
Photocatalytic Oxygenation of Heterostilbenes—Batch versus Microflow Reactor

 ,
,  ,
,  , and
, and
Abstract

1. Introduction
2. Results and Discussion
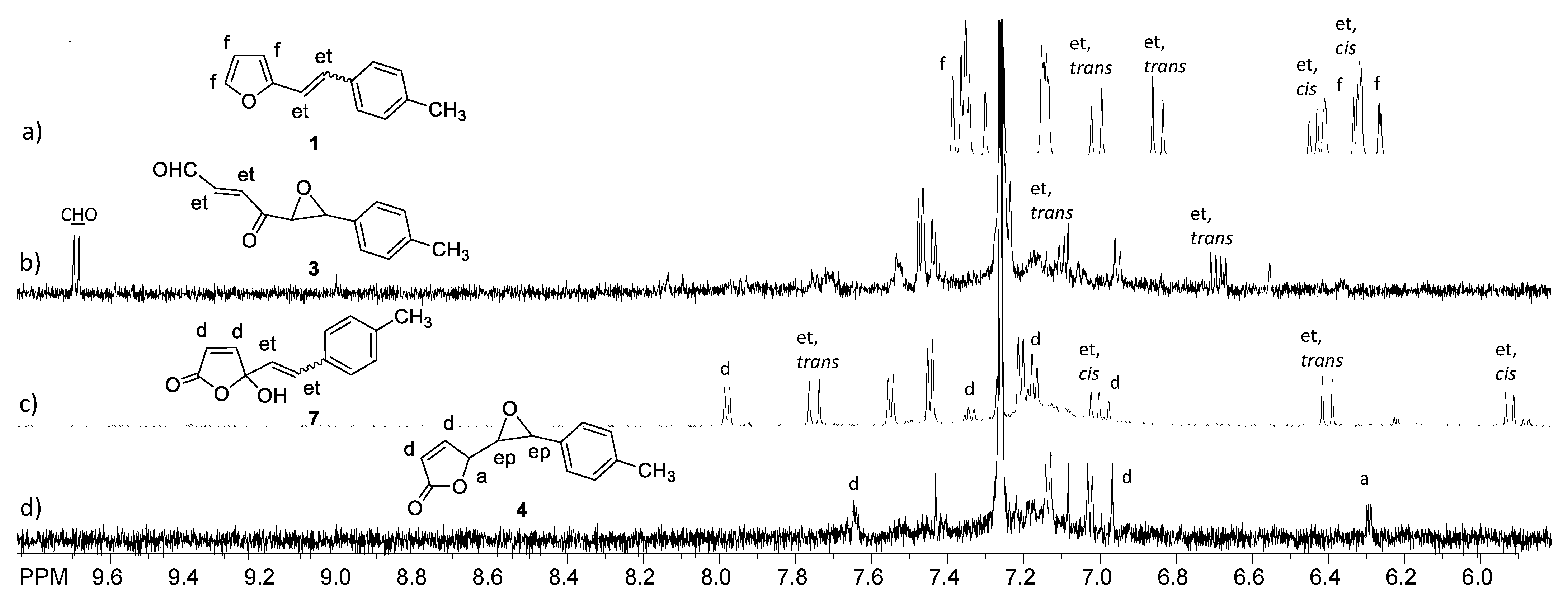
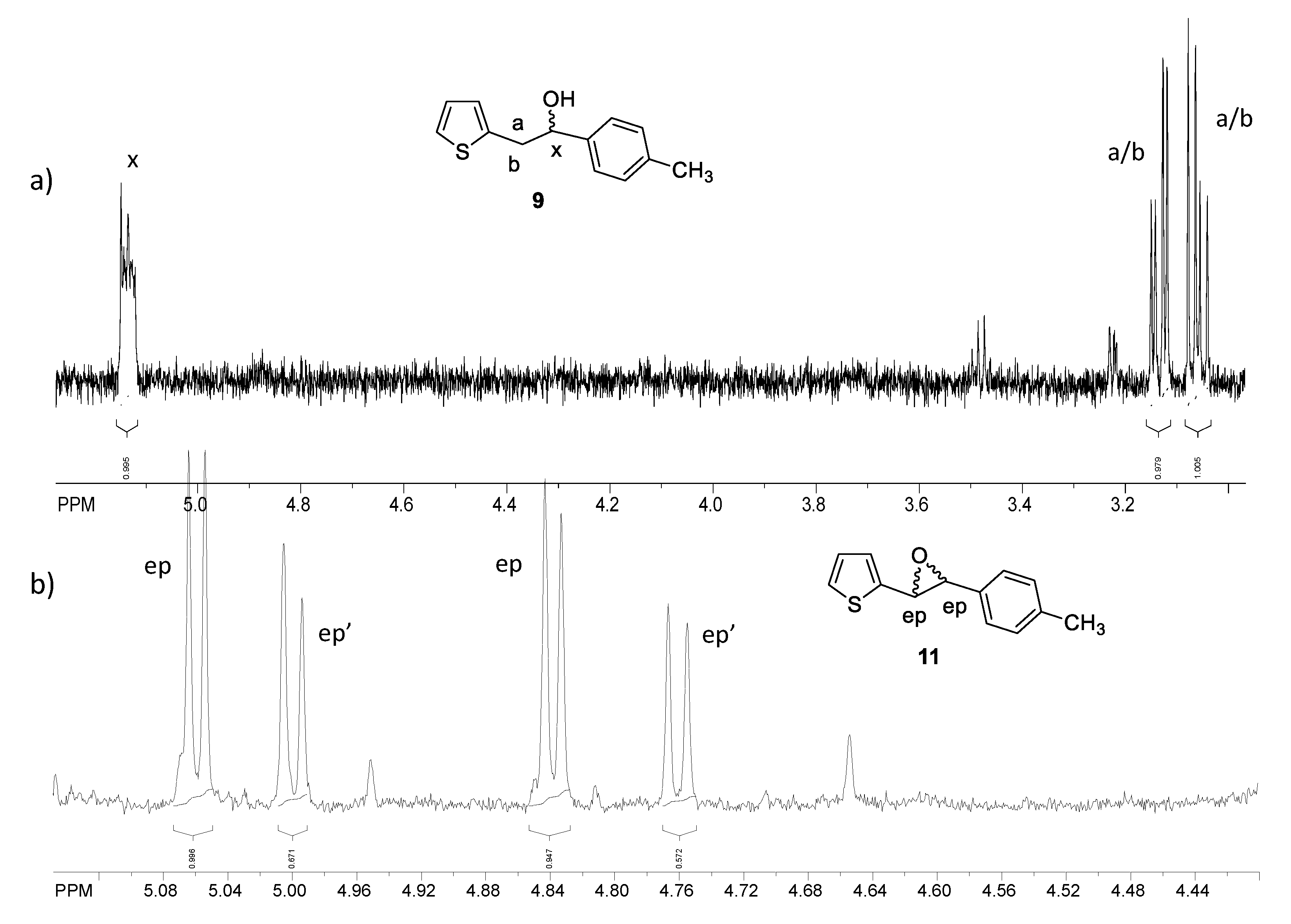
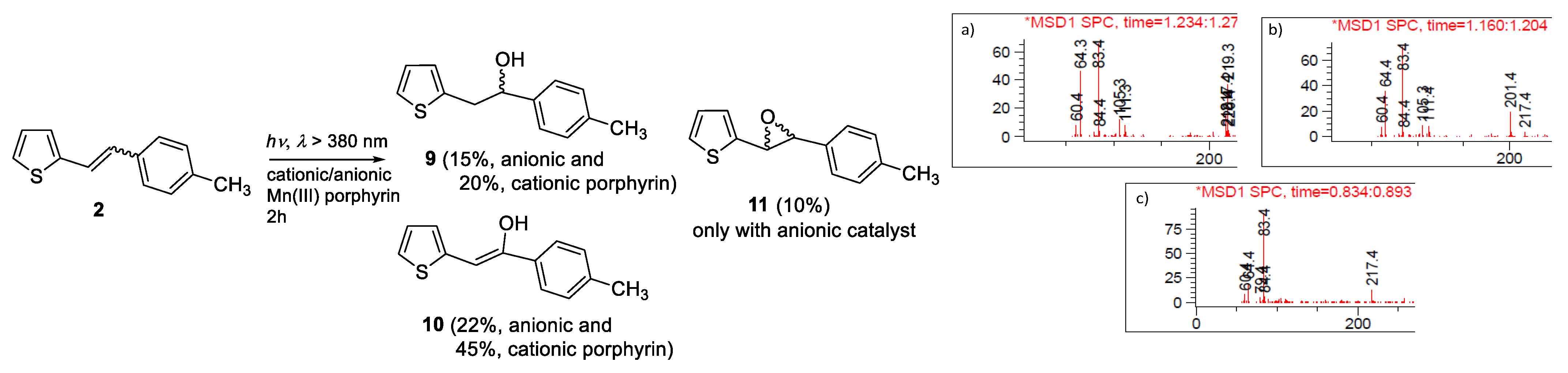
2.1. Synthesis and Identification of Photooxygenation Products
2.2. Kinetics
2.3. Synthesis in Microflow Reactors
2.4. Calculations
3. Materials and Methods
3.1. General
3.2. Typical Experimental Procedure for Photocatalytic Oxygenation in Batch Reactor
3.3. Experimental Procedure for Photocatalytic Oxygenation in Microflow Reactors
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, D.; Li, Z.; Qi, Z.; Hu, S.; Long, R.; Song, L.; Xiong, Y. Boosting photocatalytic activity in cross-coupling reactions by constructing Pd-oxide heterostructures. ChemNanoMat 2020, 6, 920–924. [Google Scholar] [CrossRef]
- Huh, S.; Kim, S.-J.; Kim, Y. Porphyrinic metal-organic frameworks from custum-designed porphyrins. CrystEngComm 2016, 18, 345–368. [Google Scholar] [CrossRef]
- Petrosyan, A.; Hauptmann, R.; Pospech, J. Heteroarene N-oxides as oxygen source in organic reactions. Eur. J. Org. Chem. 2018, 38, 5237–5252. [Google Scholar] [CrossRef]
- De Paula, R.; Simões, M.-M.-Q.; Neves, M.-G.-P.-M.-S.; Cavaleiro, J.-A.-S. Oxidation of styrene and of some derivatives with H2O2 catalyzed by novel imidazolium-containing manganese porphyrins: A mechanistic and therodynamic interpretation. J. Mol. Catal. A Chem. 2011, 345, 1–11. [Google Scholar] [CrossRef]
- Zhao, X.; Li, B.; Xia, W. Visible-light-promoted photocatalyst-free hydroacylation and diacylation of alkanes turned by NiCl2·DME. Org. Lett. 2020, 22, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Duke, K.; Scaiano, J.-C.; Lanterna, A.-E. Cobalt-molybdenum co-catalyst for heterogeneous photocatalytic H-mediated transformations. J. Catal. 2019, 379, 33–38. [Google Scholar] [CrossRef]
- Firoozi, S.; Hosseini-Sarvari, M. Photo-difunctionalization and photo-oxidative cleavage of the C–C double bond of styrenes in the presence of nanosized Cadmium sulfide (CdS) as a highly efficient photo-induced reusable nanocatalyst. Eur. J. Org. Chem. 2020, 25, 3834–3843. [Google Scholar] [CrossRef]
