
Protein Modifications: From Chemoselective Probes to Novel Biocatalysts



Abstract
:1. Introduction
2. Historical Background
3. The Challenges of Biocompatibility and Selectivity
4. Chemical Aspects of Selective Protein Modification
4.1. Cysteines
4.2. Lysines
4.3. N-Terminus
4.4. Carboxilates
5. Selective Protein Modification and Novel Semisynthetic Biocatalysts
5.1. Modification at Cysteines
5.2. Modification at Lysines
5.3. Modification at the N-Terminus
5.4. Modification at Carboxilates
6. Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Truppo, M.D. Biocatalysis in the Pharmaceutical Industry: The Need for Speed. ACS Med. Chem. Lett. 2017, 8, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouthier, C.M.; Pelletier, J.N. Expanding the Organic Toolbox: A Guide to Integrating Biocatalysis in Synthesis. Chem. Soc. Rev. 2012, 41, 1585–1605. [Google Scholar] [CrossRef]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Pyser, J.B.; Chakrabarty, S.; Romero, E.O.; Narayan, A.R.H. State-of-the-Art Biocatalysis. ACS Cent. Sci. 2021, 7, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-M.; Han, S.-S.; Kim, H.-S. Industrial Applications of Enzyme Biocatalysis: Current Status and Future Aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef]
- Sandoval, B.A.; Hyster, T.K. Emerging Strategies for Expanding the Toolbox of Enzymes in Biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. [Google Scholar] [CrossRef]
- Woodley, J.M. New Frontiers in Biocatalysis for Sustainable Synthesis. Curr. Opin. Green Sustain. Chem. 2020, 21, 22–26. [Google Scholar] [CrossRef]
- Davis, B.G. Chemical Modification of Biocatalysts. Curr. Opin. Biotechnol. 2003, 14, 379–386. [Google Scholar] [CrossRef]
- DeSantis, G.; Jones, J.B. Chemical Modification of Enzymes for Enhanced Functionality. Curr. Opin. Biotechnol. 1999, 10, 324–330. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, A.; Davis, B.G. Chemical Modification in the Creation of Novel Biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 211–219. [Google Scholar] [CrossRef]
- Kaushik, M.; Sinha, P.; Jaiswal, P.; Mahendru, S.; Roy, K.; Kukreti, S. Protein Engineering and de Novo Designing of a Biocatalyst. J. Mol. Recognit. 2016, 29, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Hamachi, I. Recent Progress in Chemical Modification of Proteins. Anal. Sci. 2019, 35, 5–27. [Google Scholar] [CrossRef] [Green Version]
- Pagar, A.D.; Patil, M.D.; Flood, D.T.; Yoo, T.H.; Dawson, P.E.; Yun, H. Recent Advances in Biocatalysis with Chemical Modification and Expanded Amino Acid Alphabet. Chem. Rev. 2021, 121, 6173–6245. [Google Scholar] [CrossRef]
- Neet, K.E.; Koshland, D.E. The Conversion of Serine at the Active Site of Subtilisin to Cysteine: A “Chemical Mutation”. Proc. Natl. Acad. Sci. USA 1966, 56, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Polgar, L.; Bender, M.L. A New Enzyme Containing a Synthetically Formed Active Site. Thiol-Subtilisin. J. Am. Chem. Soc. 1966, 88, 3153–3154. [Google Scholar] [CrossRef]
- Yokosawa, H.; Ojima, S.; Ishii, S. Thioltrypsin: Chemical Transformation of the Active-Site Serine Residue of Streptomyces griseus Trypsin to a Cysteine Residue. J. Biochem. 1977, 82, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.I.; Lowe, G. Chemical Mutations of Papain. The Preparation of Ser 25- and Gly 25-Papain. J. Chem. Soc. Chem. Commun. 1977, 24, 923–924. [Google Scholar] [CrossRef]
- Kaiser, E.T. Catalytic Activity of Enzymes Altered at Their Active Sites. Angew. Chem. Int. Ed. Engl. 1988, 27, 913–922. [Google Scholar] [CrossRef]
- Kaiser, E.T.; Lawrence, D.S. Chemical Mutation of Enzyme Active Sites. Science 1984, 226, 505–511. [Google Scholar] [CrossRef]
- Kokubo, T.; Sassa, S.; Kaiser, E.T. Flavohemoglobin: A Semisynthetic Hydroxylase Acting in the Absence of Reductase. J. Am. Chem. Soc. 1987, 109, 606–607. [Google Scholar] [CrossRef]
- Nakatsuka, T.; Sasaki, T.; Kaiser, E.T. Peptide Segment Synthesis Catalyzed by the Semisynthetic Enzyme Thiolsubtilisin. J. Am. Chem. Soc. 1987, 109, 3808–3810. [Google Scholar] [CrossRef]
- Shen, W.C.; Ryser, H.J. Conjugation of Poly-L-Lysine to Albumin and Horseradish Peroxidase: A Novel Method of Enhancing the Cellular Uptake of Proteins. Proc. Natl. Acad. Sci. USA 1978, 75, 1872–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Sigman, D.S. Chemical Conversion of a DNA-Binding Protein into a Site-Specific Nuclease. Science 1987, 237, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Inada, Y.; Takahashi, K.; Yoshimoto, T.; Ajima, A.; Matsushima, A.; Saito, Y. Application of Polyethylene Glycol-Modified Enzymes in Biotechnological Processes: Organic Solvent-Soluble Enzymes. Trends Biotechnol. 1986, 4, 190–194. [Google Scholar] [CrossRef]
