Photocatalytic Methane Reforming: Recent Advances


Abstract

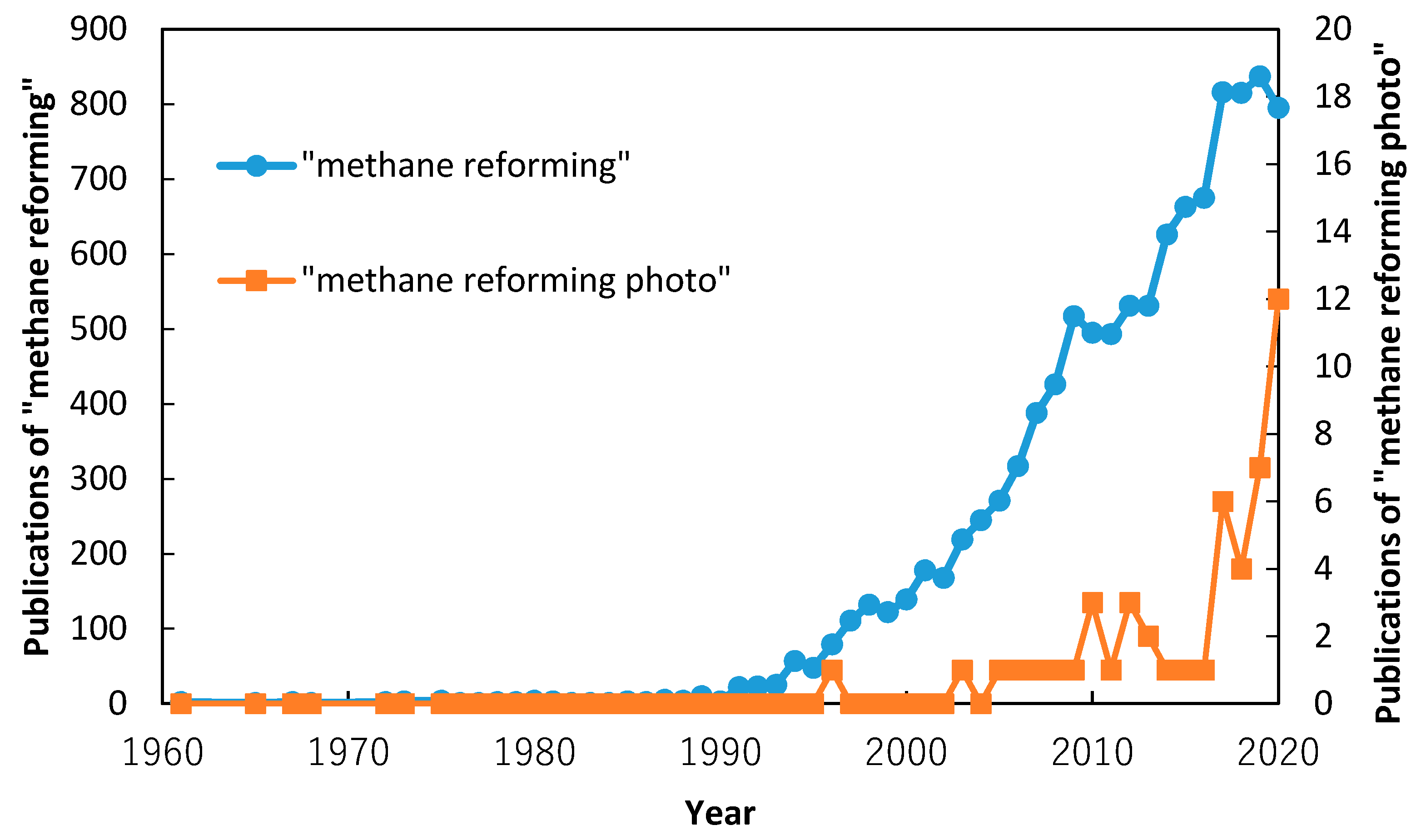
