
Preparation of Granulated Biomass Carbon Catalysts—Structure Tailoring, Characterization, and Use in Catalytic Wet Air Oxidation of Bisphenol A


 , , ,
, , ,  , and
, and 
Abstract
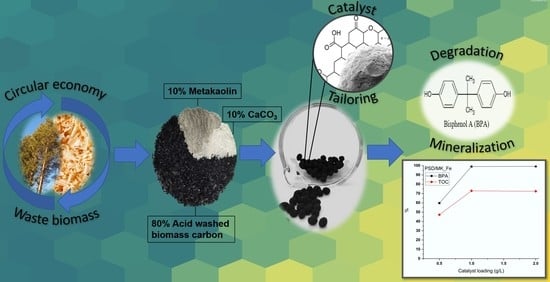
1. Introduction
2. Results and Discussion
2.1. Characterization of the Support Materials and the Catalysts
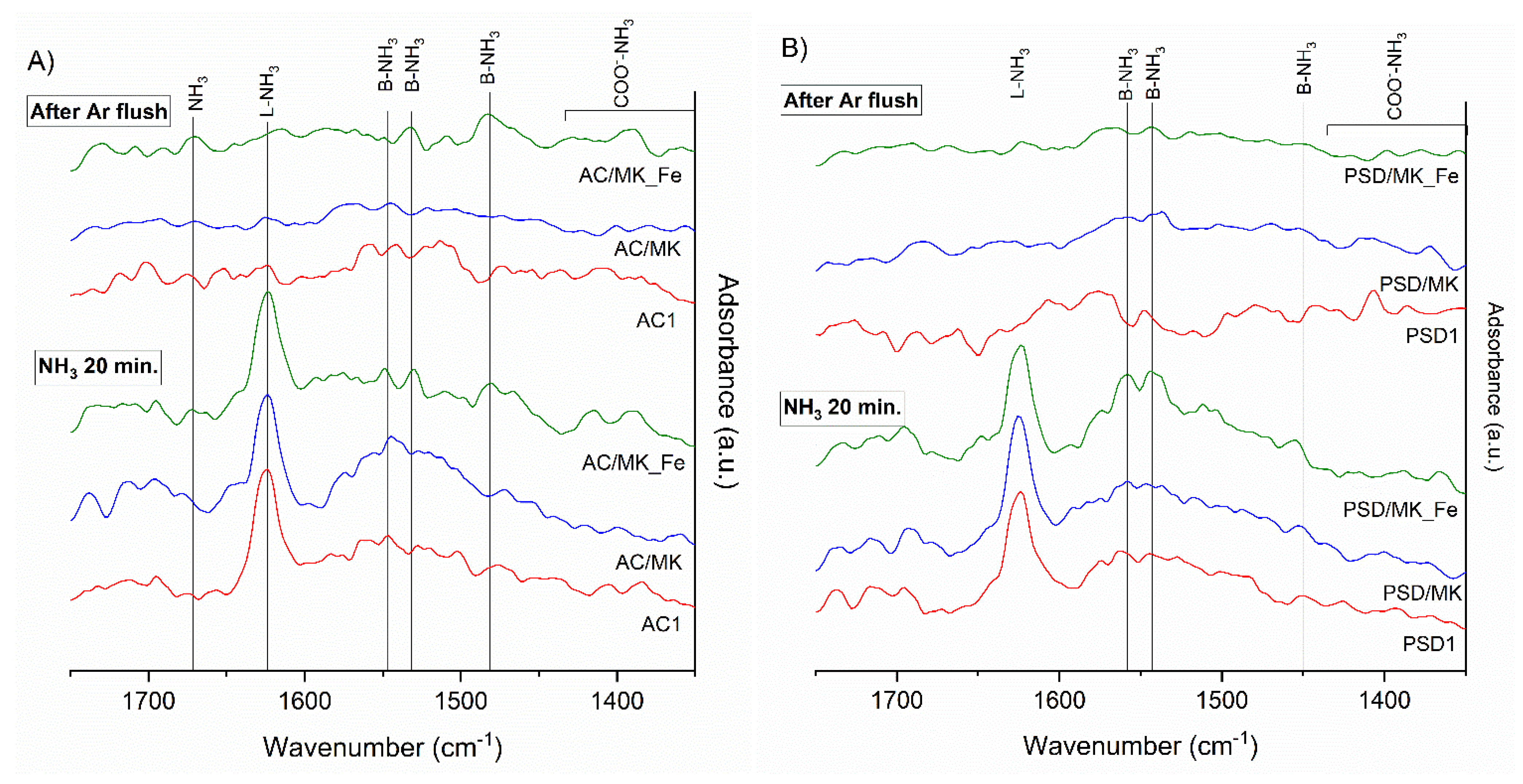
2.2. Spectroscopic Studies
2.3. Non-Catalytic Experiments
2.4. CWAO Experiments
2.5. Kinetic Modeling
2.6. Stability and Reusability of Catalysts
2.7. Analysis of By-Products
2.8. Characterization of the Spent Catalysts
3. Materials and Methods
3.1. Materials
3.2. Pretreatment of Carbon Materials
3.3. Characterization of Catalysts
3.4. Catalytic Oxidation Experiments
Analysis of Water Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerra-Que, Z.; Pérez-Vidal, H.; Torres-Torres, G.; Arévalo-Pérez, J.C.; Silahua Pavón, A.A.; Cervantes-Uribe, A.; Espinosa de los Monteros, A.; Lunagómez-Rocha, M.A. Treatment of phenol by catalytic wet air oxidation: A comparative study of copper and nickel supported on γ-alumina, ceria and γ-alumina–ceria. RSC Adv. 2019, 9, 8463–8479. [Google Scholar] [CrossRef]
- Quintanilla, A.; Casas, J.A.; Rodríguez, J.J. Catalytic wet air oxidation of phenol with modified activated carbons and Fe/activated carbon catalysts. Appl. Catal. B Environ. 2007, 76, 135–145. [Google Scholar] [CrossRef]
- Heponiemi, A.; Azalim, S.; Hu, T.; Vielma, T.; Lassi, U. Efficient removal of bisphenol A from wastewaters: Catalytic wet air oxidation with Pt catalysts supported on Ce and Ce–Ti mixed oxides. AIMS Mater. Sci. 2019, 6, 25–44. [Google Scholar] [CrossRef]
