
The Electrochemical Behaviour of Quaternary Amine-Based Room-Temperature Ionic Liquid N4111(TFSI)

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
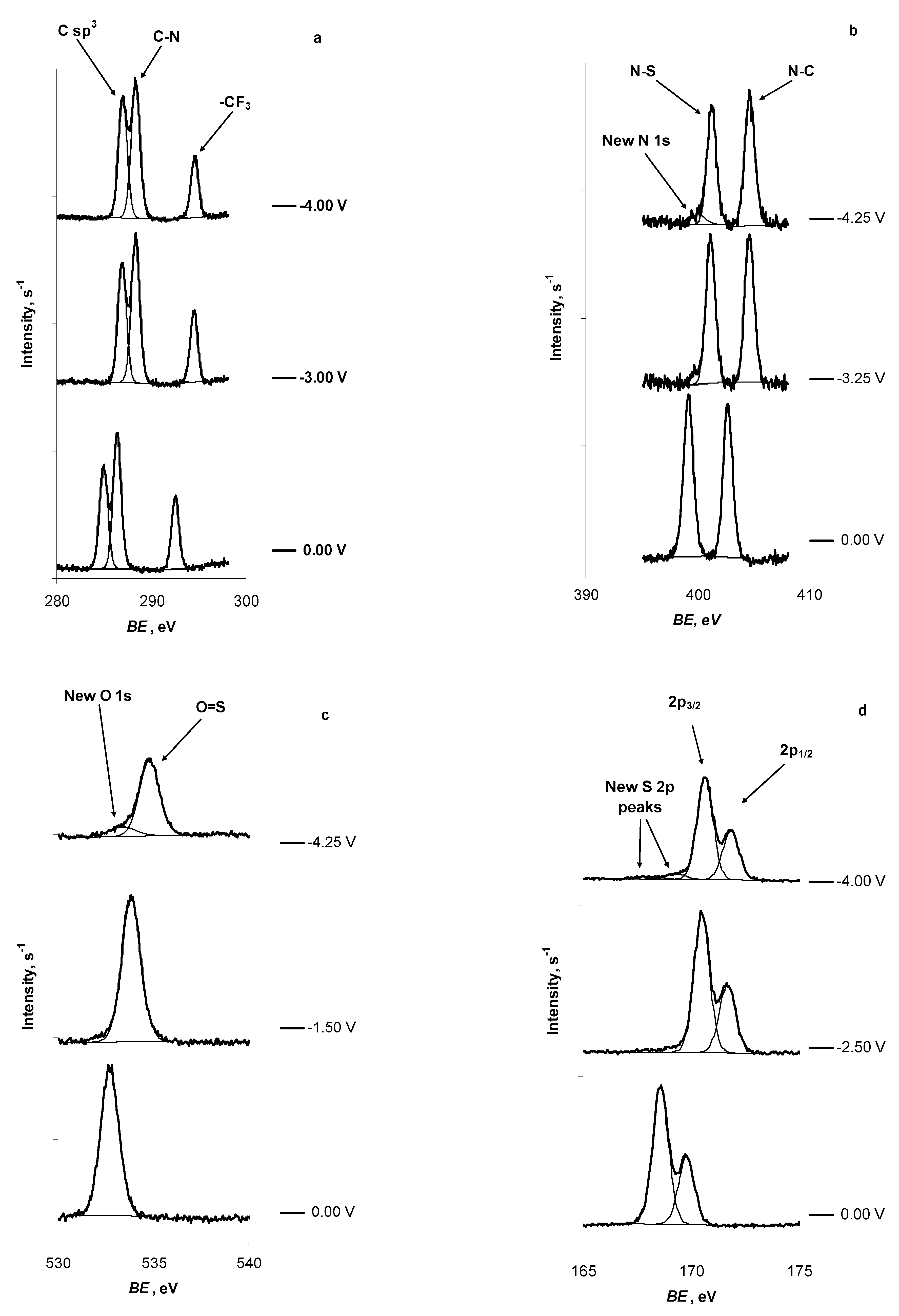
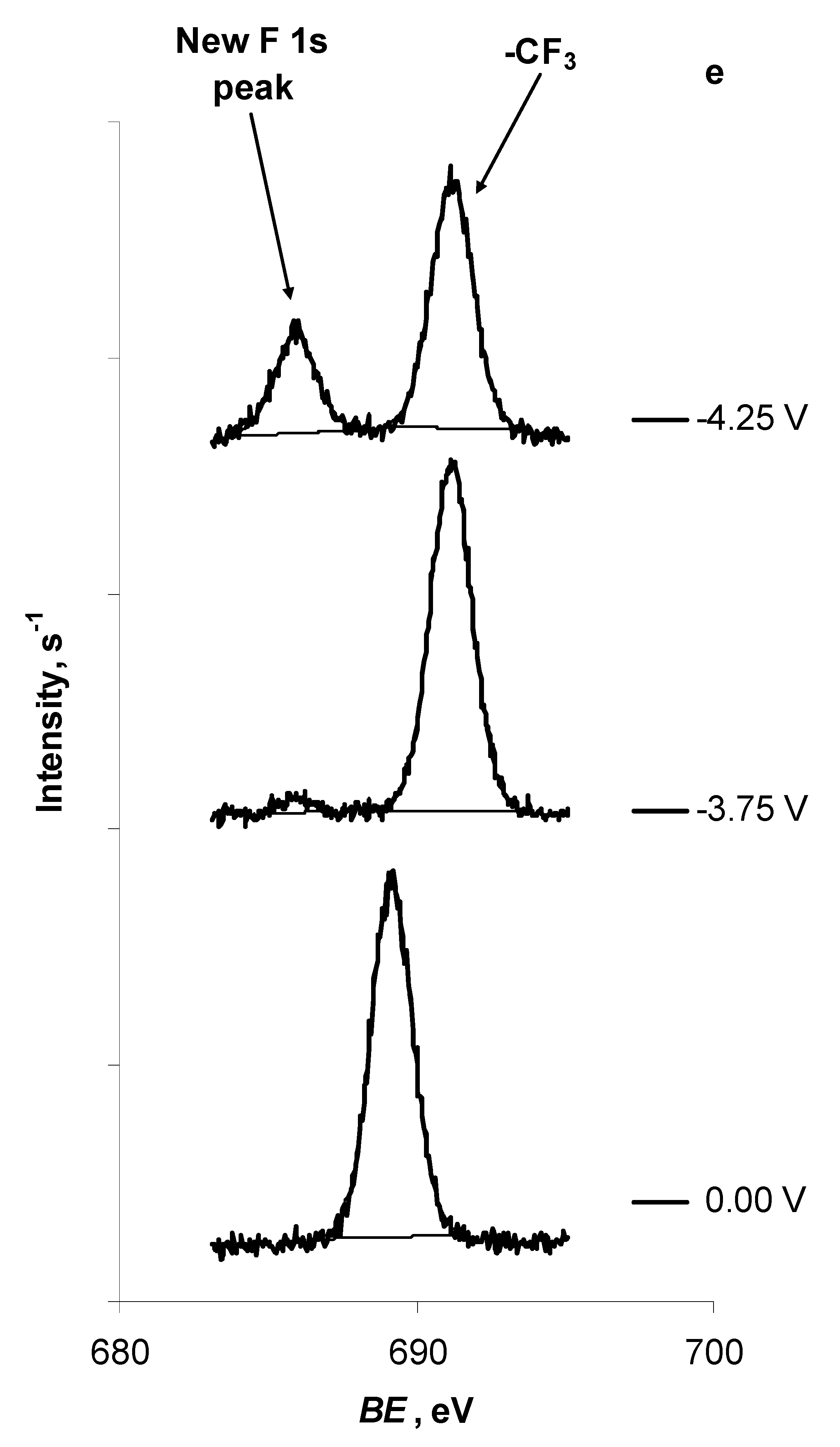
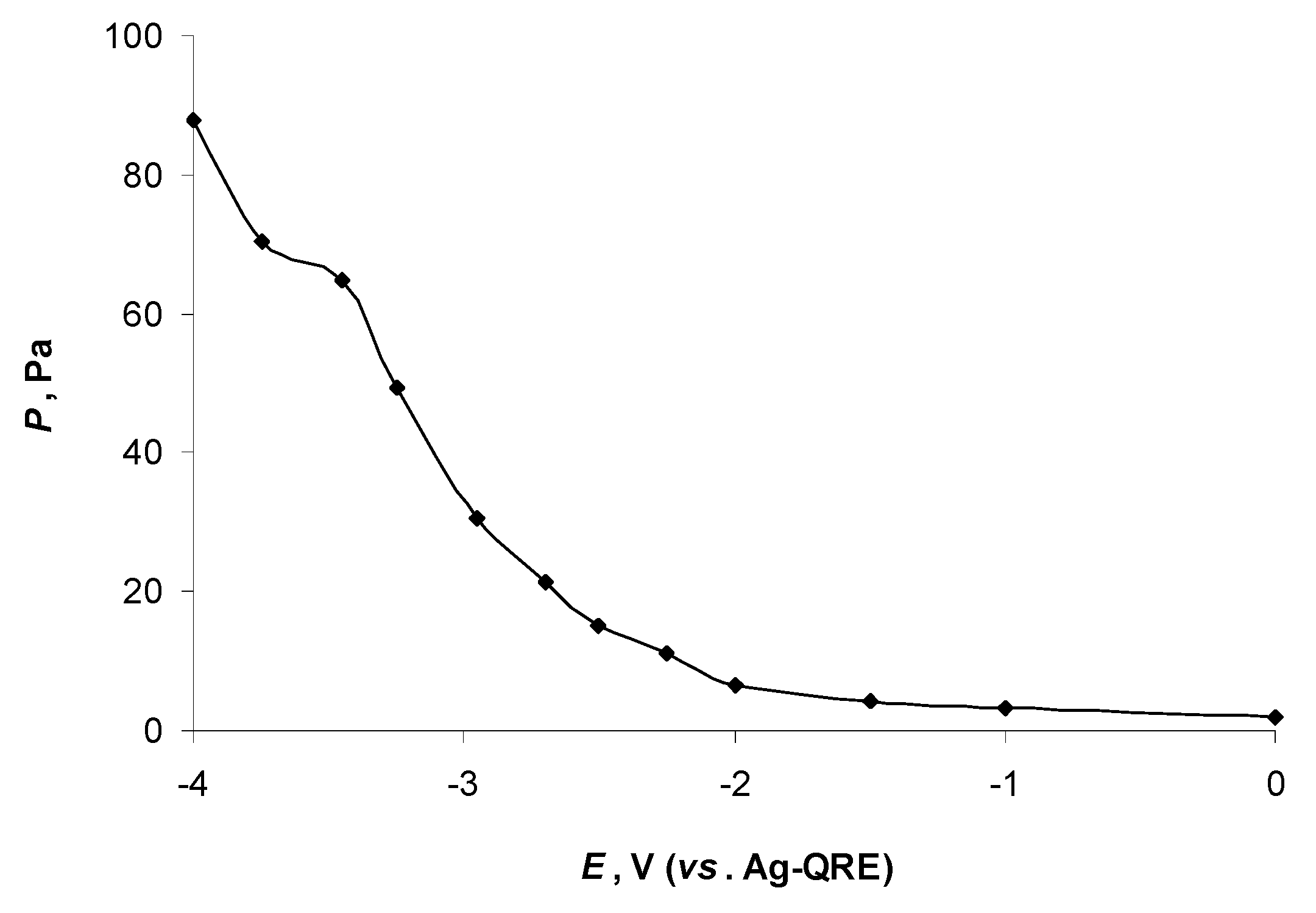
2.1. In Situ XPS Measurements of N4111(TFSI) at the Negatively Polarised mmp-C(Mo2C) Electrode
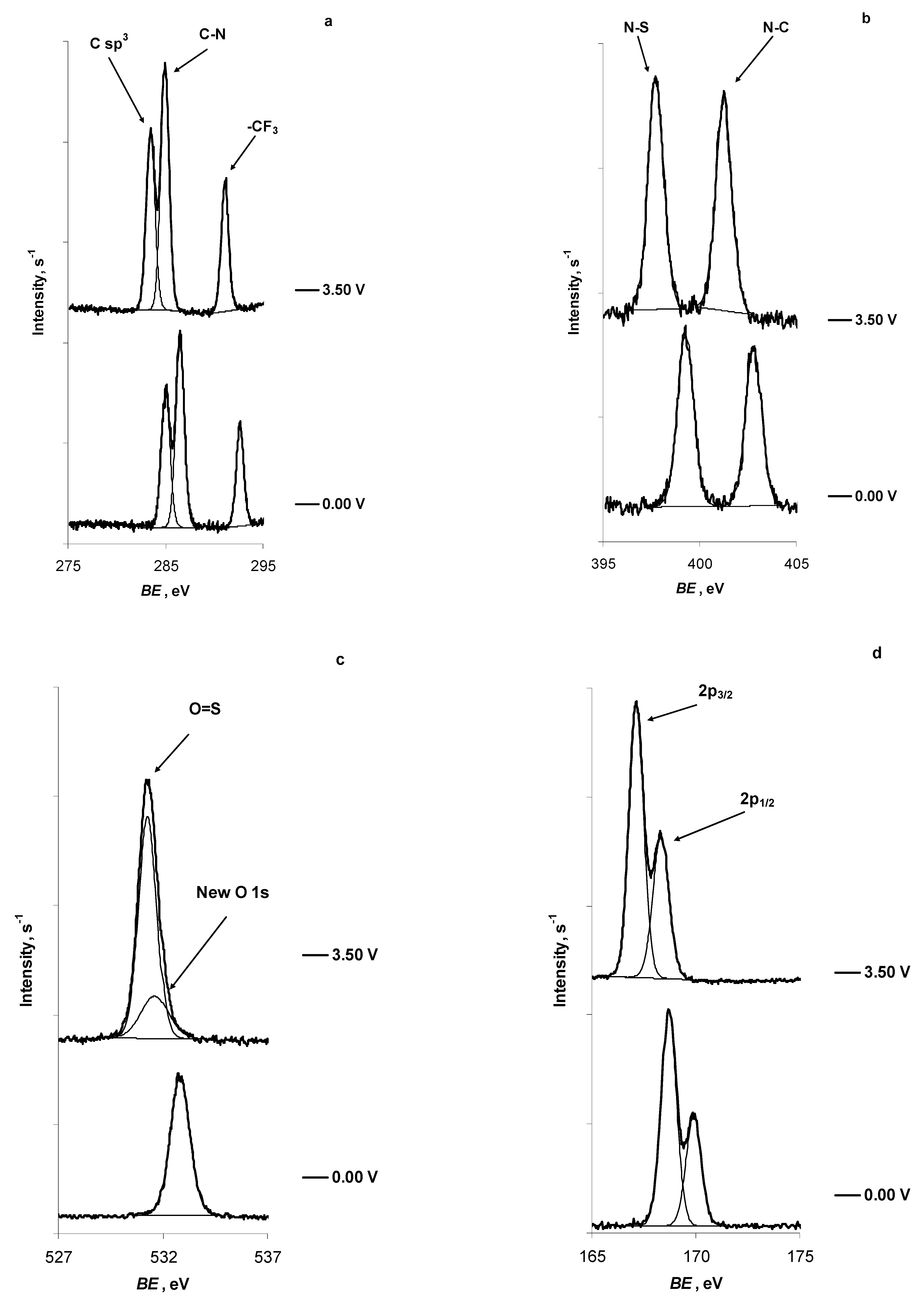
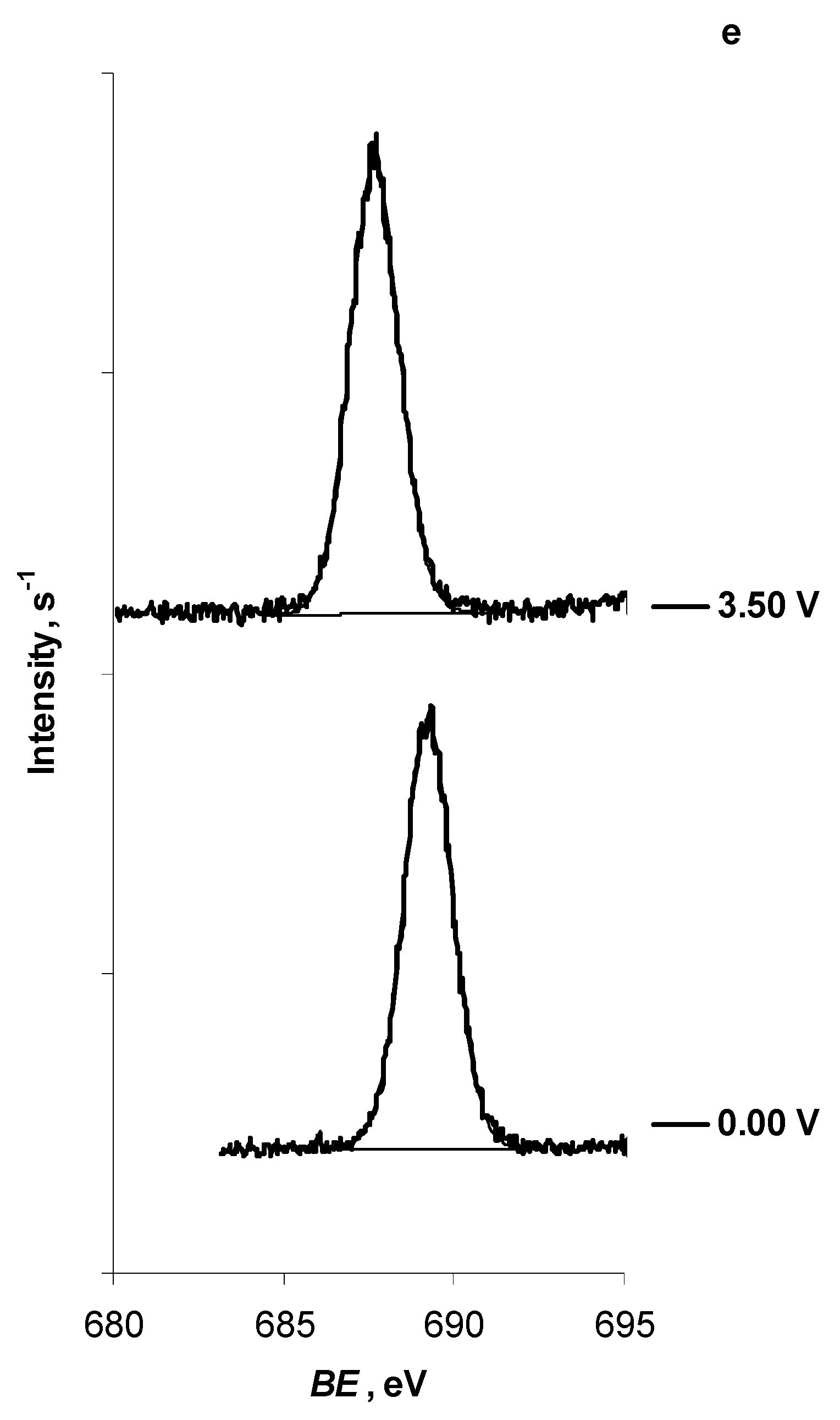
2.2. In Situ XPS Measurements of N4111(TFSI) at the Positively Polarised mmp-C(Mo2C) Electrode
2.3. Mass Spectroscopic Measurements of N4111(TFSI) at the Negatively Polarised mmp-C(Mo2C) Electrode
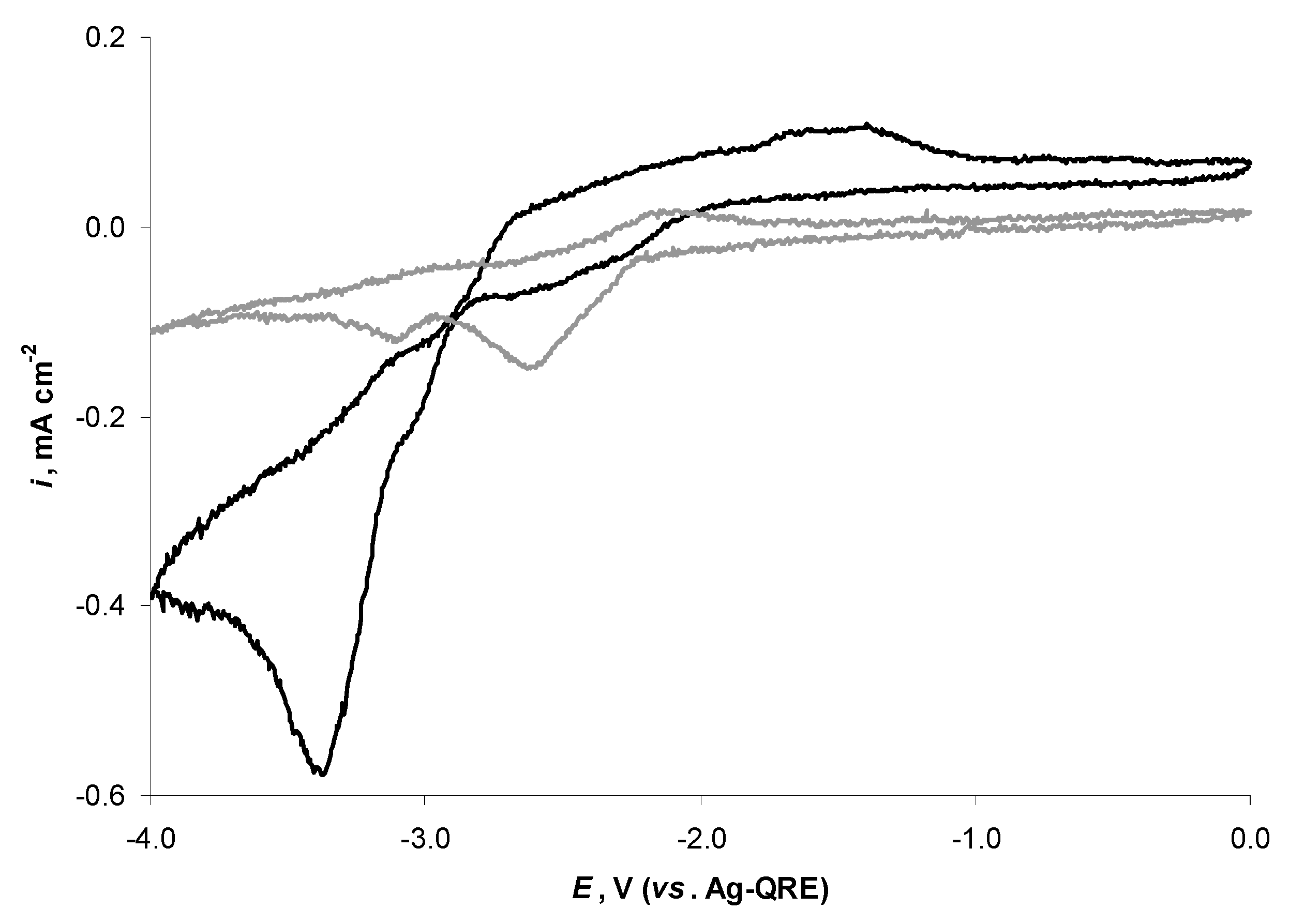
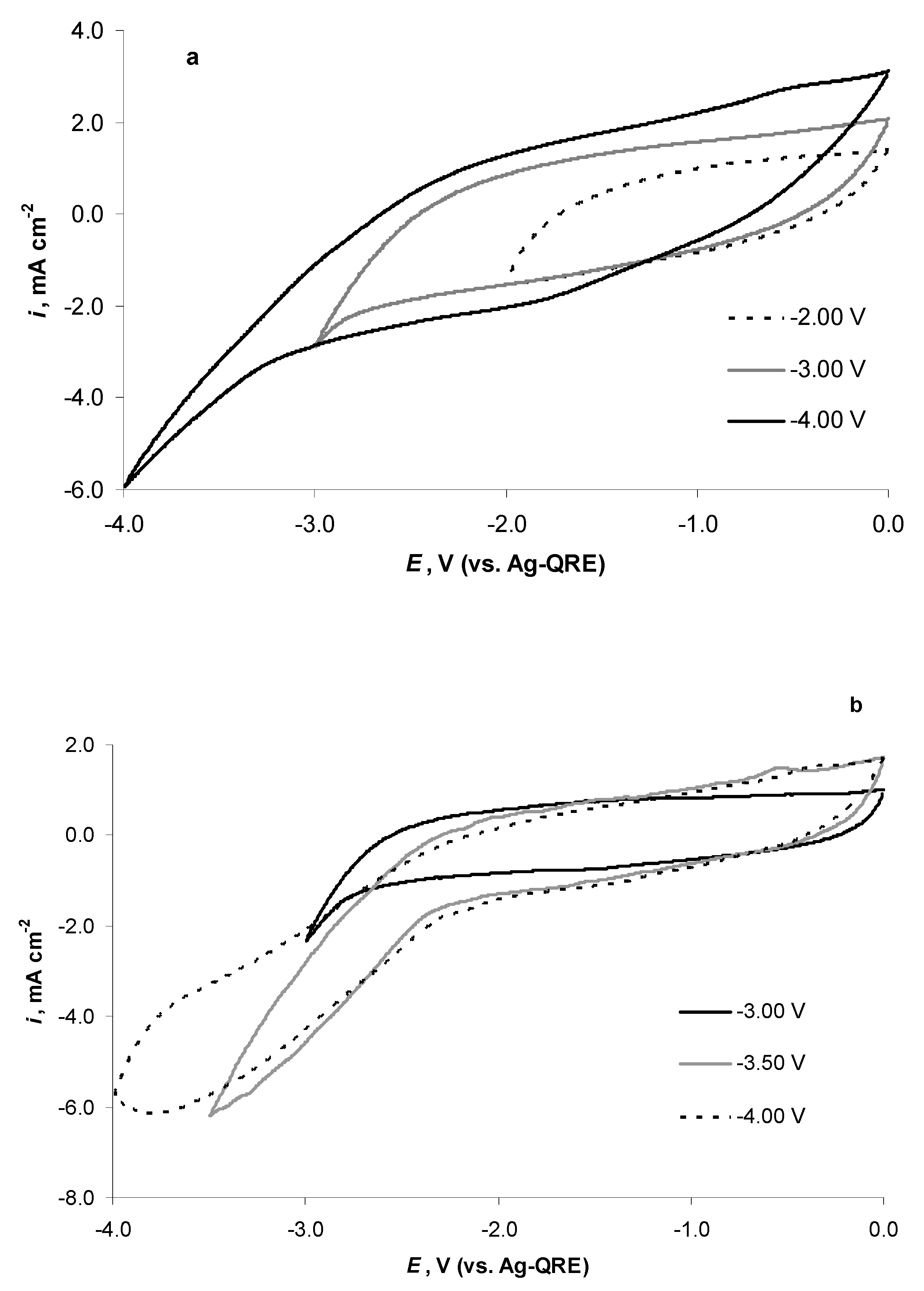
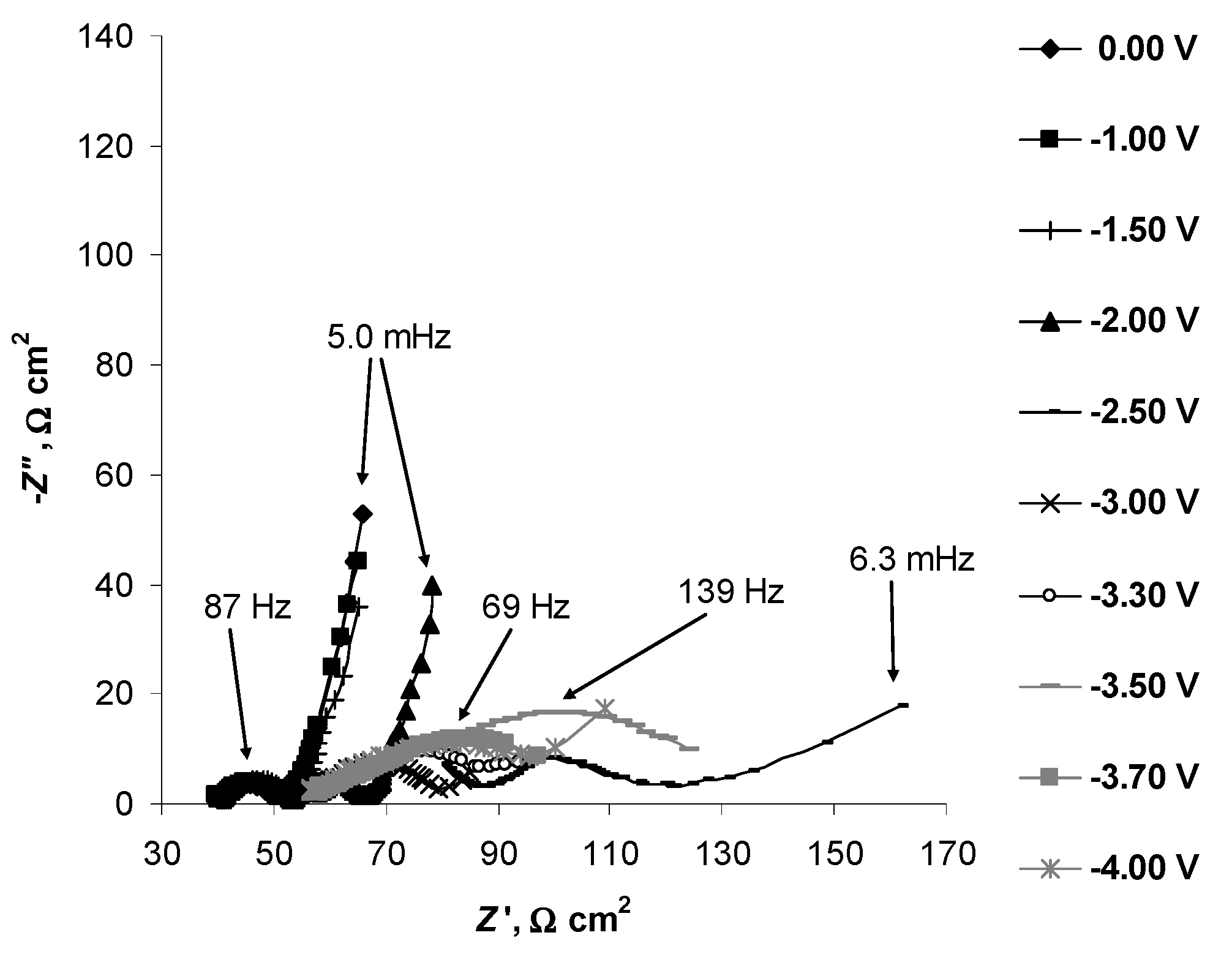
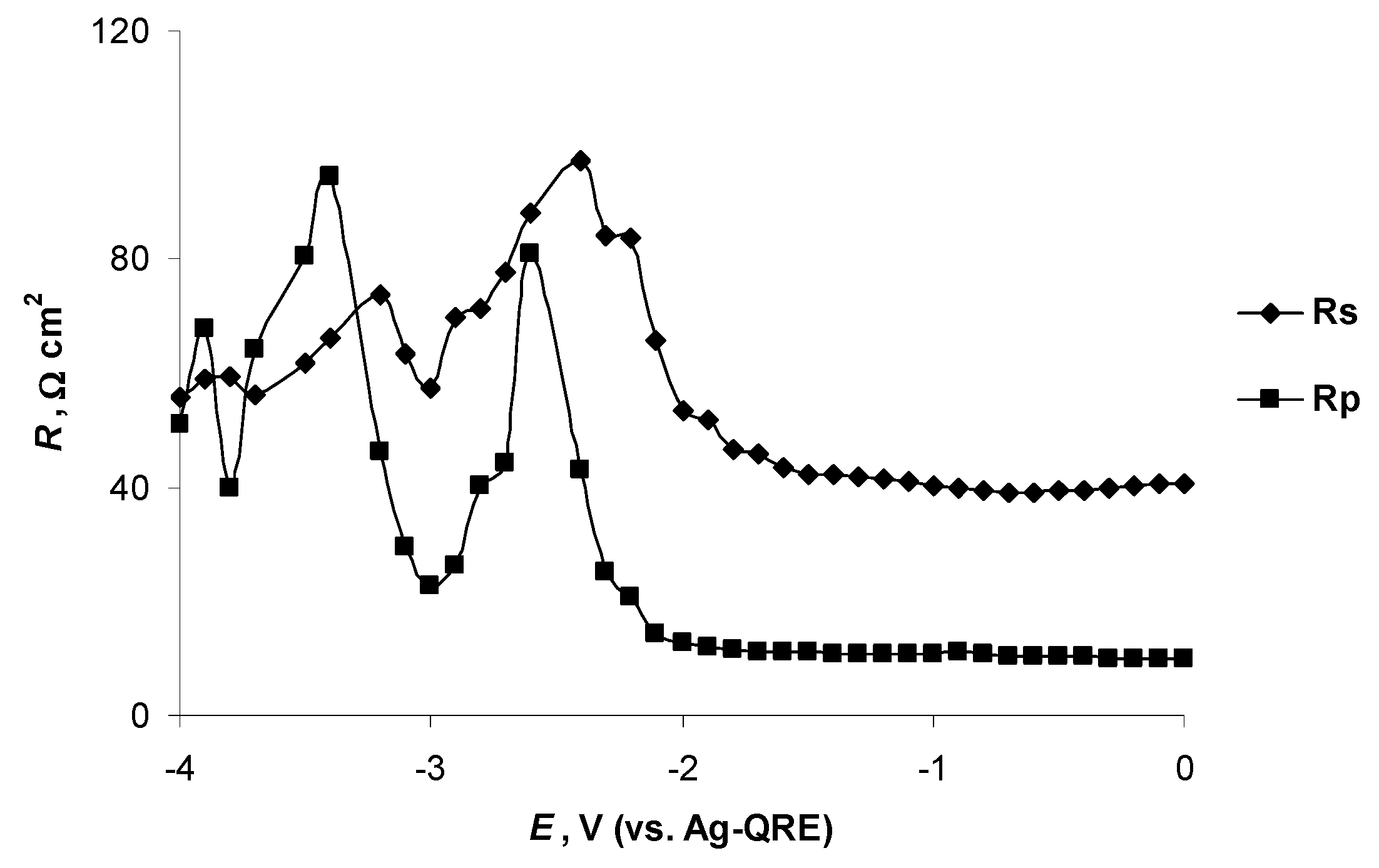
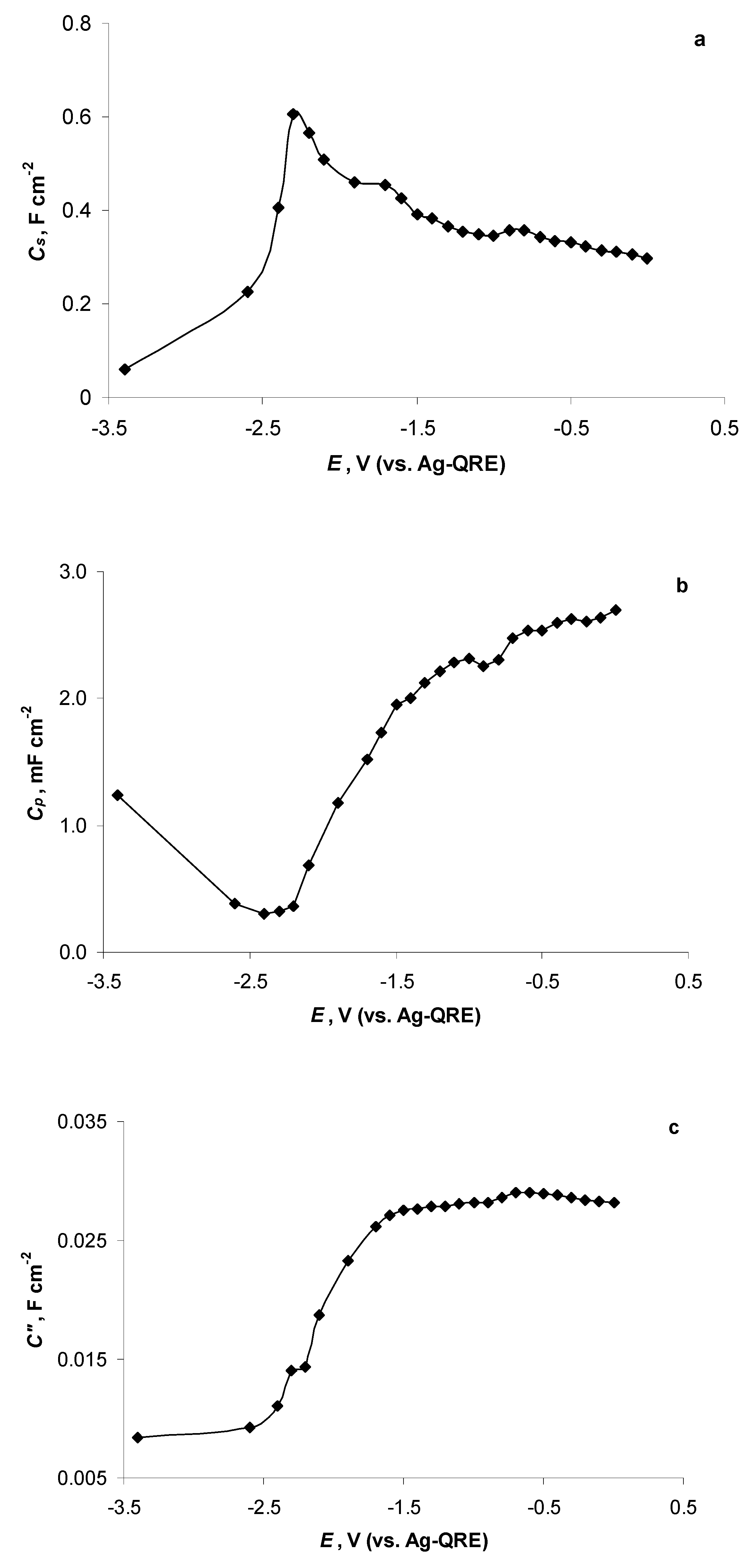
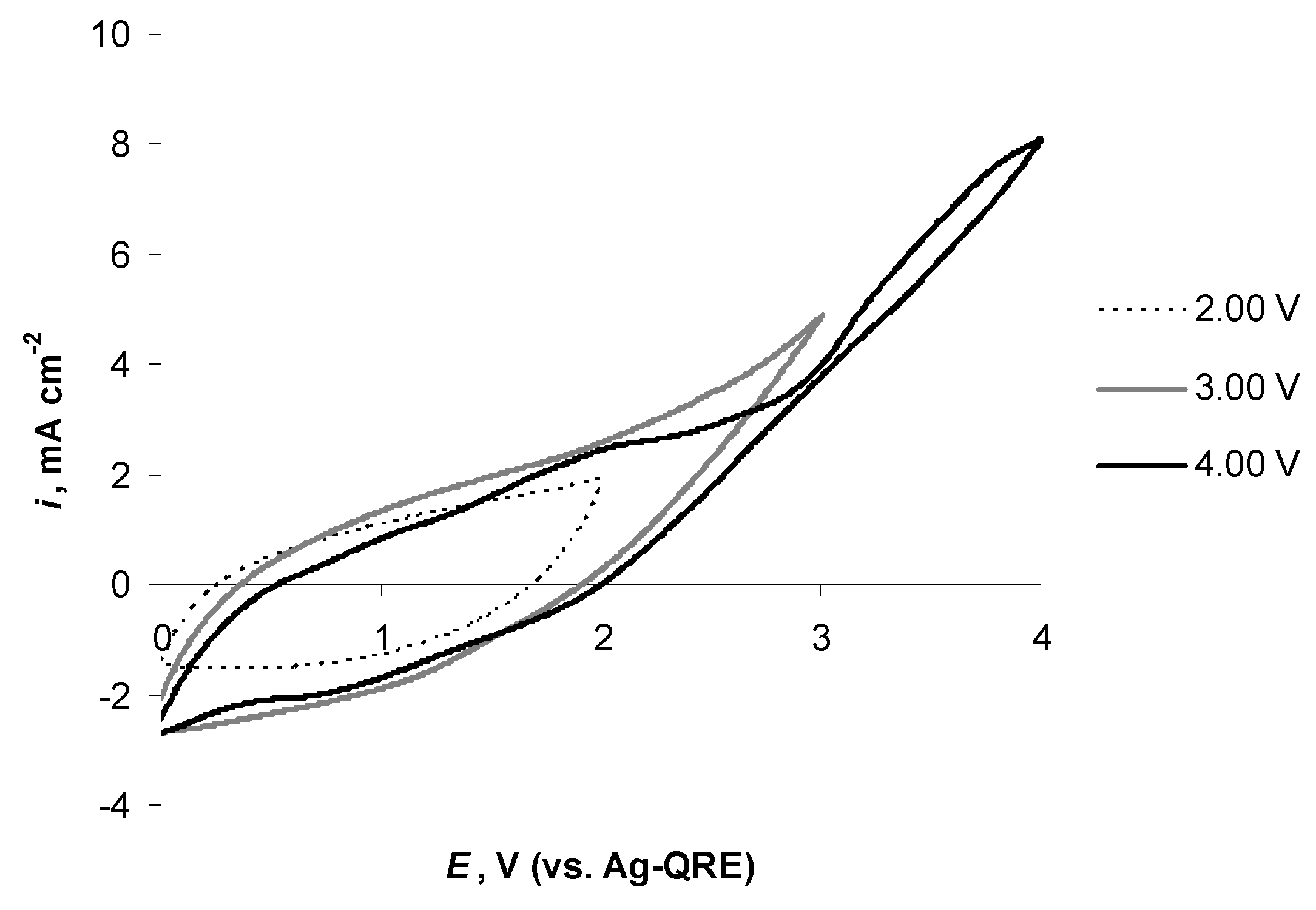
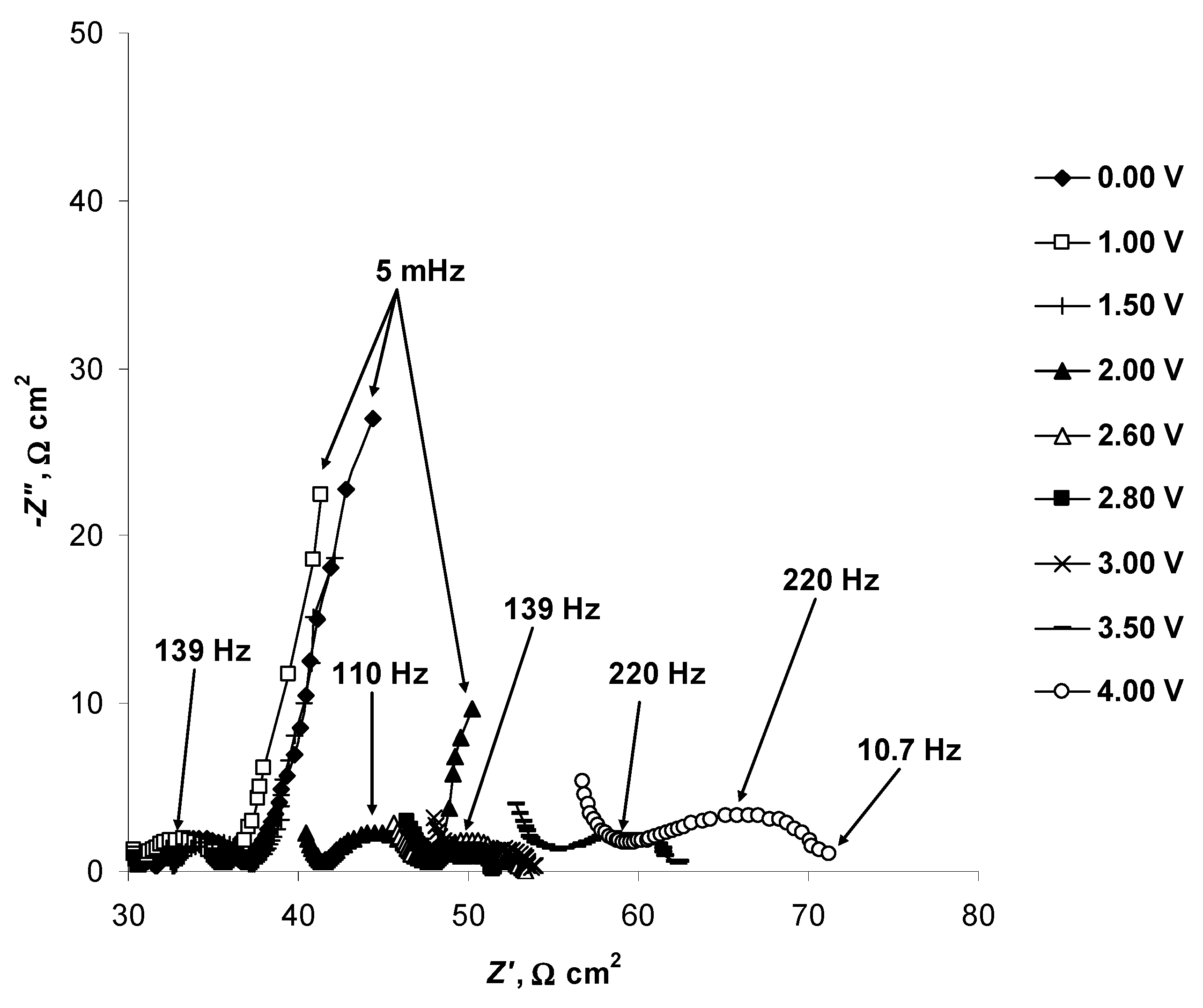
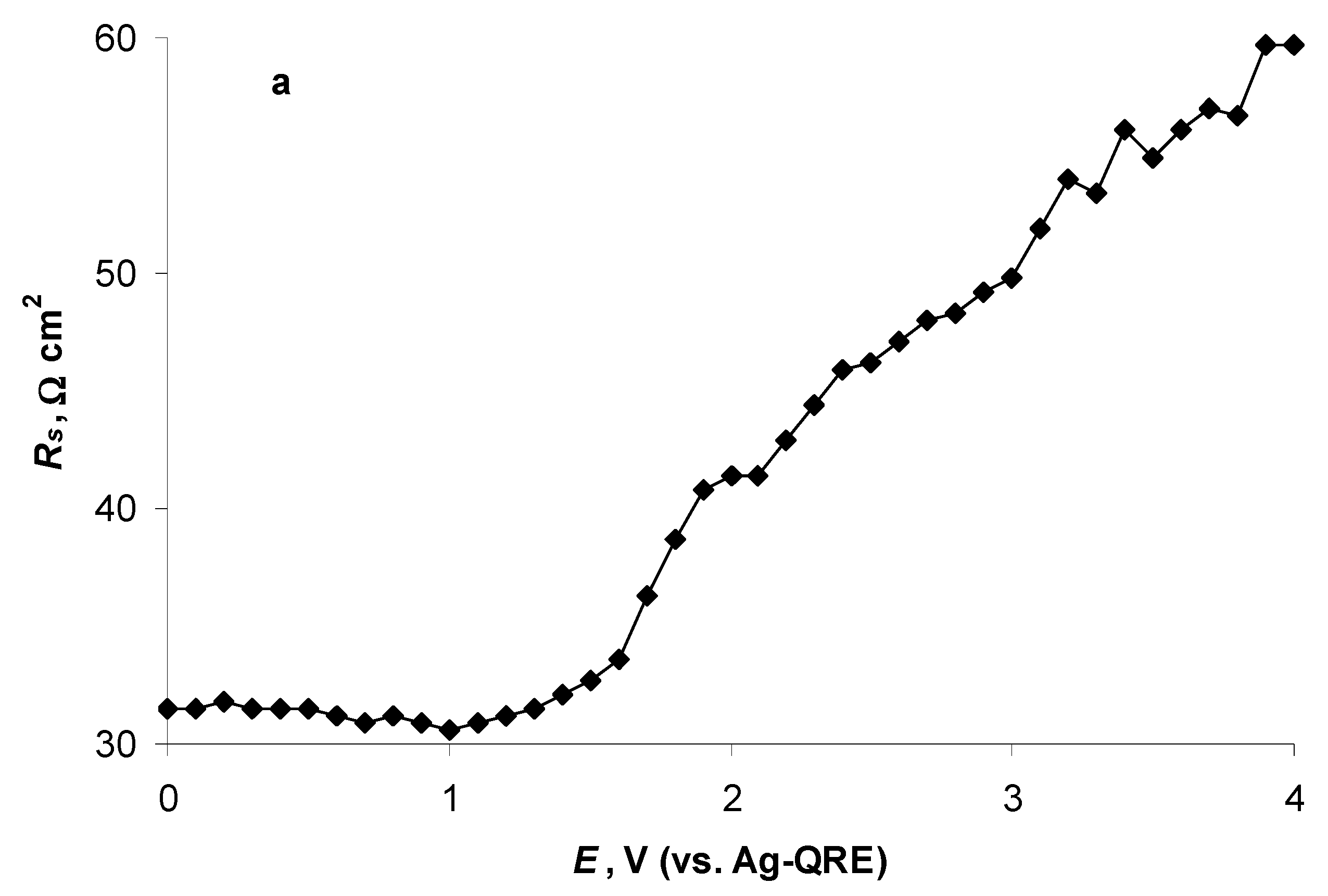
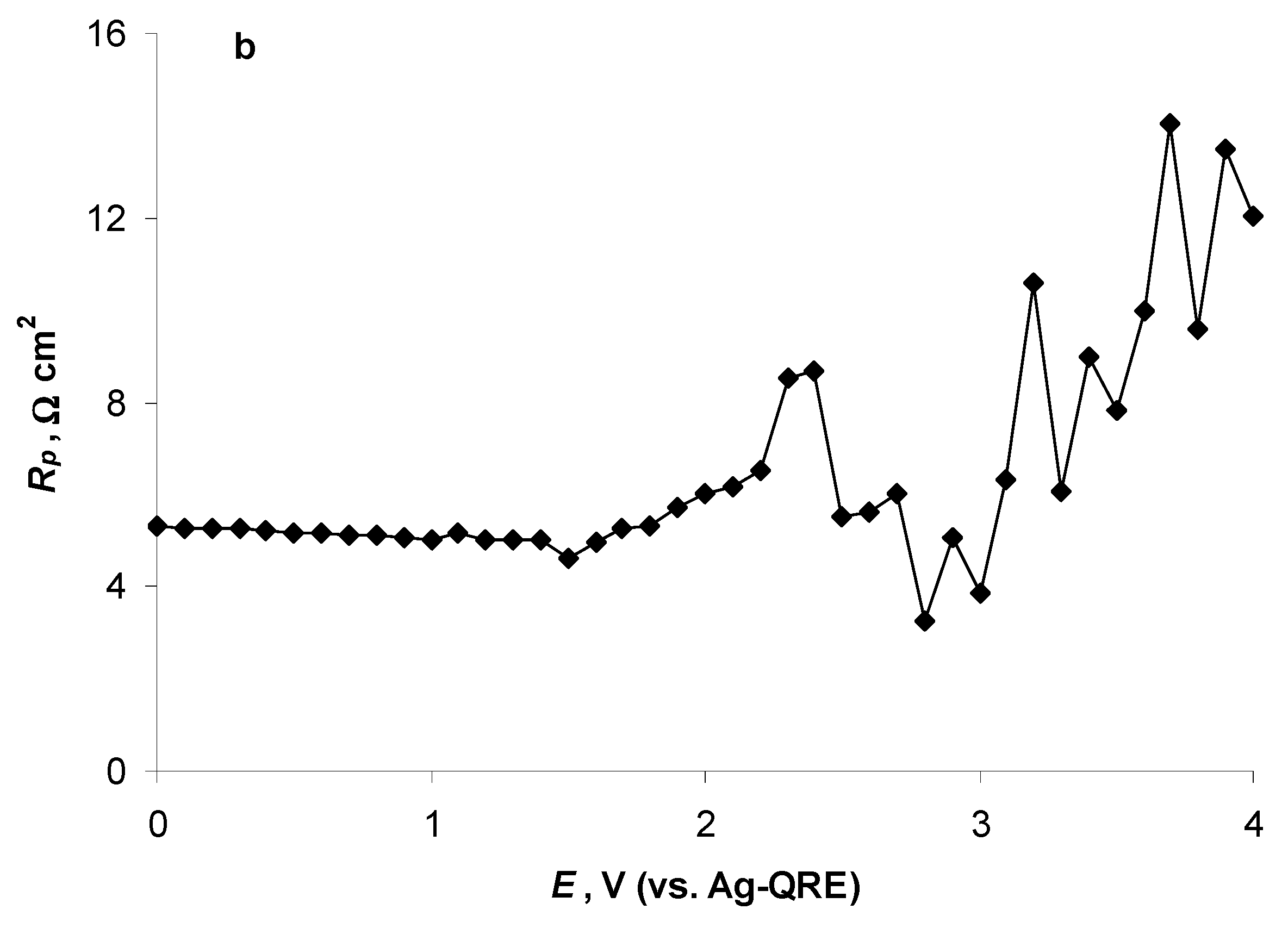
2.4. Electrochemical Behaviour of N4111(TFSI) at the Negatively Polarised mmp-C(Mo2C) Electrode
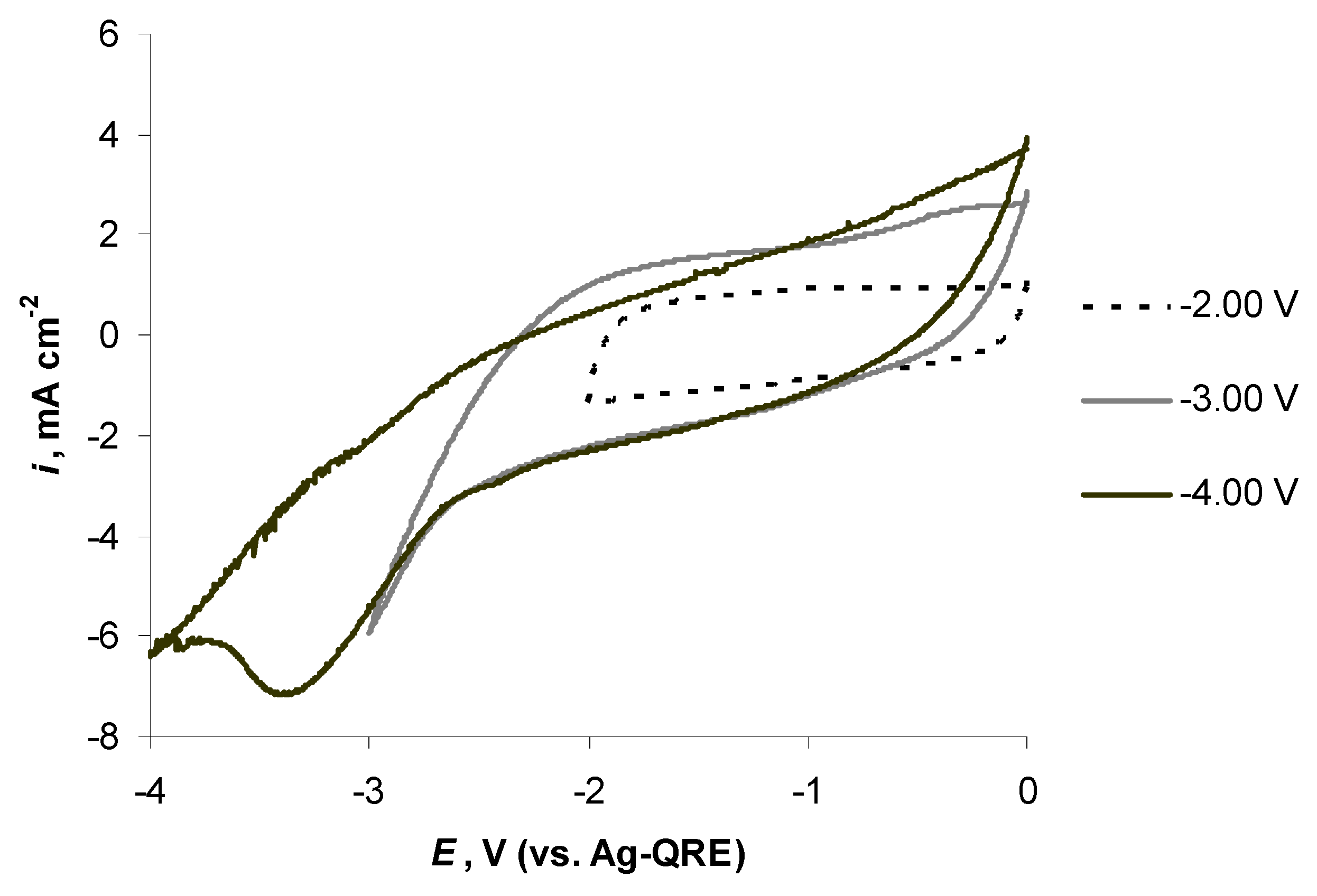
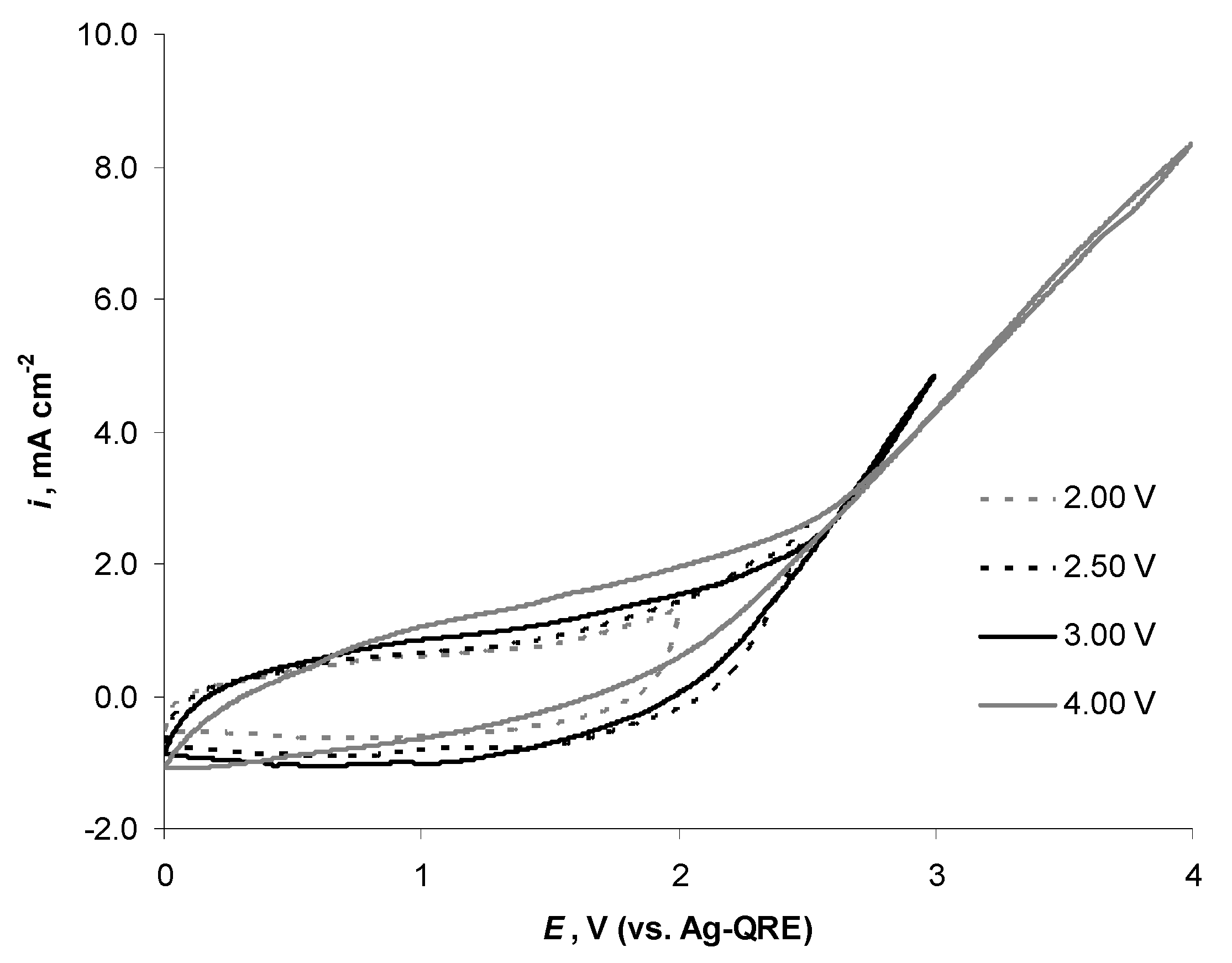
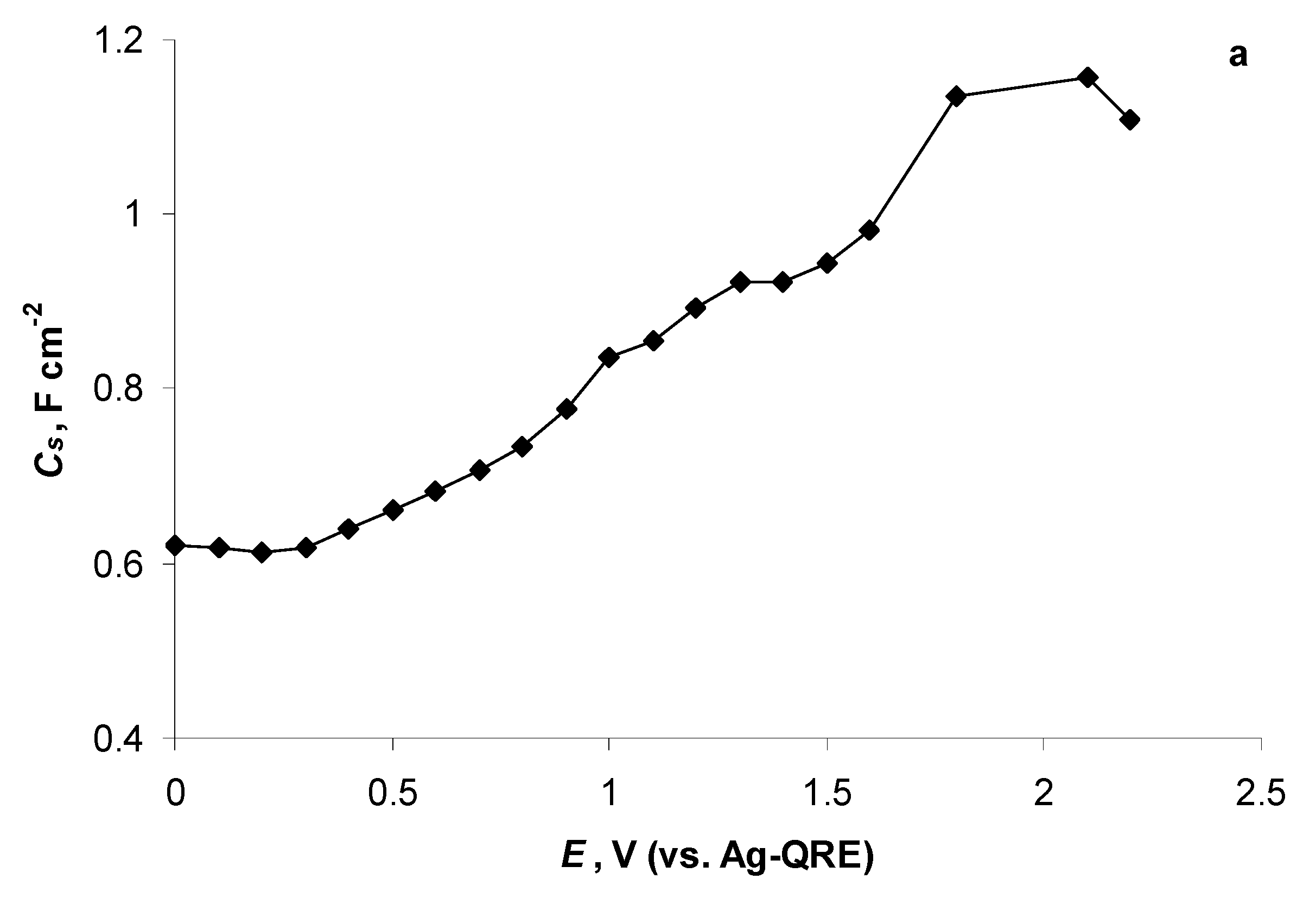
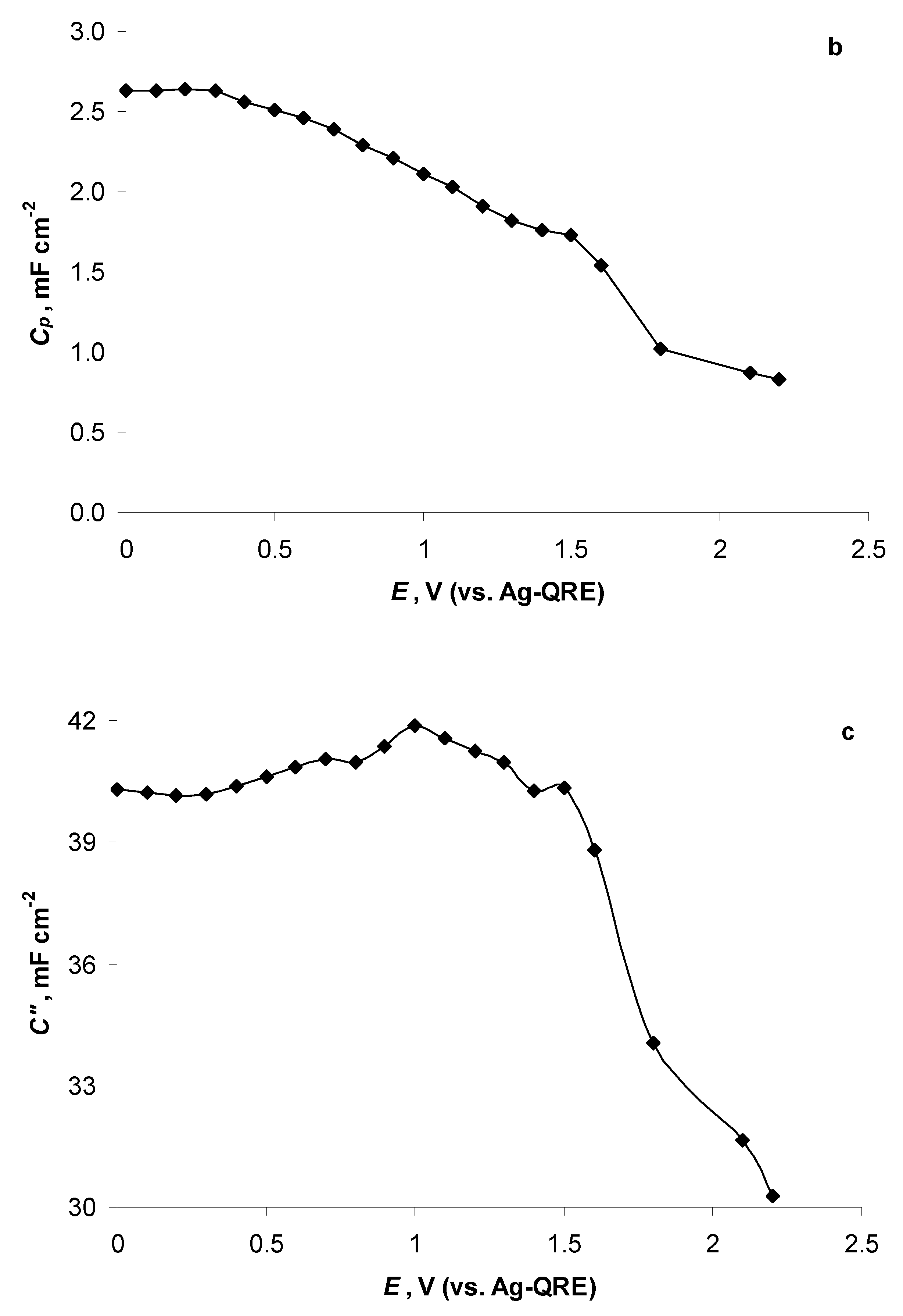
2.5. Electrochemical Behaviour of N4111(TFSI) at Positively Polarised mmp-C(Mo2C) Electrode
3. Experimental
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rao, C.J.; Venkatesan, K.A.; Nagarajan, K.; Srinivasan, T.G.; Rao, P.V. Electrodeposition of metallic uranium at near ambient conditions from room temperature ionic liquid. J. Nucl. Mater. 2011, 408, 25–29. [Google Scholar] [CrossRef]
- Wibowo, R.; Aldous, L.; Jacobs, R.M.; Manan, N.S.; Compton, R.G. Monitoring potassium metal electrodeposition from an ionic liquid using in situ electrochemical-X-ray photoelectron spectroscopy. Chem. Phys. Lett. 2011, 509, 72–76. [Google Scholar] [CrossRef]