1. Introduction
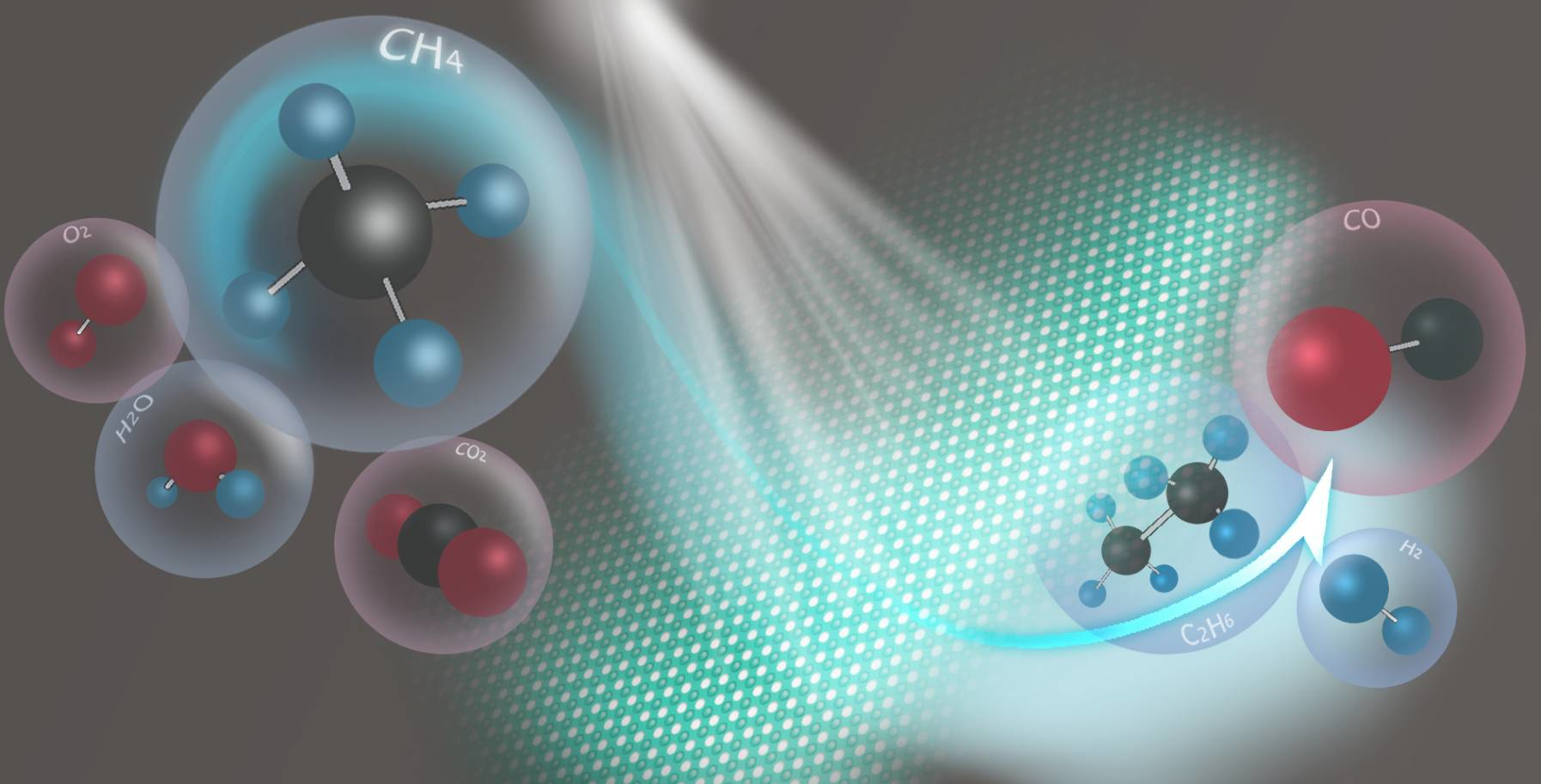
2. The Concept of Photocatalytic Methane Reforming