- Erjavec, B.; Kaplan, R.; Djinović, P.; Pintar, A. Catalytic wet air oxidation of bisphenol A model solution in a trickle-bed reactor over titanate nanotube-based catalysts. Appl. Catal. B Environ. 2013, 132–133, 342–352. [Google Scholar] [CrossRef]
- Stüber, F.; Font, J.; Fortuny, A.; Bengoa, C.; Eftaxias, A.; Fabregat, A. Carbon materials and catalytic wet air oxidation of organic pollutants in wastewater. Top. Catal. 2005, 33, 3–50. [Google Scholar] [CrossRef]
- Žerjav, G.; Kaplan, R.; Pintar, A. Catalytic wet air oxidation of bisphenol A aqueous solution in trickle-bed reactor over single TiO2 polymorphs and their mixtures. J. Environ. Chem. Eng. 2018, 6, 2148–2158. [Google Scholar] [CrossRef]
- Vaschetto, E.G.; Sicardi, M.I.; Elías, V.R.; Ferrero, G.O.; Carraro, P.M.; Casuscelli, S.G.; Eimer, G.A. Metal modified silica for catalytic wet air oxidation (CWAO) of glyphosate under atmospheric conditions. Adsorption 2019, 25, 1299–1306. [Google Scholar] [CrossRef]
- Bistan, M.; Tišler, T.; Pintar, A. Catalytic and Photocatalytic Oxidation of Aqueous Bisphenol A Solutions: Removal, Toxicity, and Estrogenicity. Ind. Eng. Chem. Res. 2012, 51, 8826–8834. [Google Scholar] [CrossRef]
- Koljonen, T.; Ruska, M.; Flyktman, M.; Forström, J.; Kiviluoma, A.J.; Kirkinen, J.; Lehtilä, A.; Ahkala, K.P. Energiaresurssit ja—Markkinat. Teknologian Tutkimuskeskus VTT Tiedotteita 2489. 2009. Available online: https://www.vttresearch.com/sites/default/files/pdf/tiedotteet/2009/T2489.pdf (accessed on 1 July 2020).
