Replacing Pyridine with Pyrazine in Molecular Cobalt Catalysts: Effects on Electrochemical Properties and Aqueous H2 Generation

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Catalyst Synthesis
2.2. X-ray Crystallography
2.3. Electrochemistry
2.4. Electron Paramagnetic Resonance (EPR) Spectroscopy
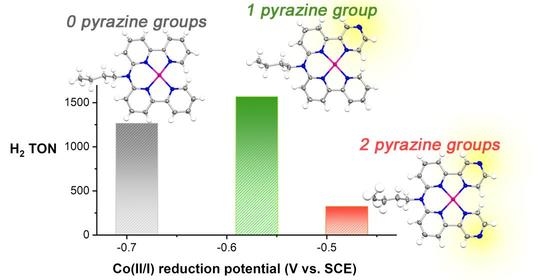
2.5. H2 Photocatalysis
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Synthesis of Complexes 1–4
4.3. Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Synthesis Schemes





References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.G. Chemistry of Personalized Solar Energy. Inorg. Chem. 2009, 48, 10001–10017. [Google Scholar] [CrossRef]
- Ashford, D.L.; Gish, M.K.; Vannucci, A.K.; Brennaman, M.K.; Templeton, J.L.; Papanikolas, J.M.; Meyer, T.J. Molecular Chromophore–Catalyst Assemblies for Solar Fuel Applications. Chem. Rev. 2015, 115, 13006–13049. [Google Scholar] [CrossRef] [PubMed]
- Matheu, R.; Garrido-Barros, P.; Gil-Sepulcre, M.; Ertem, M.Z.; Sala, X.; Gimbert-Suriñach, C.; Llobet, A. The development of molecular water oxidation catalysts. Nat. Rev. Chem. 2019, 3, 331–341. [Google Scholar] [CrossRef]
- Concepcion, J.J.; House, R.L.; Papanikolas, J.M.; Meyer, T.J. Chemical approaches to artificial photosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, W.T.; Eisenberg, R. Molecular systems for light driven hydrogen production. Dalton Trans. 2012, 41, 13004–13021. [Google Scholar] [CrossRef]
- Berardi, S.; Drouet, S.; Francàs, L.; Gimbert-Suriñach, C.; Guttentag, M.; Richmond, C.; Stoll, T.; Llobet, A. Molecular artificial photosynthesis. Chem. Soc. Rev. 2014, 43, 7501–7519. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabehere-Mallart, E.; Robert, M. Molecular catalysis of CO(2)reduction: Recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef]
- Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M. Splitting Water with Cobalt. Angew. Chem. Int. Ed. 2011, 50, 7238–7266. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 2012, 5, 6012–6021. [Google Scholar] [CrossRef]
- Nippe, M.; Khnayzer, R.S.; Panetier, J.A.; Zee, D.Z.; Olaiya, B.S.; Head-Gordon, M.; Chang, C.J.; Castellano, F.N.; Long, J.R. Catalytic proton reduction with transition metal complexes of the redox-active ligand bpy2PYMe. Chem. Sci. 2013, 4, 3934–3945. [Google Scholar] [CrossRef]
- Queyriaux, N.; Jane, R.T.; Massin, J.; Artero, V.; Chavarot-Kerlidou, M. Recent developments in hydrogen evolving molecular cobalt(II)–polypyridyl catalysts. Coord. Chem. Rev. 2015, 304–305, 3–19. [Google Scholar] [CrossRef]
- Hogue, R.W.; Schott, O.; Hanan, G.S.; Brooker, S. A Smorgasbord of 17 Cobalt Complexes Active for Photocatalytic Hydrogen Evolution. Chem.-Eur. J. 2018, 24, 9820–9832. [Google Scholar] [CrossRef]
- Dolui, D.; Khandelwal, S.; Majumder, P.; Dutta, A. The odyssey of cobaloximes for catalytic H2 production and their recent revival with enzyme-inspired design. Chem. Commun. 2020, 56, 8166–8181. [Google Scholar] [CrossRef]
- Jurss, J.W.; Khnayzer, R.S.; Panetier, J.A.; El Roz, K.A.; Nichols, E.M.; Head-Gordon, M.; Long, J.R.; Castellano, F.N.; Chang, C.J. Bioinspired design of redox-active ligands for multielectron catalysis: Effects of positioning pyrazine reservoirs on cobalt for electro- and photocatalytic generation of hydrogen from water. Chem. Sci. 2015, 6, 4954–4972. [Google Scholar] [CrossRef]