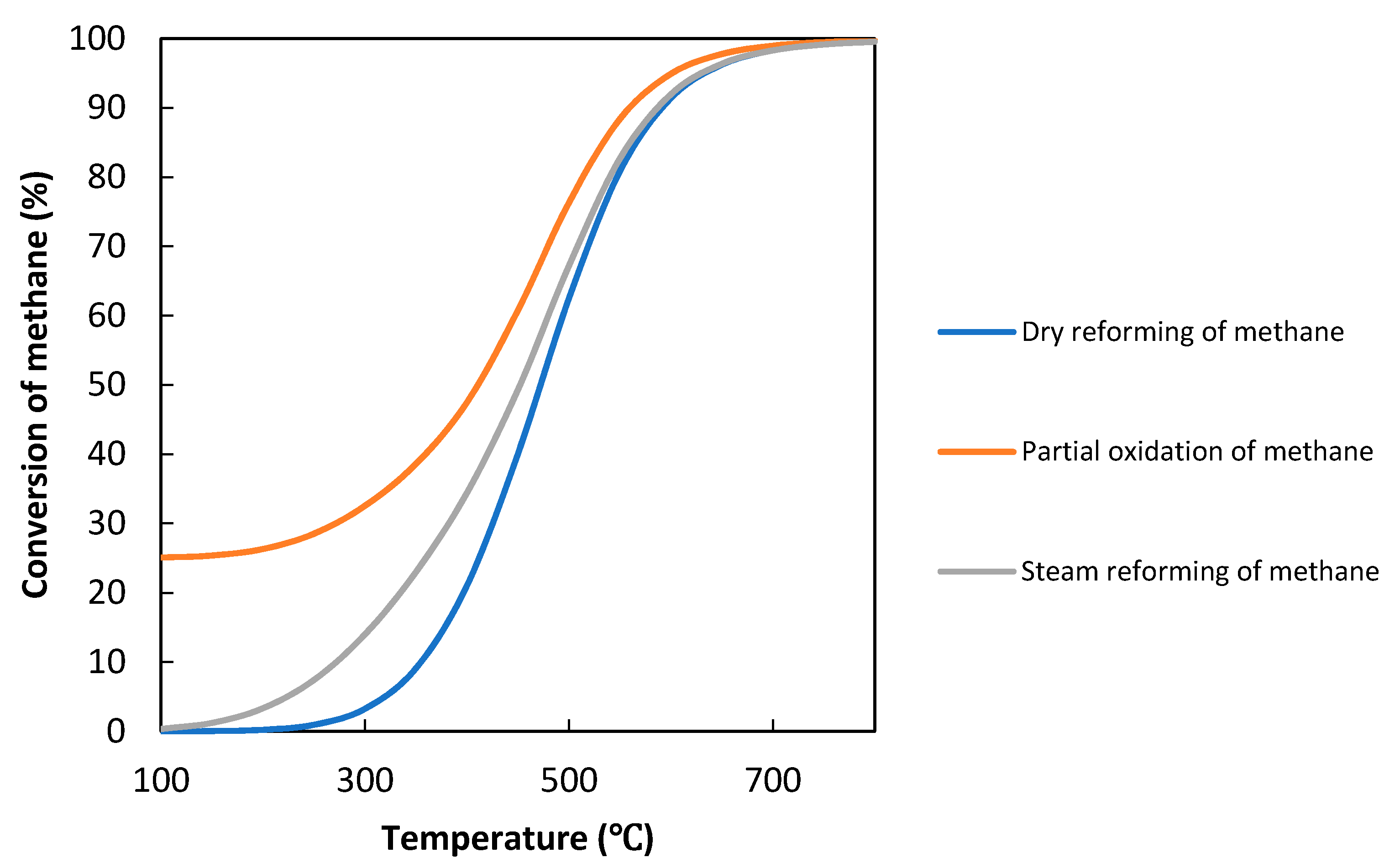
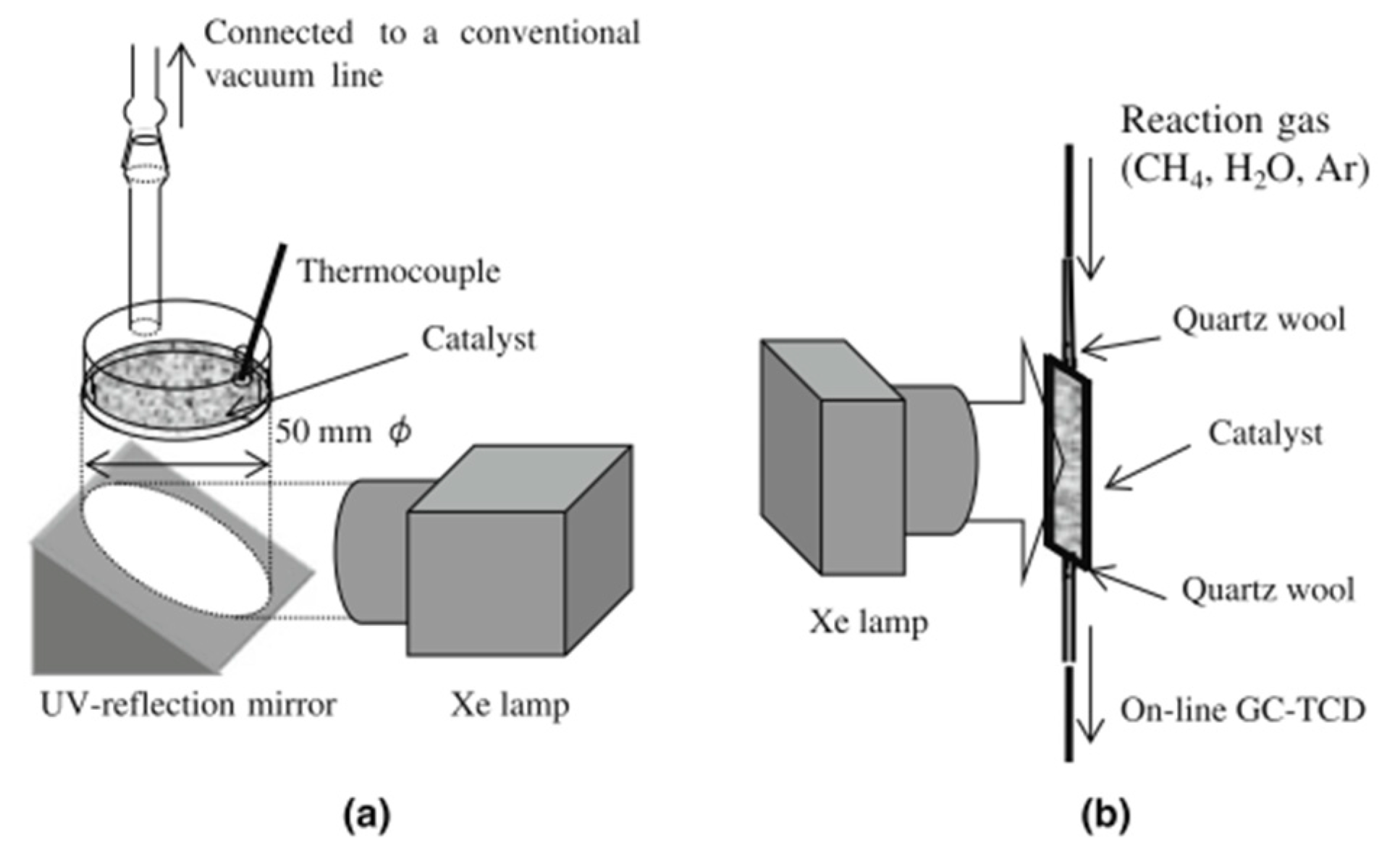