- Dadová, J.; Galan, S.R.; Davis, B.G. Synthesis of Modified Proteins via Functionalization of Dehydroalanine. Curr. Opin. Chem. Biol. 2018, 46, 71–81. [Google Scholar] [CrossRef]
- Rawale, D.G.; Thakur, K.; Adusumalli, S.R.; Rai, V. Chemical Methods for Selective Labeling of Proteins. Eur. J. Org. Chem. 2019, 2019, 6749–6763. [Google Scholar] [CrossRef]
- Shadish, J.A.; DeForest, C.A. Site-Selective Protein Modification: From Functionalized Proteins to Functional Biomaterials. Matter 2020, 2, 50–77. [Google Scholar] [CrossRef]
- Spicer, C.D.; Davis, B.G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.C.; Kumar, M.; Molla, R.; Rai, V. Chemical Methods for Modification of Proteins. Org. Biomol. Chem. 2020, 18, 4669–4691. [Google Scholar] [CrossRef]
- Tamura, T.; Hamachi, I. Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems. J. Am. Chem. Soc. 2019, 141, 2782–2799. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Reddy, N.C.; Rai, V. Chemical Technologies for Precise Protein Bioconjugation Interfacing Biology and Medicine. Chem. Commun. 2021, 57, 7083–7095. [Google Scholar] [CrossRef]
- Shannon, D.A.; Weerapana, E. Covalent Protein Modification: The Current Landscape of Residue-Specific Electrophiles. Curr. Opin. Chem. Biol. 2015, 24, 18–26. [Google Scholar] [CrossRef]
- Koniev, O.; Wagner, A. Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.K.; Turner, G.J. Structural Basis of Perturbed PKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.J.; Weerapana, E. Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol. 2013, 8, 283–296. [Google Scholar] [CrossRef]
- Bischoff, R.; Schlüter, H. Amino Acids: Chemistry, Functionality and Selected Non-Enzymatic Post-Translational Modifications. J. Proteom. 2012, 75, 2275–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, D.W.; Bechtel, T.J.; Falco, J.A.; Weerapana, E. Cysteine Reactivity across the Subcellular Universe. Curr Opin. Chem. Biol. 2019, 48, 96–105. [Google Scholar] [CrossRef]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The Cysteine Proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Miseta, A.; Csutora, P. Relationship Between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, C.; Kumar, A.; Lang, A.; Ohlenschläger, O. Cysteines and Disulfide Bonds as Structure-Forming Units: Insights From Different Domains of Life and the Potential for Characterization by NMR. Front. Chem. 2020, 8, 280. [Google Scholar] [CrossRef]
- Marino, S.M.; Gladyshev, V.N. Cysteine Function Governs Its Conservation and Degeneration and Restricts Its Utilization on Protein Surfaces. J. Mol. Biol. 2010, 404, 902–916. [Google Scholar] [CrossRef] [Green Version]
- Wible, R.S.; Sutter, T.R. Soft Cysteine Signaling Network: The Functional Significance of Cysteine in Protein Function and the Soft Acids/Bases Thiol Chemistry That Facilitates Cysteine Modification. Chem. Res. Toxicol. 2017, 30, 729–762. [Google Scholar] [CrossRef]
- Bulaj, G.; Kortemme, T.; Goldenberg, D.P. Ionization-Reactivity Relationships for Cysteine Thiols in Polypeptides. Biochemistry 1998, 37, 8965–8972. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T. Reactions of Electrophiles with Nucleophilic Thiolate Sites: Relevance to Pathophysiological Mechanisms and Remediation. Free Radic. Res. 2016, 50, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Boutureira, O.; Bernardes, G.J.L. Advances in Chemical Protein Modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Hoch, D.G.; Abegg, D.; Adibekian, A. Cysteine-Reactive Probes and Their Use in Chemical Proteomics. Chem. Commun. 2018, 54, 4501–4512. [Google Scholar] [CrossRef]
- Hoyt, E.A.; Cal, P.M.S.D.; Oliveira, B.L.; Bernardes, G.J.L. Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem. 2019, 3, 147–171. [Google Scholar] [CrossRef] [Green Version]
- Litwin, K.; Crowley, V.M.; Suciu, R.M.; Boger, D.L.; Cravatt, B.F. Chemical Proteomic Identification of Functional Cysteines with Atypical Electrophile Reactivities. Tetrahedron Lett. 2021, 67, 152861. [Google Scholar] [CrossRef] [PubMed]
- Maurais, A.J.; Weerapana, E. Reactive-Cysteine Profiling for Drug Discovery. Curr. Opin. Chem. Biol. 2019, 50, 29–36. [Google Scholar] [CrossRef]
- Ochtrop, P.; Hackenberger, C.P.R. Recent Advances of Thiol-Selective Bioconjugation Reactions. Curr. Opin. Chem. Biol. 2020, 58, 28–36. [Google Scholar] [CrossRef]
- Lahnsteiner, M.; Kastner, A.; Mayr, J.; Roller, A.; Keppler, B.K.; Kowol, C.R. Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting. Chem. Eur. J. 2020, 26, 15867–15870. [Google Scholar] [CrossRef] [PubMed]