- Wang, P.; Liang, G.; Reddy, M.R.; Long, M.; Driskill, K.; Lyons, C.; Donnadieu, B.; Bollinger, J.C.; Webster, C.E.; Zhao, X. Electronic and Steric Tuning of Catalytic H2 Evolution by Cobalt Complexes with Pentadentate Polypyridyl-Amine Ligands. J. Am. Chem. Soc. 2018, 140, 9219–9229. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-Assisted Reduction of CO2 by Cobalt Aminopyridine Macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef]
- Chapovetsky, A.; Welborn, M.; Luna, J.M.; Haiges, R.; Miller, T.F.; Marinescu, S.C. Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction. ACS Cent. Sci. 2018, 4, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, S.; Zamader, A.; Nagayach, V.; Dolui, D.; Mir, A.Q.; Dutta, A. Inclusion of Peripheral Basic Groups Activates Dormant Cobalt-Based Molecular Complexes for Catalytic H2 Evolution in Water. ACS Catal. 2019, 9, 2334–2344. [Google Scholar] [CrossRef]
- Dolui, D.; Ghorai, S.; Dutta, A. Tuning the reactivity of cobalt-based H2 production electrocatalysts via the incorporation of the peripheral basic functionalities. Coord. Chem. Rev. 2020, 416, 213335. [Google Scholar] [CrossRef]
- Kohler, L.; Niklas, J.; Johnson, R.C.; Zeller, M.; Poluektov, O.G.; Mulfort, K.L. Molecular Cobalt Catalysts for H2 Generation with Redox Activity and Proton Relays in the Second Coordination Sphere. Inorg. Chem. 2019, 58, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Reintjens, N.R.M.; Siegler, M.A.; Roubeau, O.; Bouwman, E.; Rudavskyi, A.; Havenith, R.W.A.; Bonnet, S. Stabilization of the Low-Spin State in a Mononuclear Iron(II) Complex and High-Temperature Cooperative Spin Crossover Mediated by Hydrogen Bonding. Chem.-Eur. J. 2016, 22, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Liebscher, J. Carbon−Carbon Coupling Reactions Catalyzed by Heterogeneous Palladium Catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Schrauzer, G.N.; Lee, L.P. Cobaloximes(II) and vitamin B12r as oxygen carriers. Evidence for monomeric and dimeric peroxides and superoxides. J. Am. Chem. Soc. 1970, 92, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Bakac, A.; Brynildson, M.E.; Espenson, J.H. Characterization of the structure, properties, and reactivity of a cobalt(II) macrocyclic complex. Inorg. Chem. 1986, 25, 4108–4114. [Google Scholar] [CrossRef]
- Baumgarten, M.; Lubitz, W.; Winscom, C.J. EPR and ENDOR studies of cobaloxime(II). Chem. Phys. Lett. 1987, 133, 102–108. [Google Scholar] [CrossRef]
- Niklas, J.; Mardis, K.L.; Rakhimov, R.R.; Mulfort, K.L.; Tiede, D.M.; Poluektov, O.G. The Hydrogen Catalyst Cobaloxime: A Multifrequency EPR and DFT Study of Cobaloxime’s Electronic Structure. J. Phys. Chem. B 2012, 116, 2943–2957. [Google Scholar] [CrossRef]
- Mukherjee, A.; Kokhan, O.; Huang, J.; Niklas, J.; Chen, L.X.; Tiede, D.M.; Mulfort, K.L. Detection of a charge-separated catalyst precursor state in a linked photosensitizer-catalyst assembly. Phys. Chem. Chem. Phys. 2013, 15, 21070–21076. [Google Scholar] [CrossRef][Green Version]
- Wirt, M.D.; Bender, C.J.; Peisach, J. Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy of Cobalt(II) Bis(dimethylglyoximes): Equatorial Co-N Coupling Parameters. Inorg. Chem. 1995, 34, 1663–1667. [Google Scholar] [CrossRef]
- DuBois, M.R.; DuBois, D.L. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem. Soc. Rev. 2009, 38, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Khnayzer, R.S.; Thoi, V.S.; Nippe, M.; King, A.E.; Jurss, J.W.; El Roz, K.A.; Long, J.R.; Chang, C.J.; Castellano, F.N. Towards a comprehensive understanding of visible-light photogeneration of hydrogen from water using cobalt(ii) polypyridyl catalysts. Energy Environ. Sci. 2014, 7, 1477–1488. [Google Scholar] [CrossRef]