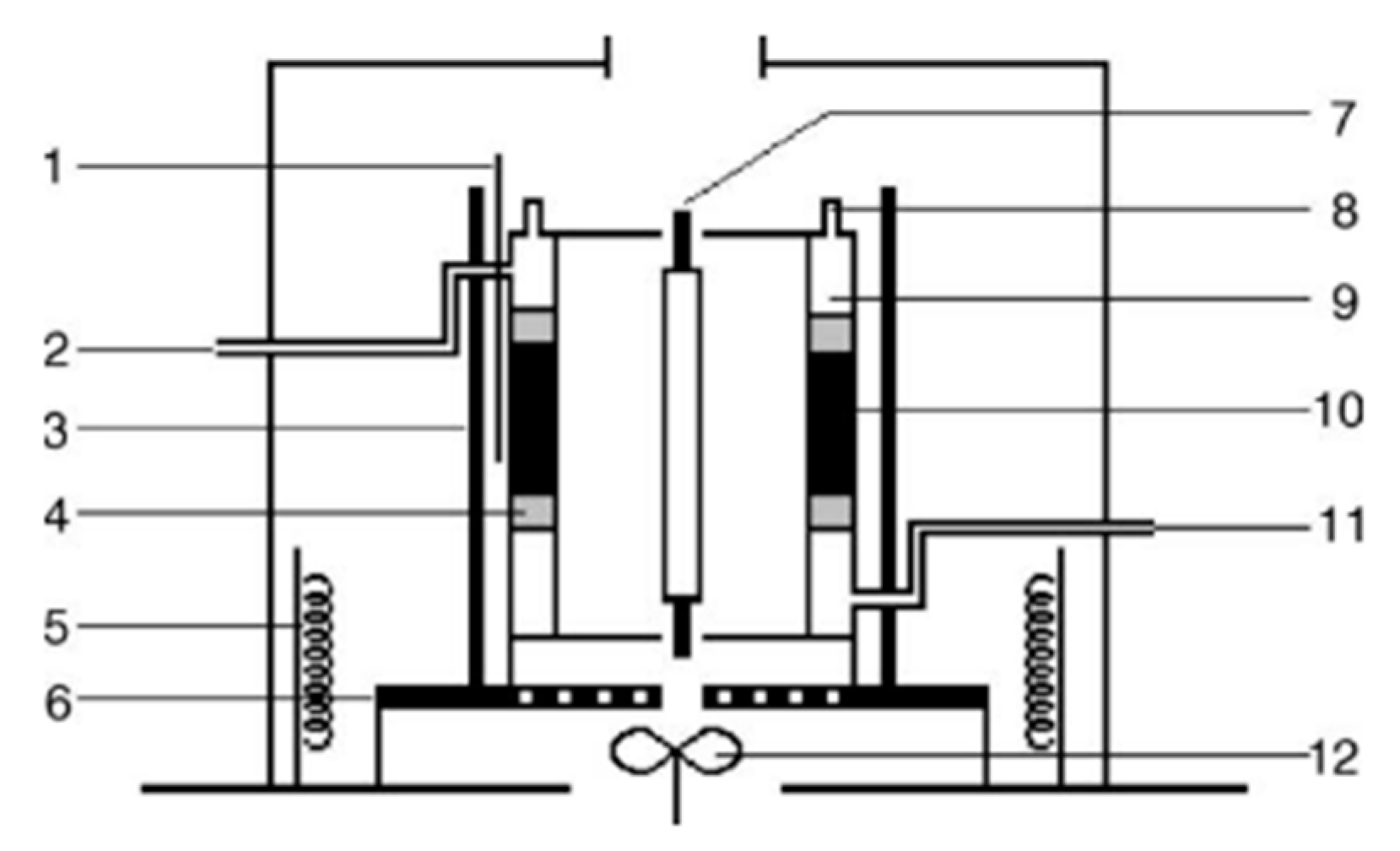
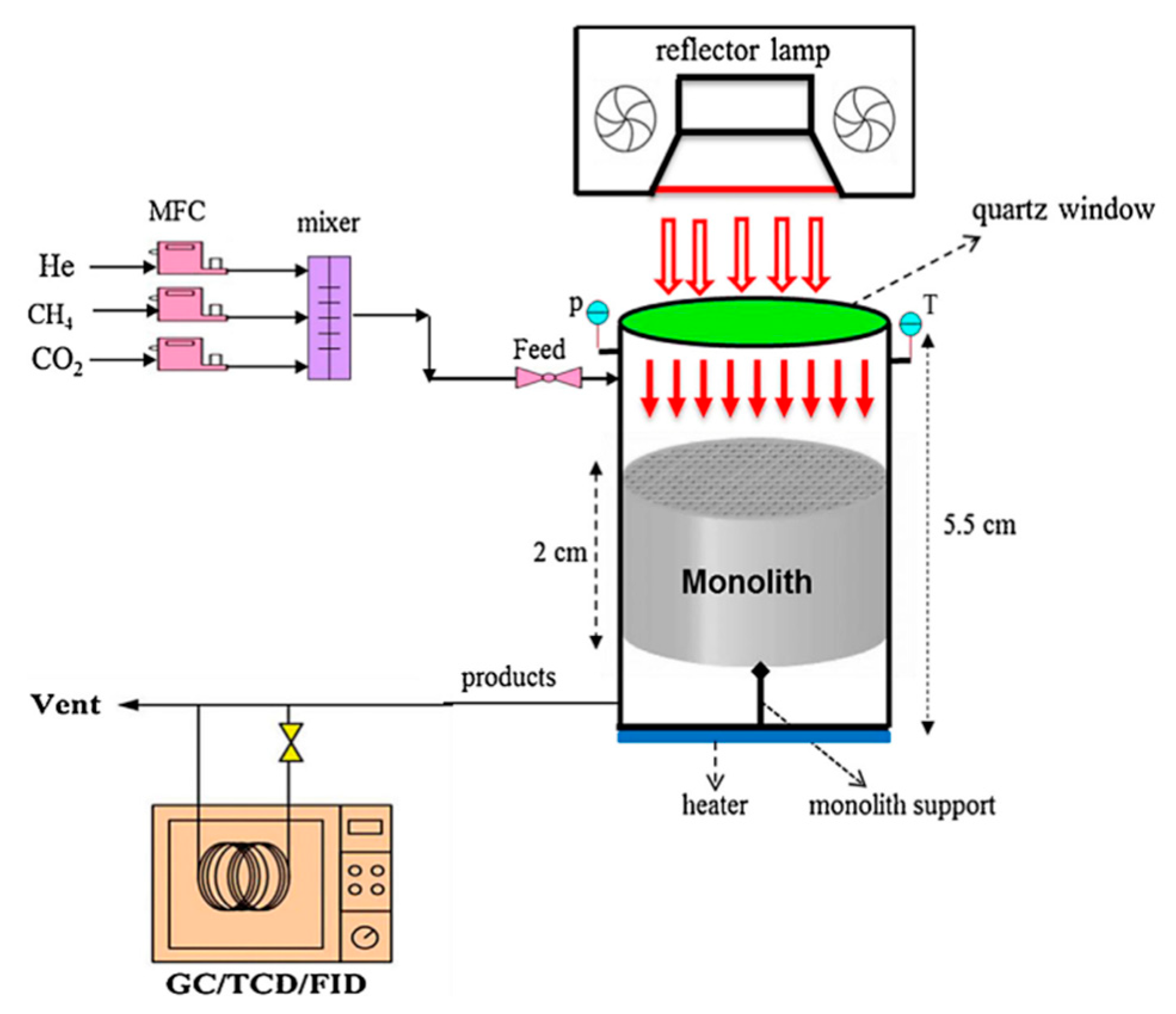
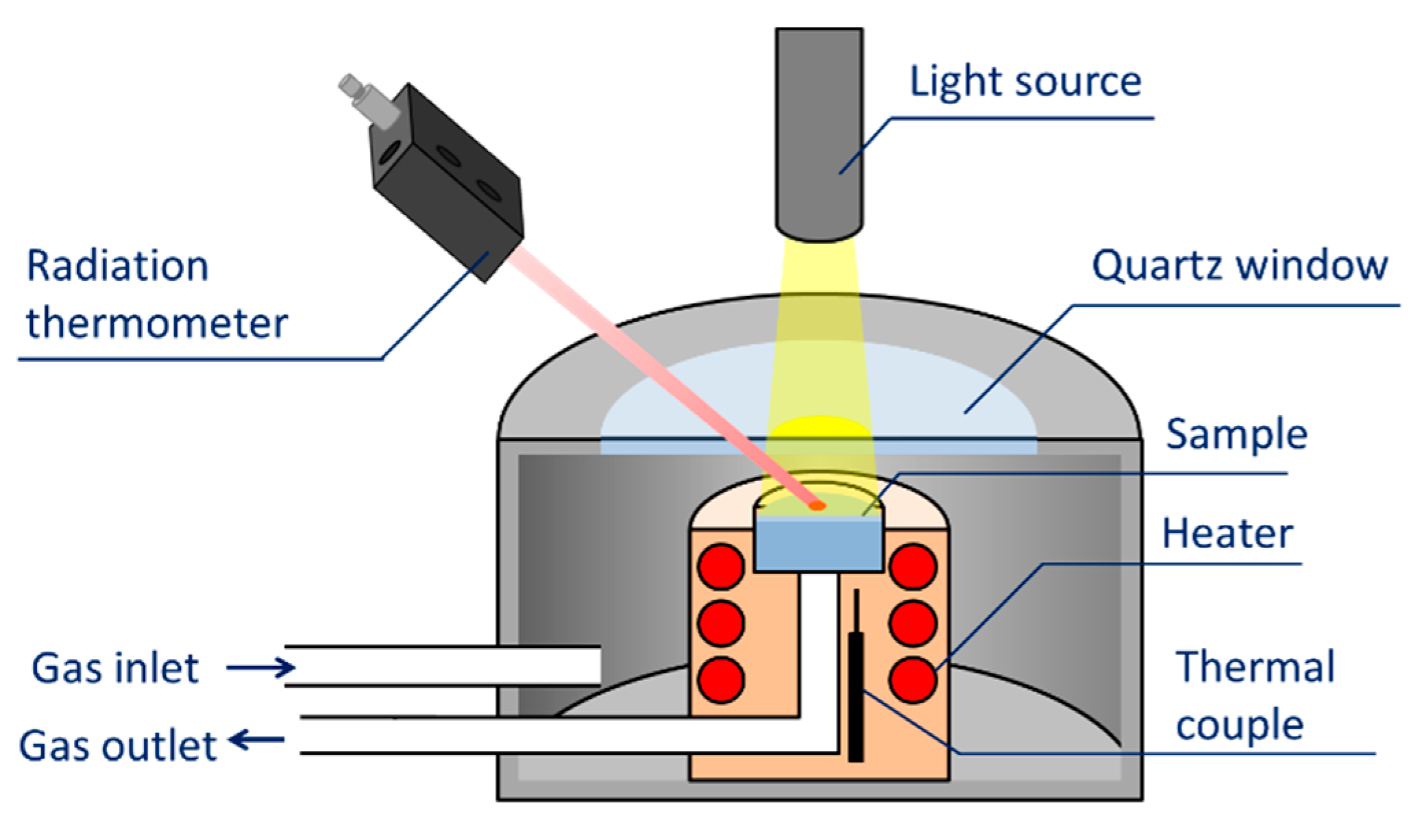
