-coated magnetic nanoparticles as an efficient and reusable catalyst for hydroformylation of olefins. N. J. Chem. 2015, 39, 7293–7299. [Google Scholar] [CrossRef]
- Wang, P.; Chen, X.; Wang, D.-L.; Li, Y.-Q.; Liu, Y. Efficient and recyclable Rh-catalytic system with involvement of phosphine-functionalized phosphonium-based ionic liquids for tandem hydroformylation–acetalization. Green Energy Environ. 2017, 2, 419–427. [Google Scholar] [CrossRef]
- Jin, X.; Zhao, K.; Cui, F.; Kong, F.; Liu, Q. Highly effective tandem hydroformylation–acetalization of olefins using a long-life Brønsted acid–Rh bifunctional catalyst in ionic liquid–alcohol systems. Green Chem. 2013, 15, 3236–3242. [Google Scholar] [CrossRef]
- Norinder, J.; Rodrigues, C.; Börner, A. Tandem hydroformylation–acetalization with a ruthenium catalyst immobilized in ionic liquids. J. Mol. Catal. A Chem. 2014, 391, 139–143. [Google Scholar] [CrossRef]
- Gorbunov, D.N.; Nenasheva, M.V.; Sinikova, N.A.; Kardasheva, Y.S.; Maksimov, A.L.; Karakhanov, E.A. Tandem Hydroformylation–acetalization using a water-soluble catalytic system: A promising procedure for preparing valuable oxygen-containing compounds from olefins and polyols. Russ. J. Appl. Chem. 2018, 91, 990–995. [Google Scholar] [CrossRef]
- Gorbunov, D.N.; Egazaryants, S.V.; Kardasheva, Y.S.; Maksimov, A.L.; Karakhanov, E.A. Synthesis of cyclic acetals by hydroformylation of oct-1-ene in the presence of polyols. Russ. Chem. Bull. 2015, 64, 943–947. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S. C-scorpionate complexes: Ever young catalytic tools. Coord. Chem. Rev. 2019, 396, 89–102. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-soluble C-scorpionate complexes – catalytic and biological applications. Eur. J. Inorg. Chem. 2016, 15–16, 2236–2252. [Google Scholar] [CrossRef]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Group 11 tris(pyrazolyl)methane complexes: Structural features and catalytic application. Dalton Trans. 2019, 48, 10772–10781. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.G.M.; Mac Leod, T.C.O.; Guedes da Silva, M.F.C.; Luzyanin, K.V.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. NiII, CuII and ZnII complexes with a sterically hindered scorpionate ligand (TpmsPh) and catalytic application in the diasteroselective nitroaldol (Henry) reaction. Dalton Trans. 2014, 43, 15192–15200. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.A.S.; Ribeiro, A.P.C.; Ferraria, A.M.; Botelho do Rego, A.M.; Martins, L.M.D.R.S. Catalytic Performance of a Magnetic Core-Shell Iron(II) C-Scorpionate under Unconventional Oxidation Conditions. Nanomaterials 2020, 10, 2111. [Google Scholar] [CrossRef]
- Matias, I.A.S.; Ribeiro, A.P.C.; Martins, L.M.D.R.S. Selective Oxidation of Ethane to Acetic Acid Catalyzed by a C-Scorpionate Iron(II) Complex: A Homogeneous vs. Heterogeneous Comparison. Molecules 2020, 25, 5642. [Google Scholar] [CrossRef]
- Ottaviania, D.; Van-Dúnemb, V.; Carvalho, A.P.; Martins, A.; Martins, L.M.D.R.S. Eco-friendly cyclohexane oxidation by a V-scorpionate complex immobilized at hierarchical MOR zeolite. Catal. Today 2020, 348, 37–44. [Google Scholar] [CrossRef]
- Ribeiro, A.P.C.; Martins, L.M.D.R.S.; Carabineiro, S.A.C.; Buijnsters, J.G.; Figueiredo, J.L.; Pombeiro, A.J.L. Heterogenized C-Scorpionate Iron(II) Complex on Nanostructured Carbon Materials as Recyclable Catalysts for Microwave-Assisted Oxidation Reactions. ChemCatChem 2018, 10, 1821–1828. [Google Scholar] [CrossRef]
- Ribeiro, A.P.C.; Martins, L.M.D.R.S.; Kuznetsov, M.L.; Pombeiro, A.J.L. Tuning Cyclohexane Oxidation: Combination of Microwave Irradiation and Ionic Liquid with the C-Scorpionate [FeCl2(Tpm)] Catalyst. Organometallics 2017, 36, 192–198. [Google Scholar] [CrossRef]