- Wibowo, R.; Aldous, L.; Jacobs, R.M.; Manan, N.S.; Compton, R.G. In situ electrochemical-X-ray Photoelectron Spectroscopy: Rubidium metal deposition from an ionic liquid in competition with solvent breakdown. Chem. Phys. Lett. 2011, 517, 103–107. [Google Scholar] [CrossRef]
- Krishna, G.M.; Suneesh, A.S.; Venkatesan, K.A.; Antony, M.P. Electrochemical behavior of zirconium (IV) in 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide ionic liquid. J. Electroanal. Chem. 2016, 776, 120–126. [Google Scholar] [CrossRef]
- Wang, Y.S.; Chen, P.Y. Electrochemical Study and Electrodeposition of Zn-Ni Alloys in an Imide-Type Hydrophobic Room-Temperature Ionic Liquid: Feasibility of Using Metal Chlorides as the Metal Sources. J. Electrochem. Soc. 2018, 165, D76–D82. [Google Scholar] [CrossRef]
- Bourbos, E.; Giannopoulou, I.; Karantonis, A.; Paspaliaris, I.; Panias, D. Reduction of light rare earths and a proposed process for Nd electrorecovery based on ionic liquids. J. Sustain. Metall. 2018, 4, 395–406. [Google Scholar] [CrossRef]
- Broder, T.L.; Silvester, D.S.; Aldous, L.; Hardacre, C.; Crossley, A.; Compton, R.G. The electrochemical oxidation and reduction of nitrate ions in the room temperature ionic liquid [C2mim][NTf2]; the latter behaves as a ‘melt’rather than an ‘organic solvent’. New J. Chem. 2007, 31, 966–972. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Kozuma, Y.; Katayama, Y.; Miura, T. Electrochemical behavior of Ni (II)/Ni in a hydrophobic amide-type room-temperature ionic liquid. Electrochim. Acta 2009, 54, 7502–7506. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Pringle, J.M.; Howlett, P.C.; Forsyth, M. Ionic liquids and reactions at the electrochemical interface. Phys. Chem. Chem. Phys. 2010, 12, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.L.; Katayama, Y.; Miura, T. Effects of Cations on Electrochemical Behavior of Ni (II)/Ni in a Hydrophobic Ionic Liquid. ECS Trans. 2015, 69, 1. [Google Scholar] [CrossRef]
- Gebresilassie Eshetu, G.; Armand, M.; Scrosati, B.; Passerini, S. Energy storage materials synthesized from ionic liquids. Angew. Chem. Int. Ed. 2014, 53, 13342–13359. [Google Scholar] [CrossRef]