- Quintanilla, A.; Casas, J.A.; Mohedano, A.F.; Rodríguez, J.J. Reaction pathway of the catalytic wet air oxidation of phenol with a Fe/activated carbon catalyst. Appl. Catal. B Environ. 2006, 67, 206–216. [Google Scholar] [CrossRef]
- Wang, X.; Liu, B.; Lu, Q.; Qu, Q. Graphene-based materials: Fabrication and application for adsorption in analytical chemistry. J. Chromatogr. A 2014, 1362, 1–15. [Google Scholar] [CrossRef]
- Boehm, H.P.; Voll, M. Basische Oberflächenoxide auf Kohlenstoff—I. Adsorption von säuren. Carbon 1970, 8, 227–240. [Google Scholar]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Donnet, J.B. The chemical reactivity of carbons. Carbon 1968, 6, 161–176. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Hogland, W.; Marques, M.; Sillanpää, M. An overview of the modification methods of activated carbon for its water treatment applications. Chem. Eng. J. 2013, 219, 499–511. [Google Scholar] [CrossRef]
- Wang, J.; Fu, W.; He, X.; Yang, S.; Zhu, W. Catalytic wet air oxidation of phenol with functionalized carbon materials as catalysts: Reaction mechanism and pathway. Int. J. Environ. Sci. 2014, 26, 1741–1749. [Google Scholar] [CrossRef]
- Ayral, C.; Lebigue, C.J.; Stüber, F.; Wilhelm, A.; Delmas, H. Catalytic Wet Air Oxidation of Phenolic Compounds and Mixtures over Activated Carbon: Conversion, Mineralization, and Catalyst Stability. Ind. Eng. Chem. 2010, 49, 10707–10714. [Google Scholar] [CrossRef]
- Serra-Pérez, E.; Álvarez-Torrellas, S.; Ismael Águeda, V.; Delgado, J.A.; Ovejero, G.; García, J. Insights into the removal of Bisphenol A by catalytic wet air oxidation upon carbon nanospheres-based catalysts: Key operating parameters, degradation intermediates and reaction pathway. Appl. Surf. Sci. 2019, 473, 726–737. [Google Scholar] [CrossRef]
- Torres, G.C.; Jablonski, E.L.; Baronetti, G.T.; Castro, A.A.; de Miguel, S.R.; Scelza, O.A.; Blanco, M.D.; Penã Jiménez, M.A.; Fierro, J.L.G. Effect of the carbon pre-treatment on the properties and performance for nitrobenzene hydrogenation of Pt/C catalysts. Appl. Catal. A Gen. 1997, 161, 213–226. [Google Scholar] [CrossRef]
- Mccrae, P.D.; Zhang, T.; Walker, D.R. Method of Making Shaped Activated Carbon. Patent No. CA2442243A1, 2002. Available online: worldwide.espacenet.com (accessed on 31 May 2020).