- Bhadra, M.; Kandambeth, S.; Sahoo, M.-K.; Adddicoat, M.; Balaraman, E.; Banerjee, R. Triazine functionalized porous covalent organic framework for photo-organocatalytic E-Z isomerization of olefins. J. Am. Chem. Soc. 2019, 141, 6152–6156. [Google Scholar] [CrossRef] [PubMed]
- Othong, J.; Boonmak, J.; Ha, J.; Leelasubcharoen, S.; Youngme, S. Thermally induced single-crystal-to-single-crystal transformation and heterogeneous catalysts for epoxidation reaction of Co(II) based metal−organic frameworks containing 1,4-phenylenediacetic acid. Cryst. Growth Des. 2017, 17, 1824–1835. [Google Scholar] [CrossRef]
- Webb, D.; Jamison, T.-F. Continuous flow multi-step organic synthesis. Chem. Sci. 2010, 1, 675–680. [Google Scholar] [CrossRef]
- Pastre, J.-C.; Browne, D.-L.; Ley, S.-V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef]
- Landgraf, S. Application of semiconductor light sources for investigations of photochemical reactions. Spectrochim. Acta Part A 2001, 57, 2029–2048. [Google Scholar] [CrossRef]
- Meščić, A.; Šalić, A.; Gregorić, T.; Zelić, B.; Raić-Malić, S. Continuous flow-ultrasonic synergy in click reactions for the synthesis of novel 1,2,3-triazolyl appended 4,5-unsaturated L-ascorbic acid derivatives. RSC Adv. 2017, 7, 791–800. [Google Scholar] [CrossRef]
- Kikaš, I.; Horváth, O.; Škorić, I. Functionalization of the benzobicyclo[3.2.1]octadiene skeleton via photocatalytic and thermal oxygenation of a furan derivative. Tetrahedron Lett. 2011, 52, 6255–6259. [Google Scholar] [CrossRef]
- Kikaš, I.; Horváth, O.; Škorić, I. Functionalization of the benzobicyclo[3.2.1]octadiene skeleton via photocatalytic oxygenation of furan and benzofuran derivatives. J. Mol. Struct. 2013, 1034, 62–68. [Google Scholar] [CrossRef]
- Vuk, D.; Kikaš, I.; Molčanov, K.; Horváth, O.; Škorić, I. Functionalization of the benzobicyclo[3.2.1]octadiene skeleton via photocatalytic oxygenation of thiophene and furan derivatives: The impact of the type and position of the heteroatom. J. Mol. Struct. 2014, 1063, 83–91. [Google Scholar] [CrossRef]