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjugate Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Martínez-Sáez, N.; Brindle, K.M.; Neves, A.A.; Corzana, F.; Bernardes, G.J.L. Site-Selective Modification of Proteins with Oxetanes. Chem. Eur. J. 2017, 23, 6483–6489. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.J.; Navo, C.D.; Hakala, T.; Ferhati, X.; Guerreiro, A.; Hartmann, D.; Bernardim, B.; Saar, K.L.; Compañón, I.; Corzana, F.; et al. Quaternization of Vinyl/Alkynyl Pyridine Enables Ultrafast Cysteine-Selective Protein Modification and Charge Modulation. Angew. Chem. Int. Ed. 2019, 58, 6640–6644. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Tao, Y.; Sheng, W.; Lu, J.; Wang, H. Dinitroimidazoles as Bifunctional Bioconjugation Reagents for Protein Functionalization and Peptide Macrocyclization. Nat. Commun. 2019, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.R.; Chung, S.F.; Leung, A.S.L.; Yip, W.M.; Yang, B.; Choi, M.C.; Cui, J.F.; Kung, K.K.Y.; Zhang, Z.; Lo, K.W.; et al. Chemoselective and Photocleavable Cysteine Modification of Peptides and Proteins Using Isoxazoliniums. Commun. Chem. 2019, 2, 93. [Google Scholar] [CrossRef]
- Smith, N.J.; Rohlfing, K.; Sawicki, L.A.; Kharkar, P.M.; Boyd, S.J.; Kloxin, A.M.; Fox, J.M. Fast, Irreversible Modification of Cysteines through Strain Releasing Conjugate Additions of Cyclopropenyl Ketones. Org. Biomol. Chem. 2018, 16, 2164–2169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zang, C.; An, G.; Shang, M.; Cui, Z.; Chen, G.; Xi, Z.; Zhou, C. Cysteine-Specific Protein Multi-Functionalization and Disulfide Bridging Using 3-Bromo-5-Methylene Pyrrolones. Nat. Commun. 2020, 11, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Stengl, A.; Ochtrop, P.; Gerlach, M.; Stoschek, T.; Schumacher, D.; Helma, J.; Penkert, M.; Krause, E.; Leonhardt, H.; et al. Ethynylphosphonamidates for the Rapid and Cysteine-Selective Generation of Efficacious Antibody–Drug Conjugates. Angew. Chem. Int. Ed. 2019, 58, 11631–11636. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Glanz, M.; Stengl, A.; Penkert, M.; Klenk, S.; Sauer, T.; Schumacher, D.; Helma, J.; Krause, E.; Cardoso, M.C.; et al. Cysteine-Selective Phosphonamidate Electrophiles for Modular Protein Bioconjugations. Angew. Chem. Int. Ed. 2019, 58, 11625–11630. [Google Scholar] [CrossRef]
- Baumann, A.L.; Schwagerus, S.; Broi, K.; Kemnitz-Hassanin, K.; Stieger, C.E.; Trieloff, N.; Schmieder, P.; Hackenberger, C.P.R. Chemically Induced Vinylphosphonothiolate Electrophiles for Thiol-Thiol Bioconjugations. J. Am. Chem. Soc. 2020, 142, 9544–9552. [Google Scholar] [CrossRef]
- Luo, M. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev. 2018, 118, 6656–6705. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. A Computational Study of Cation-π Interactions vs Salt Bridges in Aqueous Media: Implications for Protein Engineering. J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar] [CrossRef]
- Larda, S.T.; Pichugin, D.; Prosser, R.S. Site-Specific Labeling of Protein Lysine Residues and N-Terminal Amino Groups with Indoles and Indole-Derivatives. Bioconjugate Chem. 2015, 26, 2376–2383. [Google Scholar] [CrossRef]
- Al Temimi, A.H.K.; Amatdjais-Groenen, H.I.V.; Reddy, Y.V.; Blaauw, R.H.; Guo, H.; Qian, P.; Mecinović, J. The Nucleophilic Amino Group of Lysine Is Central for Histone Lysine Methyltransferase Catalysis. Commun. Chem. 2019, 2, 112. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-Translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef]
- Kozlowski, L.P. Proteome-pI: Proteome Isolectric Point Database. Nucleic Acid Res. 2016, 45, D1112–D1116. [Google Scholar] [CrossRef]
- Geoghegan, K.F. Modification of Amino Groups. Curr. Protoc. Protein Sci. 2016, 86, 15.2.1–15.2.20. [Google Scholar] [CrossRef]
- Pace, C.N.; Grimsley, G.R.; Scholtz, J.M. Protein Ionizable Groups: PK Values and Their Contribution to Protein Stability and Solubility. J. Biol. Chem. 2009, 284, 13285–13289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahari, S.; Sun, L.; Alexov, E. PKAD: A Database of Experimentally Measured PKa Values of Ionizable Groups in Proteins. Database 2019, 2019, baz024. [Google Scholar] [CrossRef] [Green Version]
- Platzer, G.; Okon, M.; McIntosh, L.P. PH-Dependent Random Coil 1H, 13C, and 15N Chemical Shifts of the Ionizable Amino Acids: A Guide for Protein PK a Measurements. J. Biomol. NMR 2014, 60, 109–129. [Google Scholar] [CrossRef]
- Pettinger, J.; Jones, K.; Cheeseman, M.D. Lysine-Targeting Covalent Inhibitors. Angew. Chem. Int. Ed. 2017, 56, 15200–15209. [Google Scholar] [CrossRef]
- Adamo, R.; Nilo, A.; Castagner, B.; Boutureira, O.; Berti, F.; Bernardes, G.J.L. Synthetically Defined Glycoprotein Vaccines: Current Status and Future Directions. Chem. Sci. 2013, 4, 2995–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, P.R.; Paquette, D.M. Comparison of Three Common Amine Reactive Fluorescent Probes Used for Conjugation to Biomolecules by Capillary Zone Electrophoresis. Bioconjugate Chem. 1995, 6, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.M.; Page, R.C.; Konkolewicz, D. Bioconjugates—From a Specialized Past to a Diverse Future. Polymer 2020, 211, 123062. [Google Scholar] [CrossRef]