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Kim, H. The mechanism of room-temperature ionic-liquid-based electrochemical CO2 reduction: A review. Molecules 2017, 22, 536. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.A.; Hod, I. Tunable Molecular-Scale Materials for Catalyzing the Low-Overpotential Electrochemical Conversion of CO2. Adv. Mater. 2018, 30, 1706238. [Google Scholar] [CrossRef] [PubMed]
- Mota-Lima, A.; Alcântara, M.L.; Pérez-Sanz, F.J.; Bazito, R.C.; Vidinha, P.; de Brito Alves, R.M.; do Nascimento, C.A.O. High-Pressure Carbon Dioxide Separation Using Ionic Liquids: A CO2-Electrocatalysis Perspective. J. Electrochem. Soc. 2021, 168, 086502. [Google Scholar] [CrossRef]
- Bengio, D.; Mendes, E.; Pellet-Rostaing, S.; Moisy, P. Electrochemical behavior of platinum and gold electrodes in the aprotic ionic liquid N, N-Trimethylbutylammonium Bis (trifluoromethanesulfonyl) imide. J. Electroanal. Chem. 2018, 823, 445–454. [Google Scholar] [CrossRef]
- Montiel, M.A.; Solla-Gullón, J.; Sanchez-Sanchez, C.M. Electrochemical reactivity and stability of platinum nanoparticles in imidazolium-based ionic liquids. J. Solid State Electrochem. 2016, 20, 1043–1052. [Google Scholar] [CrossRef]
- Howlett, P.C.; Izgorodina, E.I.; Forsyth, M.; MacFarlane, D.R. Electrochemistry at negative potentials in bis (trifluoromethanesulfonyl) amide ionic liquids. Z. Für Phys. Chem. 2006, 220, 1483–1498. [Google Scholar] [CrossRef]
- Markevich, E.; Sharabi, R.; Borgel, V.; Gottlieb, H.; Salitra, G.; Aurbach, D.; Semrau, G.; Schmidt, M.A. In situ FTIR study of the decomposition of N-butyl-N-methylpyrrolidinium bis (trifluoromethanesulfonyl) amide ionic liquid during cathodic polarization of lithium and graphite electrodes. Electrochim. Acta 2010, 55, 2687–2696. [Google Scholar] [CrossRef]
- Ismail, A.S.; El Abedin, S.Z.; Höfft, O.; Endres, F. Unexpected decomposition of the bis (trifluoromethylsulfonyl) amide anion during electrochemical copper oxidation in an ionic liquid. Electrochem. Commun. 2010, 12, 909–911. [Google Scholar] [CrossRef]