- Roy, S.; Bacchi, M.; Berggren, G.; Artero, V. A Systematic Comparative Study of Hydrogen-Evolving Molecular Catalysts in Aqueous Solutions. ChemSusChem 2015, 8, 3632–3638. [Google Scholar] [CrossRef]
- Natali, M. Elucidating the Key Role of pH on Light-Driven Hydrogen Evolution by a Molecular Cobalt Catalyst. ACS Catal. 2017, 7, 1330–1339. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Losse, S.; Vos, J.G.; Rau, S. Catalytic hydrogen production at cobalt centres. Coord. Chem. Rev. 2010, 254, 2492–2504. [Google Scholar] [CrossRef]
- Pellegrin, Y.; Odobel, F. Sacrificial electron donor reagents for solar fuel production. Comptes Rendus Chim. 2017, 20, 283–295. [Google Scholar] [CrossRef]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef]
- Andreiadis, E.S.; Jacques, P.-A.; Tran, P.D.; Leyris, A.; Chavarot-Kerlidou, M.; Jousselme, B.; Matheron, M.; Pécaut, J.; Palacin, S.; Fontecave, M.; et al. Molecular engineering of a cobalt-based electrocatalytic nanomaterial for H2 evolution under fully aqueous conditions. Nat. Chem. 2013, 5, 48–53. [Google Scholar] [CrossRef]
- Utschig, L.M.; Soltau, S.R.; Tiede, D.M. Light-driven hydrogen production from Photosystem I-catalyst hybrids. Curr. Opin. Chem. Biol. 2015, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mulfort, K.L.; Utschig, L.M. Modular Homogeneous Chromophore–Catalyst Assemblies. Acc. Chem. Res. 2016, 49, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.N.; Surendranath, Y. Molecular Control of Heterogeneous Electrocatalysis through Graphite Conjugation. Acc. Chem. Res. 2019, 52, 3432–3441. [Google Scholar] [CrossRef] [PubMed]
- Guiton, B.S.; Stefik, M.; Augustyn, V.; Banerjee, S.; Bardeen, C.J.; Bartlett, B.M.; Li, J.; López-Mejías, V.; MacGillivray, L.R.; Morris, A.; et al. Frontiers in hybrid and interfacial materials chemistry research. MRS Bull. 2020, 45, 951–964. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E1/2 (Co(II/I)) (V vs. SCE) a | E1/2 (L0/−) (V vs. SCE) | E1/2 (L−/2−) (V vs. SCE) | Initial TOF (H2/Co/h) b | TON at 4 h (H2/Co) | |
|---|---|---|---|---|---|
| O-CAT | −0.69 | −1.30 | −1.64 | 1017 ± 5 | 1268 |
| 1 | −0.54 | −1.07 | −1.51 | 450 ± 1 | 629 |
| 2 | −0.57 | −1.08 | −1.52 | 3419 ± 25 | 1569 |
| 3 | −0.42 | −0.89 | −1.38 | 376 ± 1 | 272 |
| 4 | −0.48 | −0.86 | −1.37 | 538 ± 9 | 324 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohler, L.; Potocny, A.M.; Niklas, J.; Zeller, M.; Poluektov, O.G.; Mulfort, K.L. Replacing Pyridine with Pyrazine in Molecular Cobalt Catalysts: Effects on Electrochemical Properties and Aqueous H2 Generation. Catalysts 2021, 11, 75. https://doi.org/10.3390/catal11010075
Kohler L, Potocny AM, Niklas J, Zeller M, Poluektov OG, Mulfort KL. Replacing Pyridine with Pyrazine in Molecular Cobalt Catalysts: Effects on Electrochemical Properties and Aqueous H2 Generation. Catalysts. 2021; 11(1):75. https://doi.org/10.3390/catal11010075
Chicago/Turabian StyleKohler, Lars, Andrea M. Potocny, Jens Niklas, Matthias Zeller, Oleg G. Poluektov, and Karen L. Mulfort. 2021. "Replacing Pyridine with Pyrazine in Molecular Cobalt Catalysts: Effects on Electrochemical Properties and Aqueous H2 Generation" Catalysts 11, no. 1: 75. https://doi.org/10.3390/catal11010075
APA StyleKohler, L., Potocny, A. M., Niklas, J., Zeller, M., Poluektov, O. G., & Mulfort, K. L. (2021). Replacing Pyridine with Pyrazine in Molecular Cobalt Catalysts: Effects on Electrochemical Properties and Aqueous H2 Generation. Catalysts, 11(1), 75. https://doi.org/10.3390/catal11010075