- Rodrigues, F.M.S.; Calvete, M.J.F.; Monteiro, C.J.P.; Carabineiro, S.A.C.; Maria, T.M.R.; Figueiredo, J.L.; Pereira, M.M. Hydroaminomethylation reaction as powerful tool for preparation of rhodium/phosphine-functionalized nanomaterials. Catalytic evaluation in styrene hydroformylation. Catal. Today 2020, 356, 456–463. [Google Scholar]
- Reger, D.L.; Gratan, T.C.; Brown, K.J.; Little, C.A.; Lamba, J.J.S.; Rheingold, A.L.; Sommer, R.D. Syntheses of tris(pyrazolyl)methane ligands and {[tris(pyrazolyl)methane]Mn(CO)3}SO3CF3 complexes: Comparison of ligand donor properties. J. Organomet. Chem. 2000, 607, 120–128. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Half-Sandwich Scorpionate Vanadium, Iron and Copper Complexes: Synthesis and Application in the Catalytic Peroxidative Oxidation of Cyclohexane under Mild Conditions. Adv. Synth. Catal. 2008, 350, 706–716. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Abreu, A.R.; Petöcz, G.; Bayón, J.C.; Moreno, M.J.S.M.; Kollár, L.; Pereira, M.M. New Binaphthyl-based C3-symmetric Chiral Hemilabile Monophosphite Ligands: Synthesis and Characterization of Their Platinum Complexes. Chem. Lett. 2009, 38, 844–845. [Google Scholar] [CrossRef]
- Rossi, L.M.; Vono, L.L.R.; Silva, F.P.; Kiyohara, P.K.; Duarte, E.L.; Matos, J.R. A magnetically recoverable scavenger for palladium based on thiol-modified magnetite nanoparticles. Appl. Catal. A 2007, 330, 139–144. [Google Scholar] [CrossRef]
- Jacinto, M.J.; Kiyohara, P.K.; Masunaga, S.H.; Jardim, R.F.; Rossi, L.M. Recoverable rhodium nanoparticles: Synthesis, characterization and catalytic performance in hydrogenation reactions. Appl. Catal. A 2008, 338, 52–57. [Google Scholar] [CrossRef]
- Dias, L.D.; Carrilho, R.M.B.; Henriques, C.A.; Calvete, M.J.F.; Masdeu-Bulto-Claver, A.M.C.; Rossi, L.M.; Pereira, M.M. Hybrid metalloporphyrin magnetic nanoparticlesas catalysts for sequential transformation of alkenes and CO2 into cyclic carbonates. ChemCatChem 2018, 10, 2792–2803. [Google Scholar] [CrossRef]
- Skalická, V.; Rybáčková, M.; Skalický, M.; Kvíčalová, M.; Cvačka, J.; Březinová, A.; Čejka, J.; Kvíčala, J. Polyfluoroalkylated tripyrazolylmethane ligands: Synthesis and complexes. J. Fluor. Chem. 2011, 132, 434–440. [Google Scholar] [CrossRef]
- Pace, V.; Hoyos, P.; Castoldi, L.; de Maria, P.D.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A biomass-derived solvent with broad application in organic chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J. Featuring Xantphos. Catal. Sci. Technol. 2018, 8, 26–113. [Google Scholar] [CrossRef]
- Kranenburg, M.; van der Burgt, Y.E.M.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. New diphosphine ligands based on heterocyclic aromatics inducing very high regioselectivity in rhodium-catalyzed hydroformylation: Effect of the bite angle. Organometallics 1995, 14, 3081–3089. [Google Scholar] [CrossRef]
- van Rooy, A.; Orij, E.N.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Hydroformylation with a rhodium/bulky Phosphite modified catalyst. A comparison of the catalyst behavior for oct-1-ene, cyclohexene, and styrene. Organometallics 1995, 14, 34–43. [Google Scholar] [CrossRef]
- van Rooy, A.; de Bruijn, J.N.H.; Roobeek, K.F.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M. Rhodium catalysed hydroformylation of branched 1-alkenes; bulky phosphites vs triphenylphosphine as modifying ligand. J. Organomet. Chem. 1996, 507, 69–73. [Google Scholar] [CrossRef]
- Rodrigues, F.M.S.; Kucmierczyk, P.K.; Pineiro, M.; Jackstell, R.; Franke, R.; Pereira, M.M.; Beller, M. Dual Rh-Ru catalysts for reductive hydroformylation of olefins to alcohols. ChemSusChem 2018, 11, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.N.; Carrilho, R.M.B.; Dias, L.D.; Viana, J.C.; Aquino, G.L.B.; Pineiro, M.; Pereira, M.M. Highly efficient Rh(I)/tris-binaphthyl monophosphite catalysts for hydroformylation of sterically hindered alkyl olefins. J. Mol. Catal. A 2016, 416, 73–80. [Google Scholar] [CrossRef]