- Lane, G.H. Electrochemical reduction mechanisms and stabilities of some cation types used in ionic liquids and other organic salts. Electrochim. Acta 2012, 83, 513–528. [Google Scholar] [CrossRef]
- Klug, C.L.; Bridges, N.J.; Visser, A.E.; Crump, S.L.; Villa-Aleman, E. Electrochemical degradation of butyltrimethylammonium bis (trifluoromethylsulfonyl) imide for lithium battery applications. New J. Chem. 2014, 38, 3879–3884. [Google Scholar] [CrossRef]
- Mousavi, M.P.; Kashefolgheta, S.; Stein, A.; Bühlmann, P. Electrochemical stability of quaternary ammonium cations: An experimental and computational study. J. Electrochem. Soc. 2016, 163, H74–H80. [Google Scholar] [CrossRef]
- Morozova, P.A.; Luchinin, N.D.; Rupasov, D.P.; Katorova, N.S.; Fedotov, S.S.; Nikitina, V.A.; Stevenson, K.J.; Abakumov, A.M. Electrochemical instability of bis (trifluoromethylsulfonyl) imide based ionic liquids as solvents in high voltage electrolytes for potassium ion batteries. Mendeleev Commun. 2020, 30, 679–682. [Google Scholar] [CrossRef]
- Howlett, P.C.; Brack, N.; Hollenkamp, A.F.; Forsyth, M.; MacFarlane, D.R. Characterization of the Lithium Surface in N-Methyl-N-Alkylpyrrolidinium Bis (trifluoromethanesulfonyl) amide Room-Temperature Ionic Liquid Electrolytes. J. Electrochem. Soc. 2006, 153, A595–A606. [Google Scholar] [CrossRef]
- Tułodziecki, M.; Tarascon, J.M.; Taberna, P.L.; Guéry, C. Catalytic reduction of TFSI-containing ionic liquid in the presence of lithium cations. Electrochem. Commun. 2017, 77, 128–132. [Google Scholar] [CrossRef]
- Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Tallo, I.; Romann, T.; Lust, E. Influence of the negative potential of molybdenum carbide derived carbon electrode on the in situ synchrotron radiation activated X-ray photoelectron spectra of 1-ethyl-3-methylimidazolium tetrafluoroborate. Electrochim. Acta 2016, 206, 419–426. [Google Scholar] [CrossRef]
- Kruusma, J.; Tonisoo, A.; Pärna, R.; Nõmmiste, E.; Vahtrus, M.; Siinor, L.; Tallo, I.; Romann, T.; Lust, E. Influence of iodide ions concentration on the stability of 1-Ethyl-3-methylimidazolium Tetrafluoroborate| molybdenum carbide derived carbon electrode interface. J. Electrochem. Soc. 2017, 164, A1110A–A1119. [Google Scholar] [CrossRef]
- Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Lust, E. In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate. Nanomaterials 2019, 9, 304. [Google Scholar] [CrossRef]
- Quan, T.; Härk, E.; Xu, Y.; Ahmet, I.; Höhn, C.; Mei, S.; Lu, Y. Unveiling the Formation of Solid Electrolyte Interphase and its Temperature Dependence in “Water-in-Salt” Supercapacitors. ACS Appl. Mater. Interfaces 2021, 13, 3979–3990. [Google Scholar] [CrossRef]
- Aurbach, D.; Zaban, A.; Ein-Eli, Y.; Weissman, I.; Chusid, O.; Markovsky, B.; Levi, M.; Levi, E.; Schechter, A.; Granot, E. Recent studies on the correlation between surface chemistry, morphology, three-dimensional structures and performance of Li and Li-C intercalation anodes in several important electrolyte systems. J. Power Sources 1997, 68, 91–98. [Google Scholar] [CrossRef]
- Aurbach, D.; Weissman, I.; Zaban, A.; Chusid, O. Correlation between surface chemistry, morphology, cycling efficiency and interfacial properties of Li electrodes in solutions containing different Li salts. Electrochim. Acta 1994, 39, 51–71. [Google Scholar] [CrossRef]
- Hamm, U.W.; Lazarescu, V.; Kolb, D.M. Adsorption of pyrazine on Au (111) and Ag (111) electrodes an ex situ XPS study. J. Chem. Soc. Faraday Trans. 1996, 92, 3785–3790. [Google Scholar] [CrossRef]
- Zhou, W.; Kolb, D.M. Influence of an electrostatic potential at the metal/electrolyte interface on the electron binding energy of adsorbates as probed by X-ray photoelectron spectroscopy. Surf. Sci. 2004, 573, 176–182. [Google Scholar] [CrossRef]
- Goldman, J.L.; McEwen, A.B. EMIIm and EMIBeti on aluminum anodic stability dependence on lithium salt and propylene carbonate. Electrochem. Solid State Lett. 1999, 2, 501–503. [Google Scholar] [CrossRef]
- Lust, E.; Nurk, G.; Jänes, A.; Arulepp, M.; Permann, L.; Nigu, P.; Möller, P. Electrochemical properties of nanoporous carbon electrodes. Condens. Matter Phys. 2002, 5, 307–327. [Google Scholar] [CrossRef][Green Version]