- Auer, E.; Freund, A.; Pietsch, J.; Tacke, T. Carbons as supports for industrial precious metal catalysts. Appl. Catal. A Gen. 1998, 173, 259–271. [Google Scholar] [CrossRef]
- Iveson, S.M.; Litster, J.D.; Hapgood, K.; Ennis, B.J. Nucleation, growth and breakage phenomena in agitated wet granulation processes: A review. Powder Technol. 2001, 117, 3–39. [Google Scholar] [CrossRef]
- Kadam, K.L. Granulation Technology for Bioproducts. Drying Technol 1993, 11, 675. [Google Scholar] [CrossRef]
- Pendyal, B.; Johns, M.M.; Marshall, W.E.; Ahmedna, M.; Rao, R.M. The effect of binders and agricultural by-products on physical and chemical properties of granular activated carbons. Bioresour. Technol. 1999, 68, 247–254. [Google Scholar] [CrossRef]
- Ahmedna, M.; Marshall, W.E.; Rao, R.M. Production of granular activated carbons from select agricultural by-products and evaluation of their physical, chemical and adsorption properties1Louisiana Agricultural Experiment Station manuscript 99-21-0066.1. Bioresour. Technol. 2000, 71, 113–123. [Google Scholar] [CrossRef]
- Holt, E.M. The properties and forming of catalysts and absorbents by granulation. Powder Technol. 2004, 140, 194–202. [Google Scholar] [CrossRef]
- Juhola, R.; Heponiemi, A.; Tuomikoski, S.; Hu, T.; Prokkola, H.; Romar, H.; Lassi, U. Biomass-based composite catalysts for catalytic wet peroxide oxidation of bisphenol A: Preparation and characterization studies. J. Environ. Chem. Eng. 2019, 7, 103127. [Google Scholar] [CrossRef]
- Ma, W.; Hu, J.; Yoza, B.A.; Wang, Q.; Zhang, X.; Li, Q.X.; Guo, S.; Chen, C. Kaolinite based catalysts for efficient ozonation of recalcitrant organic chemicals in water. Appl. Clay Sci. 2019, 175, 159–168. [Google Scholar] [CrossRef]
- Gao, L.; Zheng, Y.; Tang, Y.; Yu, J.; Yu, X.; Liu, B. Effect of phosphoric acid content on the microstructure and compressive strength of phosphoric acid-based metakaolin geopolymers. Heliyon 2020, 6, e03853. [Google Scholar] [CrossRef]
- Le-Ping, L.; Xue-Min, C.; Shu-Heng, Q.; Jun-Li, Y.; Lin, Z. Preparation of phosphoric acid-based porous geopolymers. Appl. Clay Sci. 2010, 50, 600–603. [Google Scholar] [CrossRef]
- Sellami, M.; Barre, M.; Toumi, M. Synthesis, thermal properties and electrical conductivity of phosphoric acid-based geopolymer with metakaolin. Appl. Clay Sci. 2019, 180, 105192. [Google Scholar] [CrossRef]
- Wang, F.; Duan, L.; Wang, F.; Chen, W. Environmental reduction of carbon nanomaterials affects their capabilities to accumulate aromatic compounds. NanoImpact 2016, 1, 21–28. [Google Scholar] [CrossRef]
- Masindi, V.; Gitari, M.W.; Tutu, H.; DeBeer, M. Efficiency of ball milled South African bentonite clay for remediation of acid mine drainage. J. Water Process. Eng. 2015, 8, 227–240. [Google Scholar] [CrossRef]
- Dydo, P.; Turek, M. Boron transport and removal using ion-exchange membranes: A critical review. Desalination 2013, 310, 2–8. [Google Scholar] [CrossRef]
- Lopez-Ramon, M.V.; Stoeckli, F.; Moreno-Castilla, C.; Carrasco-Marin, F. On the characterization of acidic and basic surface sites on carbons by various techniques. Carbon 1999, 37, 1215–1221. [Google Scholar] [CrossRef]
- Valix, M.; Cheung, W.H.; Zhang, K. Role of heteroatoms in activated carbon for removal of hexavalent chromium from wastewaters. J. Hazard. Mater. 2006, 135, 395–405. [Google Scholar] [CrossRef]
- Williams, R.P.; van Riessen, A. Determination of the reactive component of fly ashes for geopolymer production using XRF and XRD. Fuel 2010, 89, 3683–3692. [Google Scholar] [CrossRef]
- van Jaarsveld, J.G.S.; van Deventer, J.S.J. Effect of the Alkali Metal Activator on the Properties of Fly Ash-Based Geopolymers. Ind. Eng. Chem. Res. 1999, 38, 3932–3941. [Google Scholar] [CrossRef]