- Šagud, I.; Škorić, I. Photocatalytic oxygenation by water-soluble metalloporphyrins as a pathway to functionalized polycycles. Int. J. Photoenergy 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Vuk, D.; Horváth, O.; Škorić, I. New functionalized polycycles obtained by photocatalytic oxygenation using Mn(III) porphyrins in basic media. Catalysts 2019, 9, 304. [Google Scholar] [CrossRef]
- Hennig, H.; Luppa, D. Photocatalytic activation of oxygen by iron(III) porphyrins. J. Für Prakt. Chem. 1999, 341, 757–767. [Google Scholar] [CrossRef]
- Hennig, H. Homogeneous photo catalysis by transition metal complexes. Coord. Chem. Rev. 1999, 182, 101–123. [Google Scholar] [CrossRef]
- Hajimohammadi, M.; Bahadoran, F.; Davarani, S.-S.-H.; Safari, N. Selective photocatalytic epoxidation of cyclooctene by molecular oxygen in the presence of porphyrin sensitizers. React. Kinet. Mech. Catal. 2009, 99, 243–250. [Google Scholar] [CrossRef]
- Zhang, R.; Horner, J.-H.; Newcomb, M. Laser flash photolysis generation and kinetic studies of porphyrin-manganese-oxo intermediates. Rate constants for oxidations effected by porphyrin-Mn(V)-oxo species and apparent disproportionation equilibrium constants for porphyrin-Mn(IV)-oxo species. J. Am. Chem. Soc. 2005, 127, 6573–6582. [Google Scholar] [CrossRef]
- Newcomb, M.; Zhang, R.; Pan, Z.; Harischandra, D.; Chandrasena, R.; Horner, J.; Martinez II, E. Laser flash photolysis production of metal-oxo derivatives and direct kinetic studies of their oxidation reactions. Catal. Today 2006, 117, 98–104. [Google Scholar] [CrossRef]
- Zhang, R.; Newcomb, M. Laser flash photolysis generation of high-valent transition metal-oxo species: Insights from kinetic studies in real time. Acc. Chem. Res. 2008, 41, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Hennig, H.; Behling, J.; Meusinger, R.; Weber, L. Photocatalytic oxygenation of selected cycloalkenes in aqueous solutions induced bywater-soluble metal porphyrin complexes. Chem. Ber. 1995, 128, 229–234. [Google Scholar] [CrossRef]