- Naowarojna, N.; Cheng, R.; Lopez, J.; Wong, C.; Qiao, L.; Liu, P. Chemical Modifications of Proteins and Their Applications in Metalloenzyme Studies. Synth. Syst. Biothecnol. 2021, 6, 32–49. [Google Scholar] [CrossRef]
- Chen, X.; Muthoosamy, K.; Pfisterer, A.; Neumann, B.; Weil, T. Site-Selective Lysine Modification of Native Proteins and Peptides via Kinetically Controlled Labeling. Bioconjugate Chem. 2012, 23, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Adusumalli, S.R.; Rawale, D.G.; Thakur, K.; Purushottam, L.; Reddy, N.C.; Kalra, N.; Shukla, S.; Rai, V. Chemoselective and Site-Selective Lysine-Directed Lysine Modification Enables Single-Site Labeling of Native Proteins. Angew. Chem. Int. Ed. 2020, 59, 10332–10336. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Oliveira, B.L.; Martínez-Sáez, N.; Guerreiro, A.; Cal, P.M.S.D.; Bertoldo, J.; Maneiro, M.; Perkins, E.; Howard, J.; Deery, M.J.; et al. Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc. 2018, 140, 4004–4017. [Google Scholar] [CrossRef]
- De Rosa, L.; Di Stasi, R.; Romanelli, A.; D’Andrea, L.D. Exploiting Protein N-Terminus for Site-Specific Bioconjugation. Molecules 2021, 26, 3521. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Disotuar, M.M.; Xiong, X.; Wang, Y.; Chou, D.H.C. Selective N-Terminal Functionalization of Native Peptides and Proteins. Chem. Sci. 2017, 8, 2717–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, C.B.; Francis, M.B. Targeting the N Terminus for Site-Selective Protein Modification. Nat. Chem. Biol. 2017, 13, 697–705. [Google Scholar] [CrossRef]
- Schoffelen, S.; van Eldijk, M.B.; Rooijakkers, B.; Raijmakers, R.; Heck, A.J.R.; van Hest, J.C.M. Metal-Free and PH-Controlled Introduction of Azides in Proteins. Chem. Sci. 2011, 2, 701–705. [Google Scholar] [CrossRef]
- Chan, A.O.Y.; Ho, C.M.; Chong, H.C.; Leung, Y.C.; Huang, J.S.; Wong, M.K.; Che, C.M. Modification of N-Terminal α-Amino Groups of Peptides and Proteins Using Ketenes. J. Am. Chem. Soc. 2012, 134, 2589–2598. [Google Scholar] [CrossRef]
- Macdonald, J.I.; Munch, H.K.; Moore, T.; Francis, M.B. One-Step Site-Specific Modification of Native Proteins with 2-Pyridinecarboxyaldehydes. Nat. Chem. Biol. 2015, 11, 326–331. [Google Scholar] [CrossRef]
- Deng, J.R.; Lai, N.C.H.; Kung, K.K.Y.; Yang, B.; Chung, S.F.; Leung, A.S.L.; Choi, M.C.; Leung, Y.C.; Wong, M.K. N-Terminal Selective Modification of Peptides and Proteins Using 2-Ethynylbenzaldehydes. Commun. Chem. 2020, 3, 67. [Google Scholar] [CrossRef]
- McGrath, N.A.; Andersen, K.A.; Davis, A.K.F.; Lomax, J.E.; Raines, R.T. Diazo Compounds for the Bioreversible Esterification of Proteins. Chem. Sci. 2015, 6, 752–755. [Google Scholar] [CrossRef] [Green Version]
- Mix, K.A.; Raines, R.T. Optimized Diazo Scaffold for Protein Esterification. Org. Lett. 2015, 17, 2358–2361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.H.; Tan, D.; Li, Q.; Li, M.; Gong, Z.; Tang, C.; Liu, Z.; Dong, M.Q.; Lei, X. Carboxylate-Selective Chemical Cross-Linkers for Mass Spectrometric Analysis of Protein Structures. Anal. Chem. 2018, 90, 1195–1201. [Google Scholar] [CrossRef]
- Ma, N.; Hu, J.; Zhang, Z.M.; Liu, W.; Huang, M.; Fan, Y.; Yin, X.; Wang, J.; Ding, K.; Ye, W.; et al. 2 H-Azirine-Based Reagents for Chemoselective Bioconjugation at Carboxyl Residues Inside Live Cells. J. Am. Chem. Soc. 2020, 142, 6051–6059. [Google Scholar] [CrossRef]
- Rawale, D.G.; Thakur, K.; Sreekumar, P.; Sajeev, T.K.; Ramesh, A.; Adusumalli, S.R.; Mishra, R.K.; Rai, V. Linchpins Empower Promiscuous Electrophiles to Enable Site-Selective Modification of Histidine and Aspartic Acid in Proteins. Chem. Sci. 2021, 12, 6732–6736. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.; Liu, C.; Kölmel, D.K.; Qiao, J.X.; Zhang, Y.; Poss, M.A.; Ewing, W.R.; Macmillan, D.W.C. Decarboxylative Alkylation for Site-Selective Bioconjugation of Native Proteins via Oxidation Potentials. Nat. Chem. 2018, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- López-Gallego, F.; Abian, O.; Guisán, J.M. Altering the Interfacial Activation Mechanism of a Lipase by Solid-Phase Selective Chemical Modification. Biochemistry 2012, 51, 7028–7036. [Google Scholar] [CrossRef]
- Ema, T.; Inoue, H. Chemical Modification of Lipase for Rational Enhancement of Enantioselectivity. Chem. Lett. 2015, 44, 1374–1376. [Google Scholar] [CrossRef] [Green Version]
- Darby, J.F.; Atobe, M.; Firth, J.D.; Bond, P.; Davies, G.J.; O’Brien, P.; Hubbard, R.E. Increase of Enzyme Activity through Specific Covalent Modification with Fragments. Chem. Sci. 2017, 8, 7772–7779. [Google Scholar] [CrossRef] [Green Version]