- Manoj, B. A comprehensive analysis of various structural parameters of Indian coals with the aid of advanced analytical tools. Int. J. Coal. Sci. Technol. 2016, 3, 123–132. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; López-Ramón, M.V.; Carrasco-Marın, F. Changes in surface chemistry of activated carbons by wet oxidation. Carbon 2000, 38, 1995–2001. [Google Scholar] [CrossRef]
- Prati, L.; Bergna, D.; Villa, A.; Spontoni, P.; Bianchi, C.L.; Hu, T.; Romar, H.; Lassi, U. Carbons from second generation biomass as sustainable supports for catalytic systems. Catal. Today 2018, 301, 239–243. [Google Scholar] [CrossRef]
- Walczyk, M.; Świątkowski, A.; Pakuła, M.; Biniak, S. Electrochemical studies of the interaction between a modified activated carbon surface and heavy metal ions. J. Appl. Electrochem. 2005, 35, 123–130. [Google Scholar] [CrossRef]
- Kamegawa, K.; Nishikubo, K.; Kodama, M.; Adachi, Y.; Yoshida, H. Oxidative degradation of carbon blacks with nitric acid: II. Formation of water-soluble polynuclear aromatic compounds. Carbon 2002, 40, 1447–1455. [Google Scholar] [CrossRef]
- Soares, O.S.; Rocha, R.P.; Órfão, J.J.; Pereira, M.F.; Figueiredo, J.L. Mechanothermal Approach for N-, S-, P-, and B-Doping of Carbon Nanotubes: Methodology and Catalytic Performance in Wet Air Oxidation. C J. Carbon Res. 2019, 5, 30. [Google Scholar] [CrossRef]
- Kolel-Veetil, M.; Gamache, R.M.; Bernstein, N.; Goswami, R.; Qadri, S.B.; Fears, K.P.; Miller, J.B.; Glaser, E.R.; Keller, T.M. Substitution of silicon within the rhombohedral boron carbide (B4C) crystal lattice through high-energy ball-milling. J. Mater. Chem. C 2015, 3, 11705–11716. [Google Scholar] [CrossRef]
- Herreros, B.; Barr, T.L.; Barrie, P.J.; Klinowski, J. Spectroscopic Studies of 5-Coordinate Silicon Compounds. J. Phys. Chem. 1994, 98, 4570–4574. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, W.; Li, X.; Wang, J.; Zhou, Y. Multi-walled carbon nanotubes (MWNTs) as an efficient catalyst for catalytic wet air oxidation of phenol. Catal. Commun. 2007, 8, 2059–2063. [Google Scholar] [CrossRef]
- Onida, B.; Gabelica, Z.; Lourenço, J.; Garrone, E. Spectroscopic Characterization of Hydroxyl Groups in SAPO-40. 1. Study of the Template-Free Samples and Their Interaction with Ammonia. Am. J. Phys. Chem. 1996, 100, 11072–11079. [Google Scholar]
- Bandosz, T.J.; Petit, C. On the reactive adsorption of ammonia on activated carbons modified by impregnation with inorganic compounds. J. Colloid 2009, 338, 329–345. [Google Scholar] [CrossRef]
- Chen, L.; Peng, P.; Lin, L.; Yang, T.C.K.; Huang, C. Facile Preparation of Nitrogen-Doped Activated Carbon for Carbon Dioxide Adsorption. Aerosol. Air Qual. Res. 2014, 14, 916–927. [Google Scholar] [CrossRef]
- Zheng, W.; Hu, J.; Rappeport, S.; Zheng, Z.; Wang, Z.; Han, Z.; Langer, J.; Economy, J. Activated carbon fiber composites for gas phase ammonia adsorption. Micropor. Mesopor. Mater. 2016, 234, 146–154. [Google Scholar] [CrossRef]
- Przepiórski, J.; Skrodzewicz, M.; Morawski, A.W. High temperature ammonia treatment of activated carbon for enhancement of CO2 adsorption. Appl. Surf. Sci. 2004, 225, 235–242. [Google Scholar] [CrossRef]
- Gonçalves, M.; Sánchez-García, L.; Oliveira Jardim, E.d.; Silvestre-Albero, J.; Rodríguez-Reinoso, F. Ammonia Removal Using Activated Carbons: Effect of the Surface Chemistry in Dry and Moist Conditions. Environ. Sci. 2011, 45, 10605–10610. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.; Komvokis, V.G.; Singh, U.G.; Ziebarth, M.S.; Alexeev, O.S.; Amiridis, M.D. In situ FTIR characterization of NH3 adsorption and reaction with O2 and CO on Pd-based FCC emission control additives. Appl. Catal. A Gen. 2011, 391, 11–21. [Google Scholar] [CrossRef]