3. Evaluation System for Photocatalytic Methane Conversion
4. Photocatalytic Steam Reforming of Methane
4.1. Overview of the Reaction
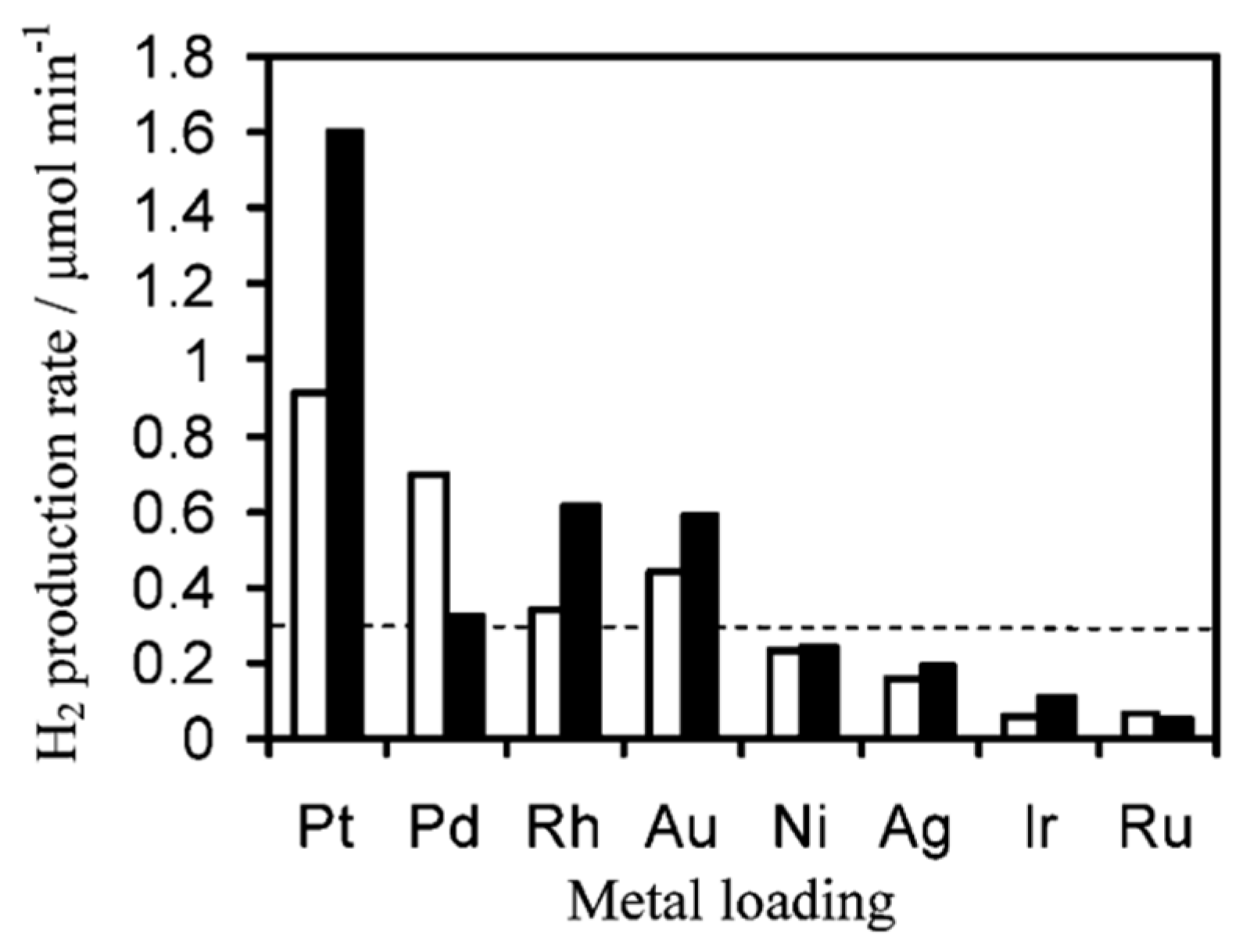
4.2. Material