- Darby, J.F.; Landström, J.; Roth, C.; He, Y.; Davies, G.J.; Hubbard, R.E. Discovery of Selective Small-Molecule Activators of a Bacterial Glycoside Hydrolase. Angew. Chem. Int. Ed. 2014, 53, 13419–13423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.J.; Yu, J.; Song, W.J. Proteins as Diverse, Efficient, and Evolvable Scaffolds for Artificial Metalloenzymes. Chem. Commun. 2020, 56, 9586–9599. [Google Scholar] [CrossRef]
- Doble, M.V.; Jarvis, A.G.; Ward, A.C.C.; Colburn, J.D.; Götze, J.P.; Bühl, M.; Kamer, P.C.J. Artificial Metalloenzymes as Catalysts for Oxidative Lignin Degradation. ACS Sustain. Chem. Eng. 2018, 6, 15100–15107. [Google Scholar] [CrossRef] [Green Version]
- Sauer, D.F.; Himiyama, T.; Tachikawa, K.; Fukumoto, K.; Onoda, A.; Mizohata, E.; Inoue, T.; Bocola, M.; Schwaneberg, U.; Hayashi, T.; et al. A Highly Active Biohybrid Catalyst for Olefin Metathesis in Water: Impact of a Hydrophobic Cavity in a β-Barrel Protein. ACS Catal. 2015, 5, 7519–7522. [Google Scholar] [CrossRef]
- Bernardes, G.J.L.; Chalker, J.M.; Errey, J.C.; Davis, B.G. Facile Conversion of Cysteine and Alkyl Cysteines to Dehydroalanine on Protein Surfaces: Versatile and Switchable Access to Functionalized Proteins. J. Am. Chem. Soc. 2008, 130, 5052–5053. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Bernardes, G.J.L.; Lin, Y.A.; Davis, B.G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Chalker, J.M.; Bernardes, G.J.L.; Davis, B.G. A “Tag-and-Modify” Approach to Site-Selective Protein Modification. Acc. Chem. Res. 2011, 44, 730–741. [Google Scholar] [CrossRef]
- Chalker, J.M.; Gunnoo, S.B.; Boutureira, O.; Gerstberger, S.C.; Fernández-González, M.; Bernardes, G.J.L.; Griffin, L.; Hailu, H.; Schofield, C.J.; Davis, B.G. Methods for Converting Cysteine to Dehydroalanine on Peptides and Proteins. Chem. Sci. 2011, 2, 1666–1676. [Google Scholar] [CrossRef]
- Jones, L.H. Dehydroamino Acid Chemical Biology: An Example of Functional Group Interconversion on Proteins. RSC Chem. Biol. 2020, 1, 298–304. [Google Scholar] [CrossRef]
- Dadová, J.; Wu, K.J.; Isenegger, P.G.; Errey, J.C.; Bernardes, G.J.L.; Chalker, J.M.; Raich, L.; Rovira, C.; Davis, B.G. Precise Probing of Residue Roles by Post-Translational β,γ-C,N Aza-Michael Mutagenesis in Enzyme Active Sites. ACS Cent. Sci. 2017, 3, 1168–1173. [Google Scholar] [CrossRef]
- Chalker, J.M.; Lercher, L.; Rose, N.R.; Schofield, C.J.; Davis, B.G. Conversion of Cysteine into Dehydroalanine Enables Access to Synthetic Histones Bearing Diverse Post-Translational Modifications. Angew. Chem. Int. Ed. 2012, 51, 1835–1839. [Google Scholar] [CrossRef] [PubMed]
- Chooi, K.P.; Galan, S.R.G.; Raj, R.; McCullagh, J.; Mohammed, S.; Jones, L.H.; Davis, B.G. Synthetic Phosphorylation of P38α Recapitulates Protein Kinase Activity. J. Am. Chem. Soc. 2014, 136, 1698–1701. [Google Scholar] [CrossRef]
- Freedy, A.M.; Matos, M.J.; Boutureira, O.; Corzana, F.; Guerreiro, A.; Akkapeddi, P.; Somovilla, V.J.; Rodrigues, T.; Nicholls, K.; Xie, B.; et al. Chemoselective Installation of Amine Bonds on Proteins Through Aza-Michael Ligation. J. Am. Chem. Soc. 2017, 139, 18365–18375. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.H.; Vallée, M.R.J.; Davis, B.G. From Chemical Mutagenesis to Post-Expression Mutagenesis: A 50 Year Odyssey. Angew. Chem. Int. Ed. 2016, 55, 5896–5903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Ha, S.; Ahn, J.; Kim, R.; Kim, S.; Lee, Y.; Kim, J.; Söll, D.; Lee, H.-Y.; Park, H.-S. A Chemical Biology Route to Site-Specific Authentic Protein Modifications. Science 2016, 354, 623–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timms, N.; Windle, C.L.; Polyakova, A.; Ault, J.R.; Trinh, C.H.; Pearson, A.R.; Nelson, A.; Berry, A. Structural Insights into the Recovery of Aldolase Activity in N-Acetylneuraminic Acid Lyase by Replacement of the Catalytically Active Lysine with γ-Thialysine by Using a Chemical Mutagenesis Strategy. ChemBioChem 2013, 14, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Windle, C.L.; Simmons, K.J.; Ault, J.R.; Trinh, C.H.; Nelson, A.; Pearson, A.R.; Berry, A. Extending Enzyme Molecular Recognition with an Expanded Amino Acid Alphabet. Proc. Natl. Acad. Sci. USA 2017, 114, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Shaw, B.F.; Schneider, G.F.; Bilgiçer, B.; Kaufman, G.K.; Neveu, J.M.; Lane, W.S.; Whitelegge, J.P.; Whitesides, G.M. Lysine Acetylation Can Generate Highly Charged Enzymes with Increased Resistance toward Irreversible Inactivation. Protein Sci. 2008, 17, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Ismaya, W.T.; Hasan, K.; Kardi, I.; Zainuri, A.; Rahmawaty, R.I.; Permanahadi, S.; el Viera, B.V.; Harinanto, G.; Gaffar, S.; Natalia, D.; et al. Chemical Modification of Saccharomycopsis Fibuligera R64 α-Amylase to Improve Its Stability against Thermal, Chelator, and Proteolytic Inactivation. Appl. Biochem. Biotechnol. 2013, 170, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Nwagu, T.N.; Okolo, B.; Aoyagi, H.; Yoshida, S. Chemical Modification with Phthalic Anhydride and Chitosan: Viable Options for the Stabilization of Raw Starch Digesting Amylase from Aspergillus Carbonarius. Int. J. Biol. Macromol. 2017, 99, 641–647. [Google Scholar] [CrossRef]