- Xu, Z.; Li, Y.; Guo, J.; Xiong, J.; Lin, Y.; Zhu, T. An efficient and sulfur resistant K-modified activated carbon for SCR denitrification compared with acid- and Cu-modified activated carbon. Chem. Eng. J. 2020, 395, 125047. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; van Bekkum, H. Amination and ammoxidation of activated carbons. Carbon 1994, 32, 1507–1516. [Google Scholar] [CrossRef]
- Yoon, Y.; Westerhoff, P.; Snyder, S.A.; Esparza, M. HPLC-fluorescence detection and adsorption of bisphenol A, 17β-estradiol, and 17α-ethynyl estradiol on powdered activated carbon. Water Res. 2003, 37, 3530–3537. [Google Scholar] [CrossRef]
- de Los Monteros, A.E.; Lafaye, G.; Cervantes, A.; Del Angel, G.; Barbier, J., Jr.; Torres, G. Catalytic wet air oxidation of phenol over metal catalyst (Ru,Pt) supported on TiO2–CeO2 oxides. Catal. Today 2015, 258, 564–569. [Google Scholar] [CrossRef]
- Moreno-Castilla, C. Adsorption of organic molecules from aqueous solutions on carbon materials. Carbon 2004, 42, 83–94. [Google Scholar] [CrossRef]
- Weber, W.J.; Morris, C.M. Kinetics of Adsorption on Carbon from Solution. J. Sanit. Eng. Div. 1963, 89, 31–60. [Google Scholar] [CrossRef]
- Radovic, L.R.; Mureno-Castilla, C.; Rivera-Utrilla, J. Carbon materials as adsorbents in aqueous solutions. In Chemistry and Physics of Carbon; Marcel-Dekker: New York, NY, USA, 2001; pp. 227–405. [Google Scholar]
- Kumari, M.; Saroha, A.K. Performance of various catalysts on treatment of refractory pollutants in industrial wastewater by catalytic wet air oxidation: A review. J. Environ. Manag. 2018, 228, 169–188. [Google Scholar]
- Rivas, F.J.; Kolaczkowski, S.T.; Beltrán, F.J.; McLurgh, D.B. Development of a model for the wet air oxidation of phenol based on a free radical mechanism. Chem. Eng. Sci. 1998, 53, 2575–2586. [Google Scholar] [CrossRef]
- Lin, S.S.; Chang, D.J.; Wang, C.; Chen, C.C. Catalytic wet air oxidation of phenol by CeO2 catalyst—Effect of reaction conditions. Water Res. 2003, 37, 793–800. [Google Scholar] [CrossRef]
- Chang, D.J.; Lin, S.S.; Chen, C.L.; Wang, S.P.; Ho, W.L. Catalytic wet air oxidation of phenol using CeO2 as the catalyst. Kinetic study and mechanism development. J. Environ. Sci. Health A Tox. Hazard. Substain. Environ. Eng. 2002, 37, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Weng, H. Liquid-phase oxidation of cyclohexane using CoAPO-5 as the catalyst. Appl. Catal. A Gen. 1993, 105, 289–308. [Google Scholar] [CrossRef]
- Anurag, G.; Alok, M. Degradation of Organic Pollutants by Wet Air Oxidation Using Nonnoble Metal-Based Catalysts. J. Hazard. Tox. Radioact. Waste 2013, 17, 89–96. [Google Scholar]
- Chang, L.; Chen, I.; Lin, S. An assessment of the suitable operating conditions for the CeO2/γ-Al2O3 catalyzed wet air oxidation of phenol. Chemosphere 2005, 58, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Levec, J.; Pintar, A. Catalytic wet-air oxidation processes: A review. Catal. Today Adv. Catal. Oxid. Process. 2007, 124, 172–184. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. The role of surface chemistry in catalysis with carbons. Catal. Today 2010, 150, 2–7. [Google Scholar] [CrossRef]
- Serp, P.; Machado, B. Doped Nanostructured Carbon Materials as Catalysts. Nanostructured Carbon Materials for Catalysis; The Royal Society of Chemistry. 2015, pp. 268–311. Available online: https://pubs.rsc.org/en/content/chapter/bk9781849739092-00268/978-1-84973-909-2 (accessed on 2 December 2020).