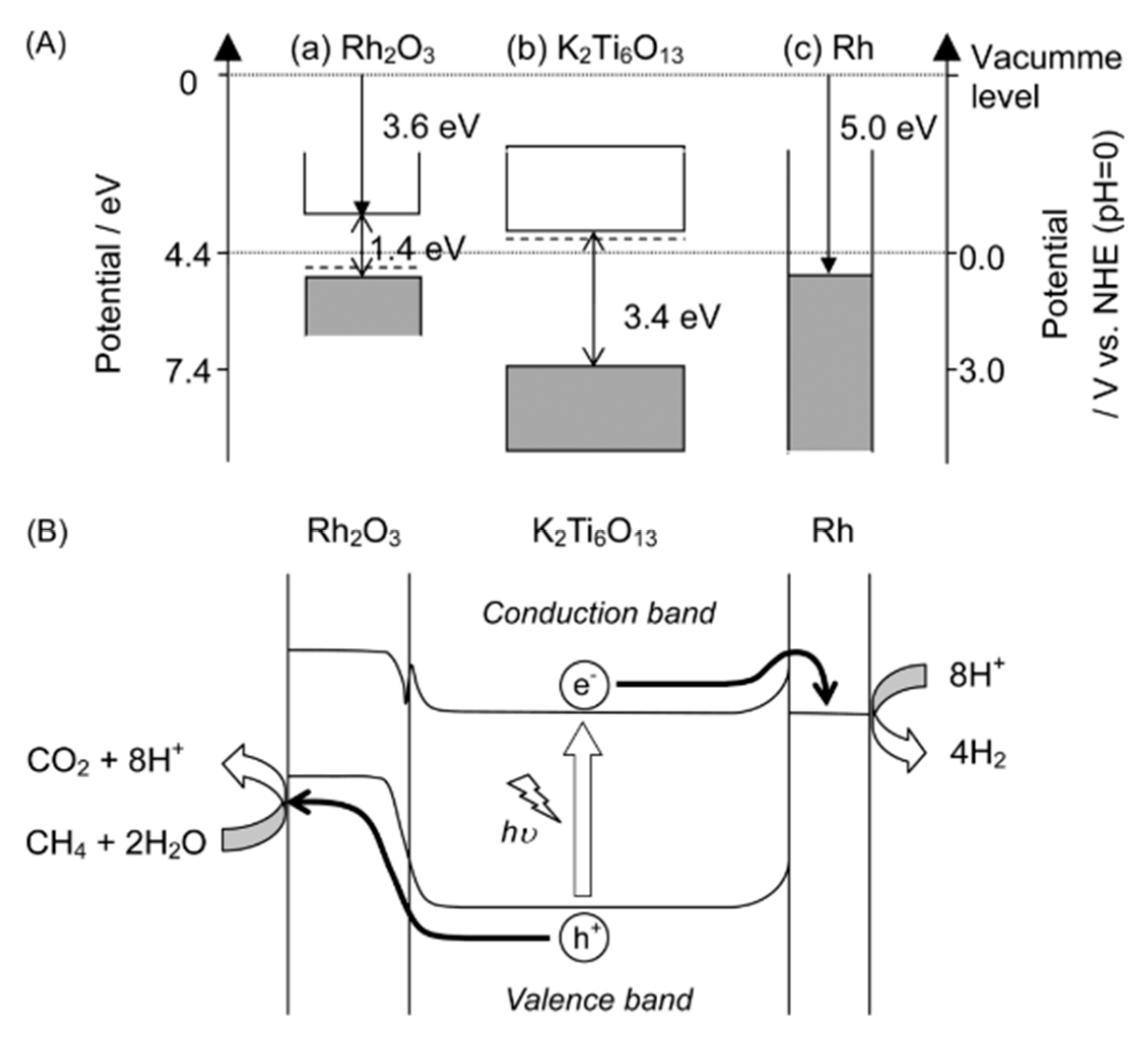
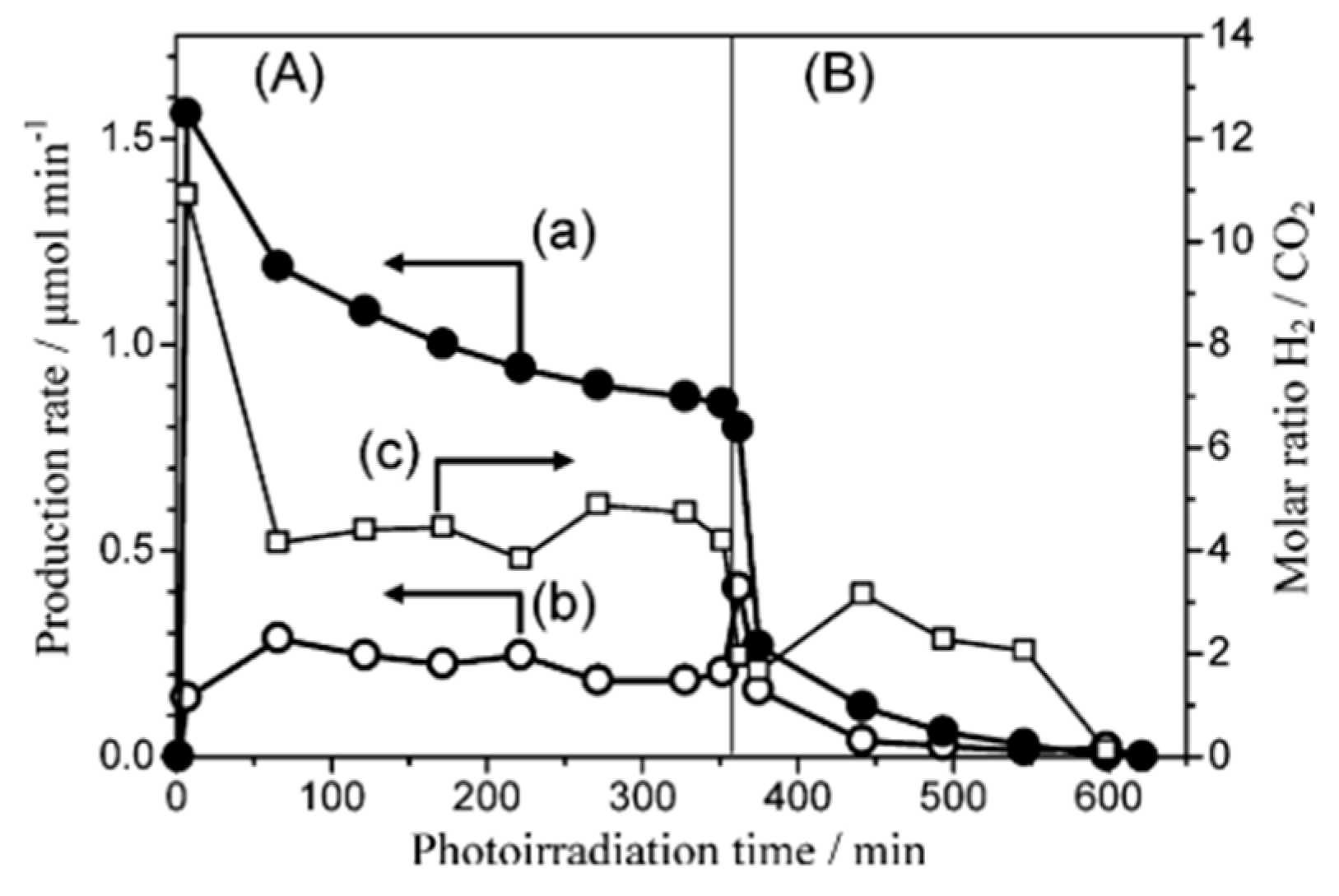
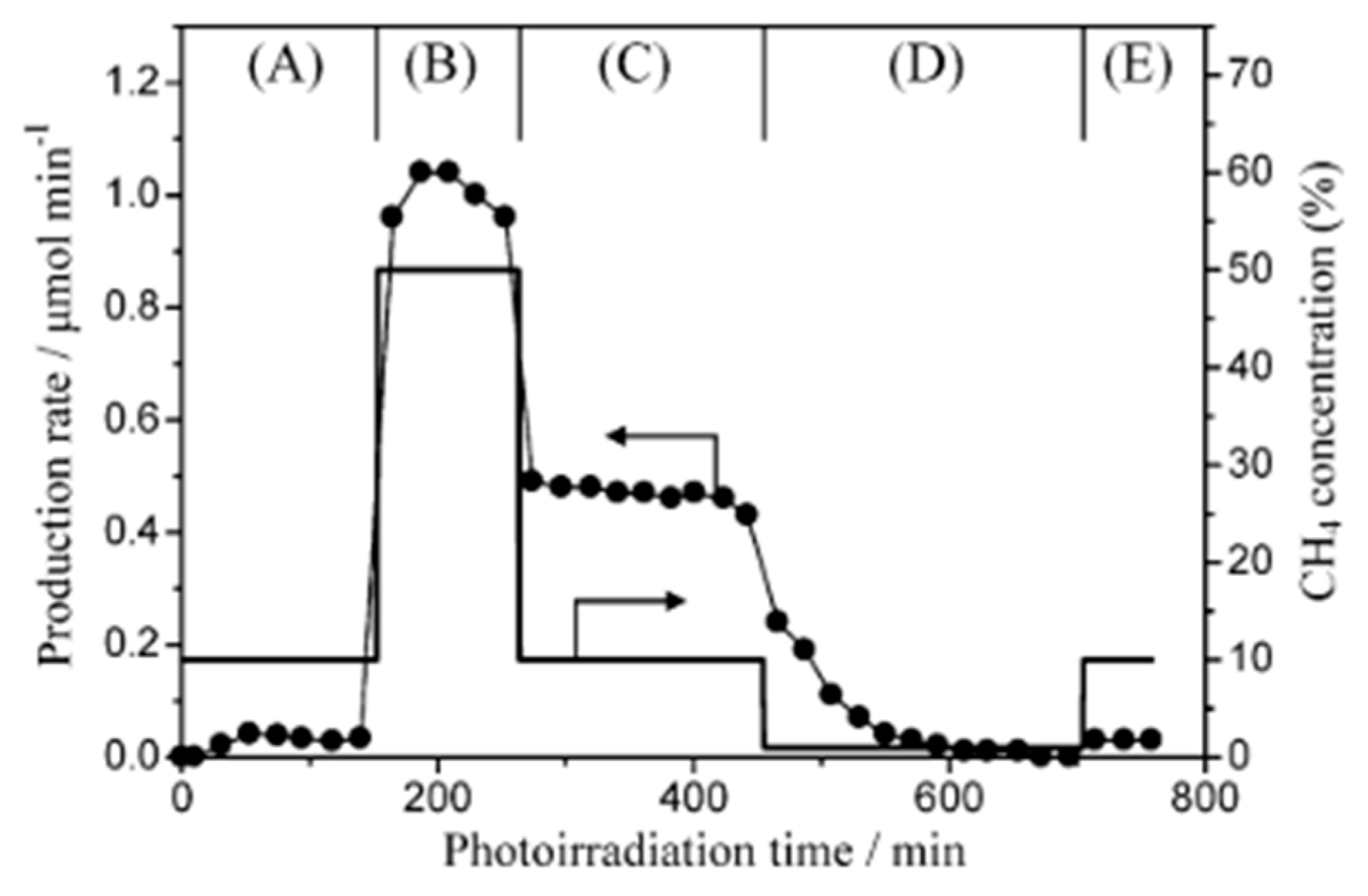