- Kamer, P.C.; Van Leeuwen, J.P.W.N.M.; Reek, J.N.H. Wide bite angle diphosphines: Xantphos ligands in transition metal complexes and catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Heck, R.F. Mechanism of arylation and carbomethoxylation of olefins with organopalladium compounds. Acc. Chem. Res. 1969, 2, 10–16. [Google Scholar] [CrossRef]
- van der Veen, L.A.; Boele, M.D.K.; Bregman, F.R.; Kamer, P.C.J.; van Leeuwen, P.W.; Goubitz, K.; Fraanje, J.; Schenck, H.; Bo, C. Electronic effect on rhodium diphosphine catalyzed hydroformylation: The bite angle effect reconsidered. J. Am. Chem. Soc. 1998, 120, 11616–11626. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Neves, A.C.B.; Lourenço, M.A.O.; Abreu, A.R.; Rosado, M.T.S.; Abreu, P.E.; Eusébio, M.E.S.; Kollar, L.; Bayón, J.C.; Pereira, M.M. Rhodium/tris-binaphthyl chiral monophosphite complexes: Efficient catalysts for the hydroformylation of disubstituted aryl olefins. J. Organomet. Chem. 2012, 698, 28–34. [Google Scholar] [CrossRef]
- Almeida, A.R.; Carrilho, R.M.B.; Peixoto, A.F.; Abreu, A.R.; Silva, A.; Pereira, M.M. Sequential reactions from catalytic hydroformylation toward the synthesis of amino compounds. Tetrahedron 2017, 73, 2389–2395. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry. | Catalytic System | Conversion (%) [b] | Selectivity (%) [b] (l:b) | ||
|---|---|---|---|---|---|
| 6 | 7 | 8 | |||
| 1 | [Rh(acac)(CO)2] | 93 | 60 | 21 (38:62) | 19 (23:77) |
| 2 | [Rh(acac)(CO)2] HC(pz)3 (1) | 95 | 46 | 34 (44:56) | 20 (18:82) |
| 3 | [Rh(acac)(CO)2] CAT1 | 99 | 36 | 47 (61:39) | 17 (27:73) |
| 4 | CAT2 CAT1 | 99 | 3 | 82 (93:7) | 15 (80:20) |
| 5 | CAT3 CAT1 | 97 | 1 | 87 (66:34) | 12 (24:76) |
| 6 | CAT4 CAT1 | 99 | - | 91 (69:31) | 9 (32:68) |
| 7 | CAT1 | 1 | - | - | - |
| 8 [c] | CAT1 | 95 | - | 100 | - |
| Entry. | Olefin | Reaction Yield (%) [a] | |
|---|---|---|---|
| Major Product (Isolated Yield) | Minor Products | ||
| 1 [b] |  |  | Oct-1-ene isomers: 4 Aldehydes: 11 Branched acetal: 6 |
| 2 [c] |  |  | Aldehydes: 20 |
| 3 [c] |  |  | Difunctionalized acetal (isomers): 33 |

| Entry | Substrate | Catalyst | Conversion (%) [b] | Selectivity (%) [b] (l:b) | ||
|---|---|---|---|---|---|---|
| 6 | 7 | 8 | ||||
| 1 [c] | Oct-1-ene | CAT5 | 99 | 3 | - | 96(98:2) |
| 2 [d] | Nonanal | CAT6 | 90 | - | 90(100:0) | 10 |
| 3 [c],[d] | Oct-1-ene | CAT5 CAT6 | 92 | 54 | - | 38(75:25) |
| 4 [e] | Nonanal | CAT6 | 90 | 2 | 87(99:1) | 10(98:2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, F.M.S.; Dias, L.D.; Calvete, M.J.F.; Maria, T.M.R.; Rossi, L.M.; Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Pereira, M.M. Immobilization of Rh(I)-N-Xantphos and Fe(II)-C-Scorpionate onto Magnetic Nanoparticles: Reusable Catalytic System for Sequential Hydroformylation/Acetalization. Catalysts 2021, 11, 608. https://doi.org/10.3390/catal11050608
Rodrigues FMS, Dias LD, Calvete MJF, Maria TMR, Rossi LM, Pombeiro AJL, Martins LMDRS, Pereira MM. Immobilization of Rh(I)-N-Xantphos and Fe(II)-C-Scorpionate onto Magnetic Nanoparticles: Reusable Catalytic System for Sequential Hydroformylation/Acetalization. Catalysts. 2021; 11(5):608. https://doi.org/10.3390/catal11050608
Chicago/Turabian StyleRodrigues, Fábio M. S., Lucas D. Dias, Mário J. F. Calvete, Teresa M. R. Maria, Liane M. Rossi, Armando J. L. Pombeiro, Luísa M. D. R. S. Martins, and Mariette M. Pereira. 2021. "Immobilization of Rh(I)-N-Xantphos and Fe(II)-C-Scorpionate onto Magnetic Nanoparticles: Reusable Catalytic System for Sequential Hydroformylation/Acetalization" Catalysts 11, no. 5: 608. https://doi.org/10.3390/catal11050608
APA StyleRodrigues, F. M. S., Dias, L. D., Calvete, M. J. F., Maria, T. M. R., Rossi, L. M., Pombeiro, A. J. L., Martins, L. M. D. R. S., & Pereira, M. M. (2021). Immobilization of Rh(I)-N-Xantphos and Fe(II)-C-Scorpionate onto Magnetic Nanoparticles: Reusable Catalytic System for Sequential Hydroformylation/Acetalization. Catalysts, 11(5), 608. https://doi.org/10.3390/catal11050608