- Fu, D.; Zhang, F.; Wang, L.; Yang, F.; Liang, X. Simultaneous removal of nitrobenzene and phenol by homogenous catalytic wet air oxidation. Chin. J. Catal. 2015, 36, 952–956. [Google Scholar] [CrossRef]
- Kim, K.; Kim, J.; Ihm, S. Wet oxidation of phenol over transition metal oxide catalysts supported on Ce0.65Zr0.35O2 prepared by continuous hydrothermal synthesis in supercritical water. J. Hazard. Mater. 2009, 167, 1158–1162. [Google Scholar] [CrossRef]
- Santos, A.; Yustos, P.; Quintanilla, A.; García-Ochoa, F.; Casas, J.A.; Rodríguez, J.J. Evolution of Toxicity upon Wet Catalytic Oxidation of Phenol. Environ. Sci. Technol. 2004, 38, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.; Yustos, P.; Rodriguez, S.; Garcia-Ochoa, F. Wet oxidation of phenol, cresols and nitrophenols catalyzed by activated carbon in acid and basic media. Appl. Catal. B Environ. 2006, 65, 269–281. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, W.; Jiang, Z.; Chen, Z.; Wang, J. The surface properties and the activities in catalytic wet air oxidation over CeO2–TiO2 catalysts. Appl. Surf. Sci. 2006, 252, 8499–8505. [Google Scholar] [CrossRef]
- Debellefontaine, H.; Chakchouk, M.; Foussard, J.N.; Tissot, D.; Striolo, P. Treatment of organic aqueous wastes: Wet air oxidation and wet peroxide oxidation®. Environ. Pollut. 1996, 92, 155–164. [Google Scholar] [CrossRef]
- Debellefontaine, H.; Foussard, J.N. Wet air oxidation for the treatment of industrial wastes. Chemical aspects, reactor design and industrial applications in Europe. J. Waste Manag. 2000, 20, 15–25. [Google Scholar] [CrossRef]
- Suarez-Ojeda, M.E.; Guisasola, A.; Baeza, J.A.; Fabregat, A.; Stüber, F.; Fortuny, A.; Font, J.; Carrera, J. Integrated catalytic wet air oxidation and aerobic biological treatment in a municipal WWTP of a high-strength o-cresol wastewater. Chemosphere 2007, 66, 2096–2105. [Google Scholar] [CrossRef]
- Quintanilla, A.; Menéndez, N.; Tornero, J.; Casas, J.A.; Rodríguez, J.J. Surface modification of carbon-supported iron catalyst during the wet air oxidation of phenol: Influence on activity, selectivity and stability. Appl. Catal. B Environ. 2008, 81, 105–114. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Seaton, N.A.; Walton, J.P. A new analysis method for the determination of the pore size distribution of porous carbons from nitrogen adsorption measurements. Carbon 1989, 27, 853–861. [Google Scholar] [CrossRef]
- Noh, J.S.; Schwarz, J.A. Estimation of the point of zero charge of simple oxides by mass titration. J. Colloid Interface Sci. 1989, 130, 157–164. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSA | Pore Size | Total Pore Volume | Micro | Meso | Macro | pH | pHzpc |