- Yoon, S.; Jang, J.H.; Bok, H.K.; Oh, S.M. Complex capacitance analysis on rate capability of electric-double layer capacitor (EDLC) electrodes of different thickness. Electrochim. Acta 2005, 50, 2255–2262. [Google Scholar] [CrossRef]
- Jänes, A.; Thomberg, T.; Tõnurist, K.; Kurig, H.; Laheäär, A.; Lust, E. Micro-and mesoporous carbide-derived carbon materials and polymer membranes for supercapacitors. ECS Trans. 2008, 16, 57–67. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, C.W.; Oh, S.M. Characterization of equivalent series resistance of electric double-layer capacitor electrodes using transient analysis. J. Power Sources 2010, 195, 4391–4399. [Google Scholar] [CrossRef]
- Härmas, R.; Palm, R.; Härmas, M.; Pohl, M.; Kurig, H.; Tallo, I.; Tee, E.; Vaas, I.; Väli, R.; Romann, T.; et al. Influence of porosity parameters and electrolyte chemical composition on the power densities of non-aqueous and ionic liquid based supercapacitors. Electrochim. Acta 2018, 283, 931–948. [Google Scholar] [CrossRef]
- Nouri-Nigjeh, E.; de Vries, M.P.; Bruins, A.P.; Bischoff, R.; Permentier, H.P. Electrochemical oxidation of quaternary ammonium electrolytes: Unexpected side reactions in organic electrochemistry. Electrochem. Commun. 2012, 21, 54–57. [Google Scholar] [CrossRef]
- Jänes, A.; Thomberg, T.; Kurig, H.; Lust, E. Nanoscale fine-tuning of porosity of carbide-derived carbon prepared from molybdenum carbide. Carbon 2009, 47, 23–29. [Google Scholar] [CrossRef]
- Kurig, H.; Jänes, A.; Lust, E. Electrochemical characteristics of carbide-derived carbon| 1-ethyl-3-methylimidazolium tetrafluoroborate supercapacitor cells. J. Electrochem. Soc. 2010, 157, A272–A279. [Google Scholar] [CrossRef]
- Thomberg, T.; Jänes, A.; Lust, E. Energy and power performance of electrochemical double-layer capacitors based on molybdenum carbide derived carbon. Electrochim. Acta 2010, 55, 3138–3143. [Google Scholar] [CrossRef]