- Nwagu, T.N.; Aoyagi, H.; Okolo, B.; Moneke, A.; Yoshida, S. Citraconylation and Maleylation on the Catalytic and Thermodynamic Properties of Raw Starch Saccharifying Amylase from Aspergillus Carbonarius. Heliyon 2020, 6, e04351. [Google Scholar] [CrossRef]
- Xue, Y.; Wu, C.Y.; Branford-White, C.J.; Ning, X.; Nie, H.L.; Zhu, L.M. Chemical Modification of Stem Bromelain with Anhydride Groups to Enhance Its Stability and Catalytic Activity. J. Mol. Catal. B Enzym. 2010, 63, 188–193. [Google Scholar] [CrossRef]
- Matsumoto, M.; Nakagawa, T.; Uchida, Y.; Seki, K.; Ohba, M.; Kondo, K. Effect of Modification of Citraconic Anhydrides on Catalytic Activity and Thermostability of Enzymes. J. Chem. Technol. Biotechnol. 2016, 91, 59–64. [Google Scholar] [CrossRef]
- Hassani, L.; Nourozi, R. Modification of Lysine Residues of Horseradish Peroxidase and Its Effect on Stability and Structure of the Enzyme. Appl. Biochem. Biotechnol. 2014, 172, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, M.B.; Yee, L.H.; Poljak, A.; Cavicchioli, R.; Kjelleberg, S.J.; Siddiqui, K.S. Enhancement of Lipase Stability and Productivity through Chemical Modification and Its Application to Latex-Based Polymer Emulsions. Process Biochem. 2017, 57, 131–140. [Google Scholar] [CrossRef]
- Solanki, K.; Gupta, M.N. A Chemically Modified Lipase Preparation for Catalyzing the Transesterification Reaction in Even Highly Polar Organic Solvents. Bioorg. Med. Chem. Lett. 2011, 21, 2934–2936. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Klok, H.A. Polymer-Protein Conjugates: An Enzymatic Activity Perspective. Polym. Chem. 2010, 1, 1352–1373. [Google Scholar] [CrossRef]
- Benson, K.R.; Gorecki, J.; Nikiforov, A.; Tsui, W.; Kasi, R.M.; Kumar, C.V. Cytochrome: C-Poly(Acrylic Acid) Conjugates with Improved Peroxidase Turnover Number. Org. Biomol. Chem. 2019, 17, 4043–4048. [Google Scholar] [CrossRef]
- Kovaliov, M.; Allegrezza, M.L.; Richter, B.; Konkolewicz, D.; Averick, S. Synthesis of Lipase Polymer Hybrids with Retained or Enhanced Activity Using the Grafting-from Strategy. Polymer 2018, 137, 338–345. [Google Scholar] [CrossRef]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine. Chem. Rev. 2017, 177, 7132–7189. [Google Scholar] [CrossRef]
- Shi, R.; Wang, Y. Dual Ionic and Organic Nature of Ionic Liquids. Sci. Rep. 2016, 6, 19644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Hu, Y.; Liu, L.; Jiang, L.; Zou, B.; Huang, H. Enhancing Catalytic Performance of Porcine Pancreatic Lipase by Covalent Modification Using Functional Ionic Liquids. ACS Catal. 2013, 3, 1976–1983. [Google Scholar] [CrossRef]
- Xu, C.; Suo, H.; Xue, Y.; Qin, J.; Chen, H.; Hu, Y. Experimental and Theoretical Evidence of Enhanced Catalytic Performance of Lipase B from Candida Antarctica Acquired by the Chemical Modification with Amino Acid Ionic Liquids. Mol. Catal. 2021, 501, 111355. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Kamiya, N.; Goto, M. Activation and Stabilization of Enzymes in Ionic Liquids. Org. Biomol. Chem. 2010, 8, 2887–2899. [Google Scholar] [CrossRef]
- Pramanik, S.; Dhoke, G.V.; Jaeger, K.E.; Schwaneberg, U.; Davari, M.D. How to Engineer Ionic Liquids Resistant Enzymes: Insights from Combined Molecular Dynamics and Directed Evolution Study. ACS Sustain. Chem. Eng. 2019, 7, 11293–11302. [Google Scholar] [CrossRef]
- Weingärtner, H.; Cabrele, C.; Herrmann, C. How Ionic Liquids Can Help to Stabilize Native Proteins. Phys. Chem. Chem. Phys. 2012, 14, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Schindl, A.; Hagen, M.L.; Muzammal, S.; Gunasekera, H.A.D.; Croft, A.K. Proteins in Ionic Liquids: Reactions, Applications, and Futures. Front. Chem. 2019, 7, 347. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liang, S.; Zong, M.H.; Lou, W.Y. Ionic Liquids for Regulating Biocatalytic Process: Achievements and Perspectives. Biotechnol. Adv. 2021, 51, 107702. [Google Scholar] [CrossRef]
- Bekhouche, M.; Blum, L.J.; Doumèche, B. Ionic Liquid-Inspired Cations Covalently Bound to Formate Dehydrogenase Improve its Stability and Activity in Ionic Liquids. ChemCatChem 2011, 3, 875–882. [Google Scholar] [CrossRef]
- Nordwald, E.M.; Brunecky, R.; Himmel, M.E.; Beckham, G.T.; Kaar, J.L. Charge Engineering of Cellulases Improves Ionic Liquid Tolerance and Reduces Lignin Inhibition. Biotechnol. Bioeng. 2014, 111, 1541–1549. [Google Scholar] [CrossRef]