|---|---|---|---|---|---|---|---|---|
| (m2/g) | (nm) | (cm3/g) | Porosity (%) | - | - | |||
| AC0 | 1031 | 2.58 | 0.45 | 69.4 | 30.4 | 0.2 | - | 10.1 |
| AC1 | 878 | 2.60 | 0.38 | 70.2 | 29.6 | 0.3 | 3.4 | 2.9 |
| AC/MK | 397 | 2.43 | 0.18 | 69.8 | 30.2 | - | 3.7 | 2.4 |
| AC/MK_Fe | 293 | 2.92 | 0.12 | 66.1 | 33.1 | 0.8 | 3.5 | 2.3 |
| PSD0 | 808 | 3.13 | 0.38 | 64.6 | 35.4 | - | 9.4 | 9.5 |
| PSD1 | 845 | 3.25 | 0.42 | 60.0 | 40.0 | - | 3.8 | 3.4 |
| PSD/MK | 212 | 3.54 | 0.12 | 50.0 | 50.0 | - | 3.9 | 2.6 |
| PSD/MK_Fe | 299 | 3.23 | 0.16 | 52.6 | 47.4 | - | 3.8 | 2.4 |
| Sample | ICP-OES | Elemental Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Al | Fe | Ca | P | Si | B | Si/Al | C | H | N | O | |
| (wt%) | (wt%) | (wt%) | (wt%) | (wt%) | (wt%) | - | (wt%) | (wt%) | (wt%) | (wt%) | |
| AC1 | - | - | - | - | - | - | - | 73.9 | 7.6 | 1.2 | 8.1 |
| AC/MK | 1.0 | 0.3 | 0.2 | 0.9 | 4.3 | 2.3 | 4.3 | 60.9 | 18.1 | 0.9 | 11.1 |
| AC/MK_Fe | 0.9 | 0.5 | 0.3 | 1.6 | 3.8 | 3.8 | 4.2 | 55.7 | 22.7 | 0.8 | 13.5 |
| PSD1 | - | - | - | - | - | - | - | 77.3 | 10.4 | 1.1 | 9.7 |
| PSD/MK | 1.2 | 0.5 | 0.3 | 0.7 | 3.6 | 2.7 | 3.0 | 59.4 | 21.9 | 0.6 | 13.1 |
| PSD/MK_Fe | 0.7 | 0.8 | 0.2 | 0.8 | 2.9 | 2.1 | 4.1 | 63.2 | 20.1 | 0.7 | 12.5 |
| Constant [Unit] | AC/MK_Fe | PSD/MK_Fe | WAO |
|---|---|---|---|
| k [1/min] | 0.055 | 0.019 | 0.008 |
| Average relative error [%] | 0.166 | 2.422 | 15.641 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juhola, R.; Heponiemi, A.; Tuomikoski, S.; Hu, T.; Huuhtanen, M.; Bergna, D.; Lassi, U. Preparation of Granulated Biomass Carbon Catalysts—Structure Tailoring, Characterization, and Use in Catalytic Wet Air Oxidation of Bisphenol A. Catalysts 2021, 11, 251. https://doi.org/10.3390/catal11020251
Juhola R, Heponiemi A, Tuomikoski S, Hu T, Huuhtanen M, Bergna D, Lassi U. Preparation of Granulated Biomass Carbon Catalysts—Structure Tailoring, Characterization, and Use in Catalytic Wet Air Oxidation of Bisphenol A. Catalysts. 2021; 11(2):251. https://doi.org/10.3390/catal11020251
Chicago/Turabian StyleJuhola, Riikka, Anne Heponiemi, Sari Tuomikoski, Tao Hu, Mika Huuhtanen, Davide Bergna, and Ulla Lassi. 2021. "Preparation of Granulated Biomass Carbon Catalysts—Structure Tailoring, Characterization, and Use in Catalytic Wet Air Oxidation of Bisphenol A" Catalysts 11, no. 2: 251. https://doi.org/10.3390/catal11020251
APA StyleJuhola, R., Heponiemi, A., Tuomikoski, S., Hu, T., Huuhtanen, M., Bergna, D., & Lassi, U. (2021). Preparation of Granulated Biomass Carbon Catalysts—Structure Tailoring, Characterization, and Use in Catalytic Wet Air Oxidation of Bisphenol A. Catalysts, 11(2), 251. https://doi.org/10.3390/catal11020251