- Kooser, K.; Kivimäki, A.; Turunen, P.; Pärna, R.; Reisberg, L.; Kirm, M.; Valden, M.; Huttula, M.; Kukk, E. Gas-phase endstation of electron, ion and coincidence spectroscopies for diluted samples at the FinEstBeAMS beamline of the MAX IV 1.5 GeV storage ring. J. Synchrotron Radiat. 2020, 27, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Blundell, R.K.; Licence, P. Quaternary ammonium and phosphonium based ionic liquids: A comparison of common anions. Phys. Chem. Chem. Phys. 2014, 16, 15278–15288. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Ejigu, A.; Licence, P.; Walsh, D.A. Hydrogen oxidation and oxygen reduction at platinum in protic ionic liquids. J. Phys. Chem. C 2012, 116, 18048–18056. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C 1s | -CF3 | C sp3 | C-N | New Peak |
|---|---|---|---|---|
| E = 0.00 V | 292.55 | 284.95 | 286.40 | — |
| E = −3.00 V | 294.50 | 286.90 | 288.30 | — |
| E = −4.00 V | 294.60 | 286.95 | 288.35 | — |
| N 1s | N-C | N-S | New peak | |
| E = 0.00 V | 402.70 | 399.20 | — | |
| E = −4.25 V | 404.65 | 401.25 | 400.15 | |
| O 1s | O=S | New peak | ||
| E = 0.00 V | 532.75 | — | ||
| E = −4.25 V | 534.80 | 533.40 | ||
| S 2p | 2p3/2 | 2p1/2 | New peak 1 | New peak 2 |
| E = 0.00 V | 168.60 | 169.80 | — | — |
| E = −4.00 V | 170.65 | 171.85 | 167.95 | 169.35 |
| F 1s | -CF3 | New peak | ||
| E = 0.00 V | 689.15 | — | ||
| E = −3.75 V | 691.15 | 685.90 | ||
| E = −4.25 V | 691.20 | 685.90 |
| C 1s | -CF3 | C sp3 | C-N |
|---|---|---|---|
| −2.00 V < E < 0.00 V | −0.90 | −0.91 | −0.90 |
| −4.00 V < E < −2.00 V | −0.13 | −0.12 | −0.08 |
| N 1s | N-C | N-S | |
| −2.00 V < E < 0.00 V | −0.89 | −0.90 | |
| −4.00 V < E < −2.00 V | −0.09 | −0.13 | |
| O 1s | O=S | ||
| −2.00 V < E < 0.00 V | −0.89 | ||
| −4.00 V < E < −2.00 V | −0.13 | ||
| S 2p | 2p3/2 | 2p1/2 | |
| −2.00 V < E < 0.00 V | −0.92 | −0.92 | |
| −4.00 V < E < −2.00 V | −0.12 | −0.12 | |
| F 1s | -CF3 | ||
| −2.00 V < E < 0.00 V | −0.92 | ||
| −4.00 V < E < −2.00 V | −0.11 |
| C 1s | -CF3 | C sp3 | C-N | New Peak |
| E = 0.00 V | 292.65 | 285.05 | 286.50 | — |
| E = 3.50 V | 291.10 | 283.50 | 284.95 | — |