- Perez-Rizquez, C.; Lopez-Tejedor, D.; Plaza-Vinuesa, L.; de las Rivas, B.; Muñoz, R.; Cumella, J.; Palomo, J.M. Chemical Modification of Novel Glycosidases from Lactobacillus Plantarum Using Hyaluronic Acid: Effects on High Specificity against 6-Phosphate Glucopyranoside. Coatings 2019, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Brogan, A.P.S.; Bui-Le, L.; Hallett, J.P. Non-Aqueous Homogenous Biocatalytic Conversion of Polysaccharides in Ionic Liquids Using Chemically Modified Glucosidase. Nat. Chem. 2018, 10, 859–865. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Mundra, S.; Belur, P.D. Improving the Catalytic Efficiency of Fibrinolytic Enzyme from Serratia Marcescens Subsp. Sakuensis by Chemical Modification. Process Biochem. 2018, 72, 79–85. [Google Scholar] [CrossRef]
- Wang, C.; Weerapana, E.; Blewett, M.M.; Cravatt, B.F. A Chemoproteomic Platform to Quantitatively Map Targets of Lipid-Derived Electrophiles. Nat. Methods 2014, 11, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.D.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 2010, 468, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Soylu, I.; Marino, S.M. Cy-Preds: An Algorithm and a Web Service for the Analysis and Prediction of Cysteine Reactivity. Proteins 2016, 84, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.; Li, C.; Liu, Y.; Yang, F.; Wang, C. Sequence-Based Prediction of Cysteine Reactivity Using Machine Learning. Biochemistry 2018, 57, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Liu, R.; Shen, J. Predicting Reactive Cysteines with Implicit-Solvent-Based Continuous Constant PH Molecular Dynamics in Amber. J. Chem. Theory Comput. 2020, 16, 3689–3698. [Google Scholar] [CrossRef]
- Adakkattil, R.; Thakur, K.; Rai, V. Reactivity and Selectivity Principles in Native Protein Bioconjugation. Chem. Rec. 2021, 21, 1941–1956. [Google Scholar] [CrossRef]
- Purushottam, L.; Adusumalli, S.R.; Chilamari, M.; Rai, V. Chemoselective and Site-Selective Peptide and Native Protein Modification Enabled by Aldehyde Auto-Oxidation. Chem. Commun. 2017, 53, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Chilamari, M.; Purushottam, L.; Rai, V. Site-Selective Labeling of Native Proteins by a Multicomponent Approach. Chem. Eur. J. 2017, 23, 3819–3823. [Google Scholar] [CrossRef] [PubMed]
- Chilamari, M.; Kalra, N.; Shukla, S.; Rai, V. Single-Site Labeling of Lysine in Proteins through a Metal-Free Multicomponent Approach. Chem. Commun. 2018, 54, 7302–7305. [Google Scholar] [CrossRef] [PubMed]
- Garreau, M.; Le Vaillant, F.; Waser, J. C-Terminal Bioconjugation of Peptides through Photoredox Catalyzed Decarboxylative Alkynylation. Angew. Chem. Int. Ed. 2019, 58, 8182–8186. [Google Scholar] [CrossRef] [PubMed]
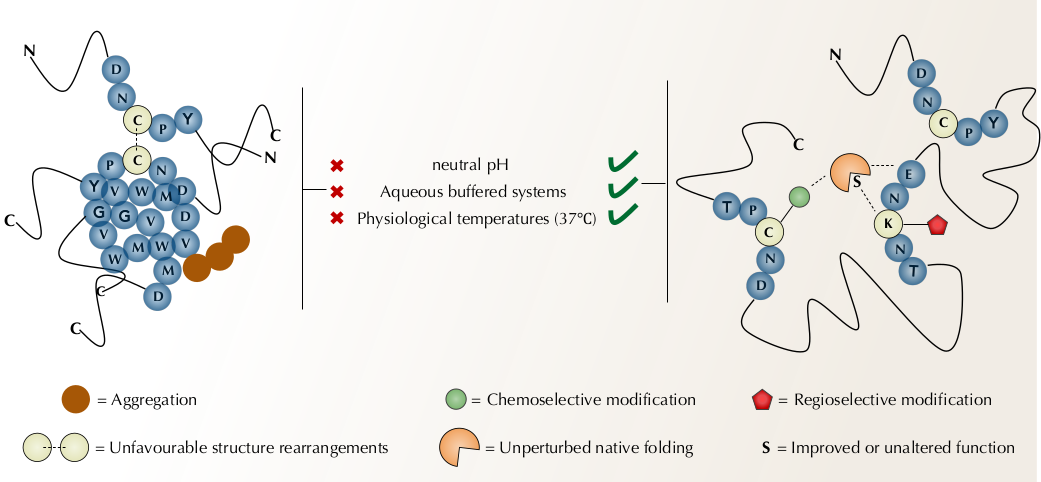


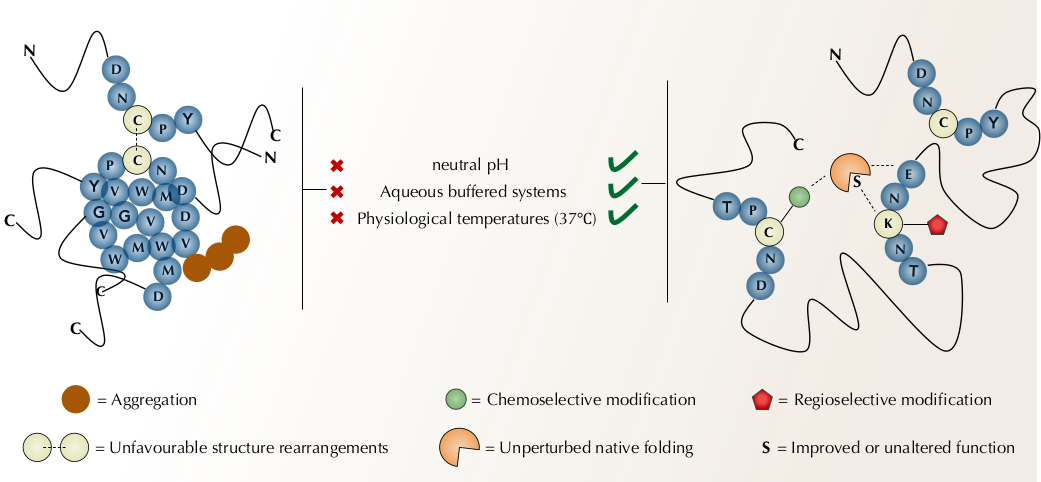{kind=link}
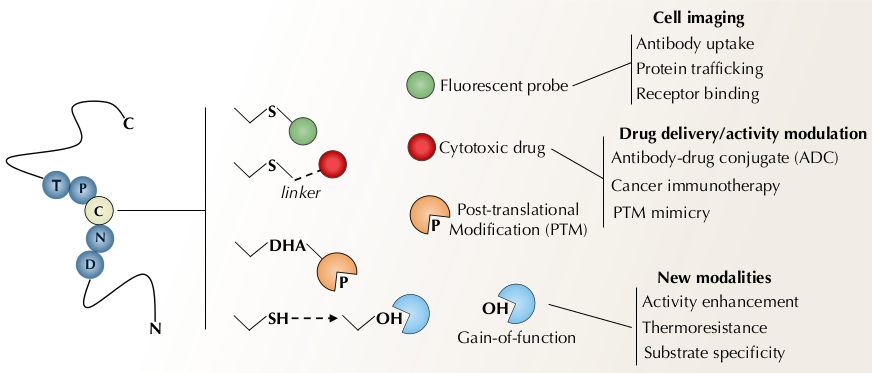{kind=link}
| Modification Strategy and Reagents | Target Group | Protein Scaffold | Functional Implications | Ref. |
|---|---|---|---|---|
| Activation of catalytic serine with PMSF and SN2 displacement with thioacetate | -SH | Subtilisin | Conversion of serine to cysteine. Impaired hydrolytic activity | [15,16] |
| Alkylation with 2-Bromo-2′,4′-dimethoxyacetophenone, photolysis (λ > 320 nm) and reduction with NaBH4 | -SH | Papain | Conversion of cysteine to serine, glycine, or formyl-glycine. Loss of activity | [18] |
| Activation of catalytic serine with PMSF and SN2 displacement with thioacetate | -SH | Trypsin (Streptomyces griseus) | Conversion of serine to cysteine. Impaired hydrolytic activity | [17] |
| 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide activation and conjugation to poly-L-lysine | -COOH | Human Serum Albumin Horseradish peroxidase | Increased cell uptake | [23] |
| Flavin incorporation through alkylation with brominated isoalloxazines | -SH | Papain | Conversion to an oxidoreductase capable of oxidizing dehydronicotinamides and dithiols | [20] |
| PEGylation with 2,4-bis (O-methoxyPEG)-6-chloro-s-triazine (Activated PEG2) | -NH2 | Lipase (Pseudomonas fluorescens) Bovine Chymotrypsin Bovine Catalase Horseradish Peroxidase | Active enzymes in organic solvents | [25] |
| Labeling with 2-iminothiolane hydrochloride, alkylation with 5-iodoacetamido-1,10-phenantroline and coordination of copper ions | -NH2 | Tryptophan repressor (Escherichia coli) | Site-specific oxidative cleavage of DNA | [24] |
| Flavin incorporation with a N-3-alkyl-7-cyanoisoalloxazine | -SH | Hemoglobin | Reductase-independent aniline hydroxylase | [21] |
| Target Residue | Modification Strategy | Protein Scafold | Functional Implications | Ref. |
|---|---|---|---|---|