- Baglia, R.-A.; Zaragoza, J.-P.-T.; Goldberg, D.-P. Biomimetic reactivity of oxygen-derived manganese and iron porphyrinoid complexes. Chem. Rev. 2017, 117, 13320–13352. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.-K.; Keshwani, M.-M. Measurement of enzyme activity. In Guide to Protein Purification, 2nd ed.; Burgess, R.-R., Deutcher, M.-P., Eds.; Biochemistry & Molecular Biology, University of Miami School of Medicine: Miami, FL, USA; Elsevier: San Diego, CA, USA, 2009; Volume 463, pp. 57–71. [Google Scholar]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Talla, A.; Driessen, B.; Straathof, N.J.W.; Milroy, L.-G.; Brunsveld, L.; Hessel, V.; Noel, T. Metal-free photocatalytic aerobic oxidation of thiols to disulfides in batch and continuous-flow. Adv. Synth. Catal. 2015, 357, 2180–2186. [Google Scholar] [CrossRef]
- Maeda, H.; Mukae, H.; Mizuno, K. Enhanced efficiency and regioselectivity of intramolecular (2π + 2π) photocycloaddition of 1- cyanonaphthalene derivative using microreactors. Chem. Lett. 2005, 34, 66–67. [Google Scholar] [CrossRef]
- Finn, P.B.; Kulyk, S.; Sieburth, S.M. Formation and isomerization of polycyclic 1,5-enynes. Tetrahedron Lett. 2015, 56, 3567–3570. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Terao, K.; Yamaguchi, H.; Yoshimura, S.; Morimoto, T.; Kakiuchi, K.; Fukuyama, T.; Ryu, I. Diastereoselective [2 + 2] photocycloaddition of chiral cyclic enone and cyclopentene using a microflow reactor system. Chem. Lett. 2010, 39, 828–829. [Google Scholar] [CrossRef]
- Terao, K.; Nishiyama, Y.; Aida, S.; Tanimoto, H.; Morimoto, T.; Kakiuchi, K. Diastereodifferentiating [2 + 2] photocycloaddition of chiral cyclohexenone carboxylates with cyclopentene by a microreactor. J. Photochem. Photobiol. A 2012, 242, 13–19. [Google Scholar] [CrossRef]
- Qu, J.; Cao, C.T.; Cao, C. Determining the excited-state substituent constants of furyl and thienyl groups. J. Phys. Org. Chem. 2018, 31, 3799. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Substrate (S) | c (mol dm−3) | ccat (mol dm−3) | r (mol dm−3 h−1) | Confidence Interval | k (h−1) |
|---|---|---|---|---|---|---|
| Cationic | 1 | 0.00113 | 11 × 10−5 | 8.61 × 10−5 | 8.31 × 10−6 | 0.045 |
| 1 | 0.00225 | 11 × 10−5 | 1.38 × 10−4 | 1.05 × 10−5 | ||
| 1 | 0.00450 | 11 × 10−5 | 1.36 × 10−4 | 1.69 × 10−5 | ||
| 2 | 0.00120 | 11 × 10−5 | 1.59 × 10−3 | 1.19 × 10−4 | 1.338 | |