| N 1s | N-C | N-S | New peak | |
| E = 0.00 V | 402.80 | 399.30 | — | |
| E = 3.50 V | 401.25 | 397.50 | — | |
| O 1s | O=S | New peak | ||
| E = 0.00 V | 532.85 | — | ||
| E = 3.50 V | 531.30 | 531.60 | ||
| S 2p | 2p3/2 | 2p1/2 | New peak | |
| E = 0.00 V | 168.7 | 169.90 | — | |
| E = 3.50 V | 167.15 | 168.30 | — | |
| F 1s | -CF3 | New peak | ||
| E = 0.00 V | 689.25 | — | ||
| E = 3.50 V | 687.65 | — |
| C 1s | -CF3 | C sp3 | C-N |
|---|---|---|---|
| 0.00 V < E < 1.80 V | −0.67 | −0.66 | −0.66 |
| 1.80 V < E < 3.50 V | −0.23 | −0.24 | −0.24 |
| N 1s | N-C | N-S | |
| 0.00 V < E < 1.80 V | −0.65 | −0.65 | |
| 1.80 V < E < 3.50 V | −0.24 | −0.24 | |
| O 1s | O=S | ||
| 0.00 V < E < 1.80 V | −0.64 | ||
| 1.80 V < E < 3.50 V | −0.27 | ||
| S 2p | 2p3/2 | 2p1/2 | |
| 0.00 V < E < 1.80 V | −0.67 | −0.67 | |
| 1.80 V < E < 3.50 V | −0.24 | −0.24 | |
| F 1s | 0 | ||
| 0.00 V < E < 1.80 V | −0.63 | ||
| 1.80 V < E < 3.50 V | −0.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruusma, J.; Tõnisoo, A.; Pärna, R.; Thomberg, T.; Kook, M.; Romann, T.; Kisand, V.; Lust, E. The Electrochemical Behaviour of Quaternary Amine-Based Room-Temperature Ionic Liquid N4111(TFSI). Catalysts 2021, 11, 1315. https://doi.org/10.3390/catal11111315
Kruusma J, Tõnisoo A, Pärna R, Thomberg T, Kook M, Romann T, Kisand V, Lust E. The Electrochemical Behaviour of Quaternary Amine-Based Room-Temperature Ionic Liquid N4111(TFSI). Catalysts. 2021; 11(11):1315. https://doi.org/10.3390/catal11111315
Chicago/Turabian StyleKruusma, Jaanus, Arvo Tõnisoo, Rainer Pärna, Thomas Thomberg, Mati Kook, Tavo Romann, Vambola Kisand, and Enn Lust. 2021. "The Electrochemical Behaviour of Quaternary Amine-Based Room-Temperature Ionic Liquid N4111(TFSI)" Catalysts 11, no. 11: 1315. https://doi.org/10.3390/catal11111315
APA StyleKruusma, J., Tõnisoo, A., Pärna, R., Thomberg, T., Kook, M., Romann, T., Kisand, V., & Lust, E. (2021). The Electrochemical Behaviour of Quaternary Amine-Based Room-Temperature Ionic Liquid N4111(TFSI). Catalysts, 11(11), 1315. https://doi.org/10.3390/catal11111315