| Cysteine | Activation of cysteines with DTNB and tethering of alkane-thiols through disulfide exchange | Lipase BTL2 (Geobacillus thermocatenulatus) | Interfacial activation mimic. Increased activity, shifted substrate specificity and improve enantioselectivity | [98] |
| Conversion of cysteine to dehydroalanine with 2,5-dibromohexanediamide followed by the conjugate addition of thiols | N-acetylneuraminic acid lyase (NAL) (Staphylococcus aureus) | Novel substrate specificity | [116,117] | |
| Cysteine alkylation with 2-Iodo-N-phenylacetamide | Lipase (Burkholderia cepacia) | Improved enantioselectivity in the resolution of secondary alcohols | [99] | |
| Anchoring of Grubbs-Hoveyda type catalyst through a maleimide linker | Nitrobindin | Olefin metathesis in aqueous media | [104] | |
| Conjugate addition of a small molecule activator (4-etoxyquinazoline) containing an acrylamide functional group | Glycoside hydrolase BtG84 (Bacteroides thetaiotaomicron) | Improved substrate affinity and catalytic efficiency | [100] | |
| Tethering of iron-binding nitrogen cofactor containing a maleimide linker | Steroid Carrier Protein 2L | Oxidation of β-O-4 linkage in benzylic alcohols | [103] | |
| Lysine | Acetylation with anhydrides | Amylases (Aspergillus carbonarius) (Bacillus licheniformis) (Saccharomycopsis fibuligera) Bromelain (Ananas comosus) Bovine α-chymotrypsin Horseradish peroxidase Lipases (Candida antarctica) (Candida rugosa) (Humicola lanuginosa) | Improved overall stability and resistance to detergents and solvents. Effect on enzyme activity depends on the scaffold | [118,119,120,121,122,123,124,125,126] |
| Grafting of hydroxylated ionic liquids analogous cations with carbonyldiimidazole as a linking agent | Fumarate dehydrogenase (Candida boidinii) | Improved catalytic efficiency in the ionic liquid [MMIm][MeSO4] (30%) | [139] | |
| Grafting of carbonyldiimidazole-activated ionic liquids | Porcine pancreatic lipases | Improved thermostability and enantioselectivity | [132] | |
| Succinylation with succinic anhydride | Trichoderma reesei cellulase cocktail | Improved cellulose conversion in the presence of the ionic liquid [BMIm][Cl] (15%) | [140] | |
| NHS-ester mediated conjugation to N-(iso-butoxymethyl)acrylamide monomers, followed by photoinduced polymerization | Lipase B (Candida antarctica) Lipase (Thermomyces lanuginosus) | Increased enzymatic activity | [129] | |
| Conjugation of carbodiimide-activated polyacrylic acid polymers | Cytochrome C | Increased peroxidase turnover | [128] | |
| Grafting proline-based ionic liquids through carbonyldiimidazole activation | Lipase B (Candida antarctica) | Increased hydrolytic activity and tolerance to organic solvents (methanol and DMSO) | [133] | |
| N-terminus | Copper-free diazotransfer of imidazole-1-sulfonylazide | Lipase B (Candida antarctica) | Labeled enzymes retained their activity and gained an azide functional group for further modification | [88] |
| Alkyne containing ketene reagents | Lysozyme | Labeled enzymes retained their activity and gained an alkyne functional group for further modification | [89] | |
| Conjugation to NHS-containing hyaluronic acids | Glycosidases (Lactobacillus plantarum) | Reduced hydrolytic activity and altered substrate specificity for p-nitrophenylglicosides | [141] | |
| 2-ethynylbenzaldehydes for the formation of isoquinolinium heterocycles | Arginase (Bacillus caldovelox) | Labeled enzymes retained their activity and cytotoxic properties | [91] | |
| Carboxylates | Carbodiimide activation and cationization with a diamine reagent, followed by electrostatic conjugation to glycolic acid ethoxylate lauril ether | Glycosidase (Aspergillus niger) | Improved thermal stability and activity in near anhydrous ionic liquids. | [142] |
| Labeling with 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide | Fibrinolytic enzyme (Serratia marcescens) | Improved affinity for the substrate and increased enzyme activity | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pessatti, T.B.; Terenzi, H.; Bertoldo, J.B. Protein Modifications: From Chemoselective Probes to Novel Biocatalysts. Catalysts 2021, 11, 1466. https://doi.org/10.3390/catal11121466
Pessatti TB, Terenzi H, Bertoldo JB. Protein Modifications: From Chemoselective Probes to Novel Biocatalysts. Catalysts. 2021; 11(12):1466. https://doi.org/10.3390/catal11121466
Chicago/Turabian StylePessatti, Tomás Bohn, Hernán Terenzi, and Jean Borges Bertoldo. 2021. "Protein Modifications: From Chemoselective Probes to Novel Biocatalysts" Catalysts 11, no. 12: 1466. https://doi.org/10.3390/catal11121466
APA StylePessatti, T. B., Terenzi, H., & Bertoldo, J. B. (2021). Protein Modifications: From Chemoselective Probes to Novel Biocatalysts. Catalysts, 11(12), 1466. https://doi.org/10.3390/catal11121466