| 2 | 0.00245 | 11 × 10−5 | 1.55 × 10−3 | 4.29 × 10−4 | ||
| 2 | 0.00490 | 11 ×10−5 | 7.17 × 10−3 | 6.02 × 10−4 | ||
| Anionic | 1 | 0.00113 | 9.8 × 10−5 | 5.56 × 10−5 | 9.57 × 10−7 | 0.052 |
| 1 | 0.00225 | 9.8 × 10−5 | 1.04 × 10−4 | 1.41 × 10−5 | ||
| 1 | 0.00450 | 9.8 × 10−5 | 2.50 × 10−4 | 5.79 × 10−5 | ||
| 2 | 0.00120 | 9.8 × 10−5 | 2.83 × 10−3 | 7.14 × 10−4 | 1.503 | |
| 2 | 0.00245 | 9.8 × 10−5 | 9.93 × 10−3 | 2.60 × 10−4 | ||
| 2 | 0.00490 | 9.8 × 10−5 | 6.57 × 10−3 | 6.53 × 10−4 |
| Microflow Reactor Geometry | Reaction | ||
|---|---|---|---|
| c(1) = 0.00225 mol dm−3 ccationic = 11 × 10−5 mol dm−3 | c(2) = 0.00245 mol dm−3 canionic = 9.8 × 10−5 mol dm−3 | ||
| X (%) | X (%) | ||
| Winding channel | 8.92 | 85.66 | |
| Streight channel | I. | 4.84 | 82.49 |
| II. | 4.61 | 81.64 | |
| III. | 3.89 | 80.94 | |
| Microflow Reactor Geometry | Channel Cross Section | Dimensions | |||
|---|---|---|---|---|---|
Winding |  | Hight (µm) | 50 | ||
| Width (µm) | 250 | ||||
| Length (mm) | 332 | ||||
| Volume (µL) | 4.15 | ||||
Straight |  | I. | II. | III. | |
| Radius (µm) | 138.09 | 197.07 | 248.67 | ||
| Length (mm) | 166 | 166 | 166 | ||
| Volume (µL) | 9.92 | 20.25 | 32.33 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakić, M.; Šalić, A.; Bačić, M.; Zelić, B.; Šagud, I.; Horváth, O.; Škorić, I. Photocatalytic Oxygenation of Heterostilbenes—Batch versus Microflow Reactor. Catalysts 2021, 11, 395. https://doi.org/10.3390/catal11030395
Mlakić M, Šalić A, Bačić M, Zelić B, Šagud I, Horváth O, Škorić I. Photocatalytic Oxygenation of Heterostilbenes—Batch versus Microflow Reactor. Catalysts. 2021; 11(3):395. https://doi.org/10.3390/catal11030395
Chicago/Turabian StyleMlakić, Milena, Anita Šalić, Matea Bačić, Bruno Zelić, Ivana Šagud, Ottó Horváth, and Irena Škorić. 2021. "Photocatalytic Oxygenation of Heterostilbenes—Batch versus Microflow Reactor" Catalysts 11, no. 3: 395. https://doi.org/10.3390/catal11030395
APA StyleMlakić, M., Šalić, A., Bačić, M., Zelić, B., Šagud, I., Horváth, O., & Škorić, I. (2021). Photocatalytic Oxygenation of Heterostilbenes—Batch versus Microflow Reactor. Catalysts, 11(3), 395. https://doi.org/10.3390/catal11030395