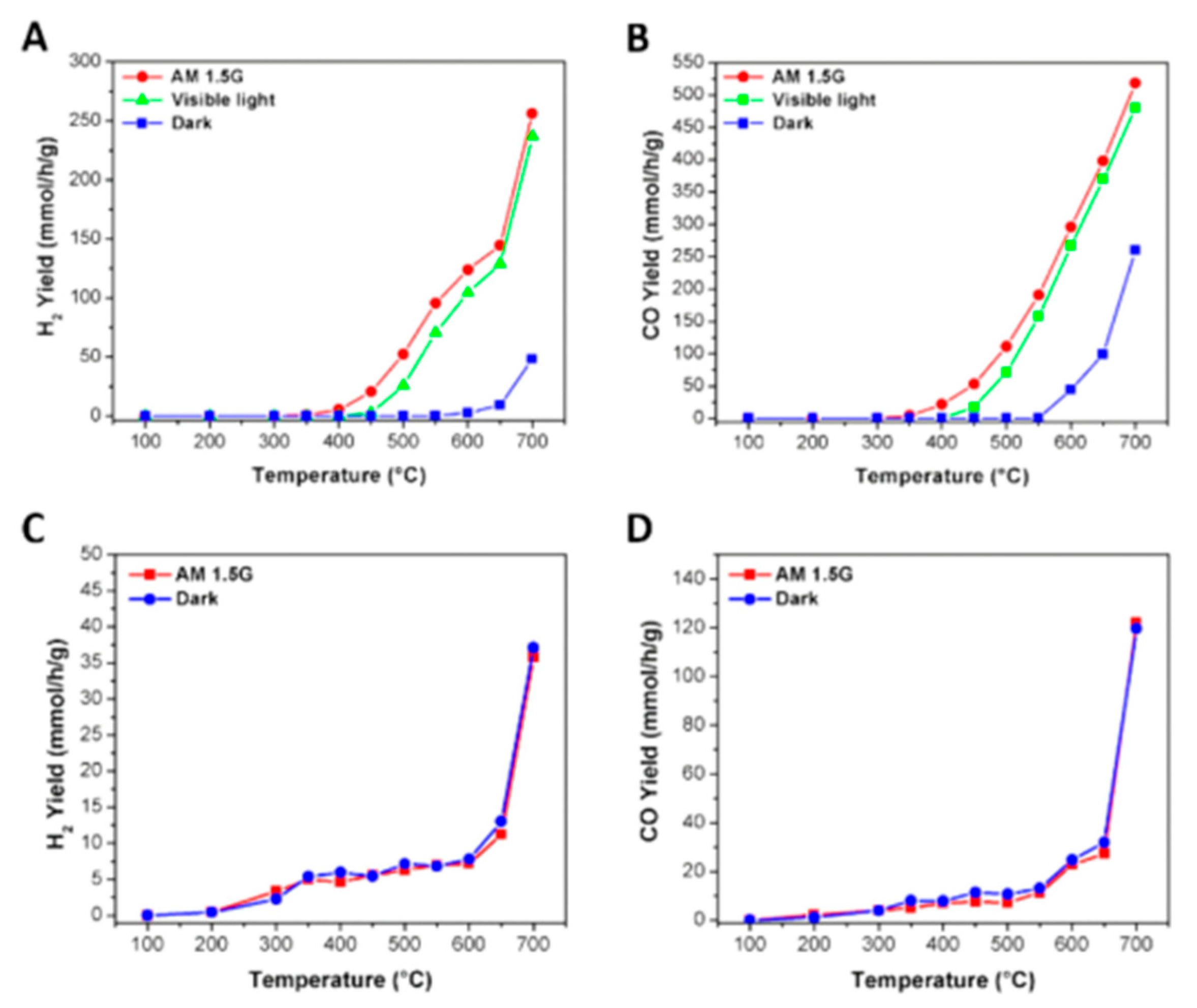
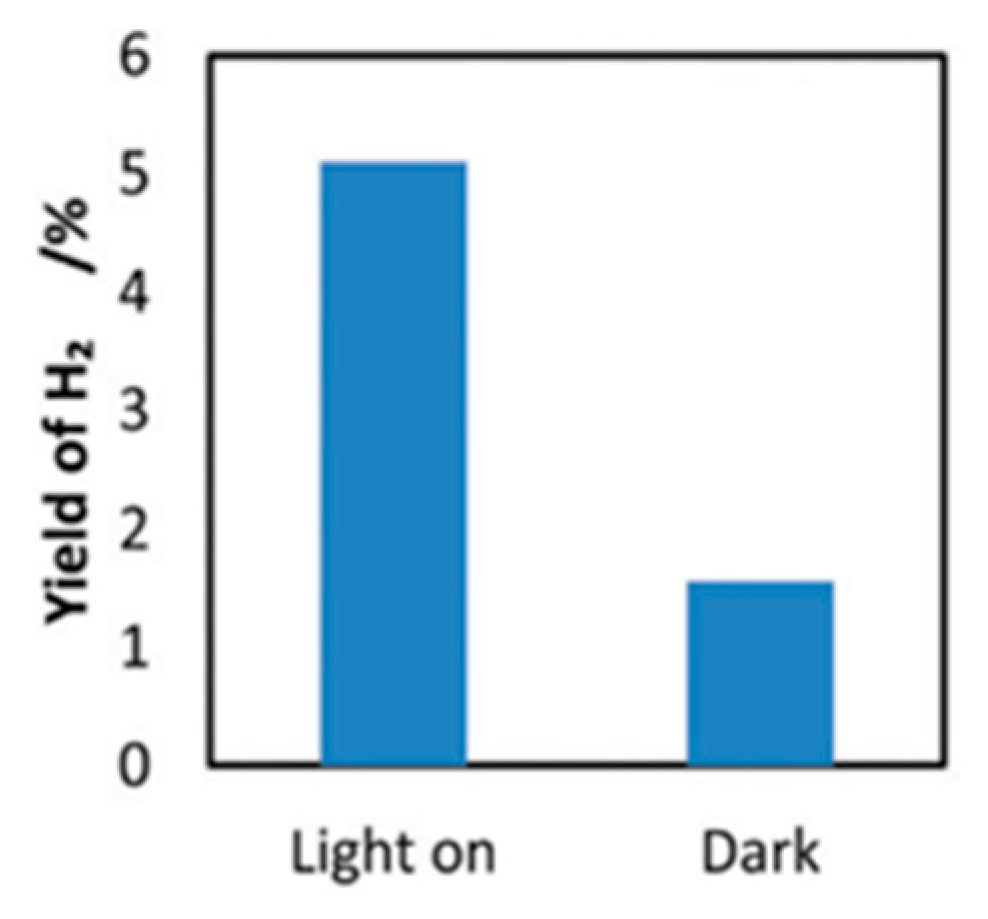
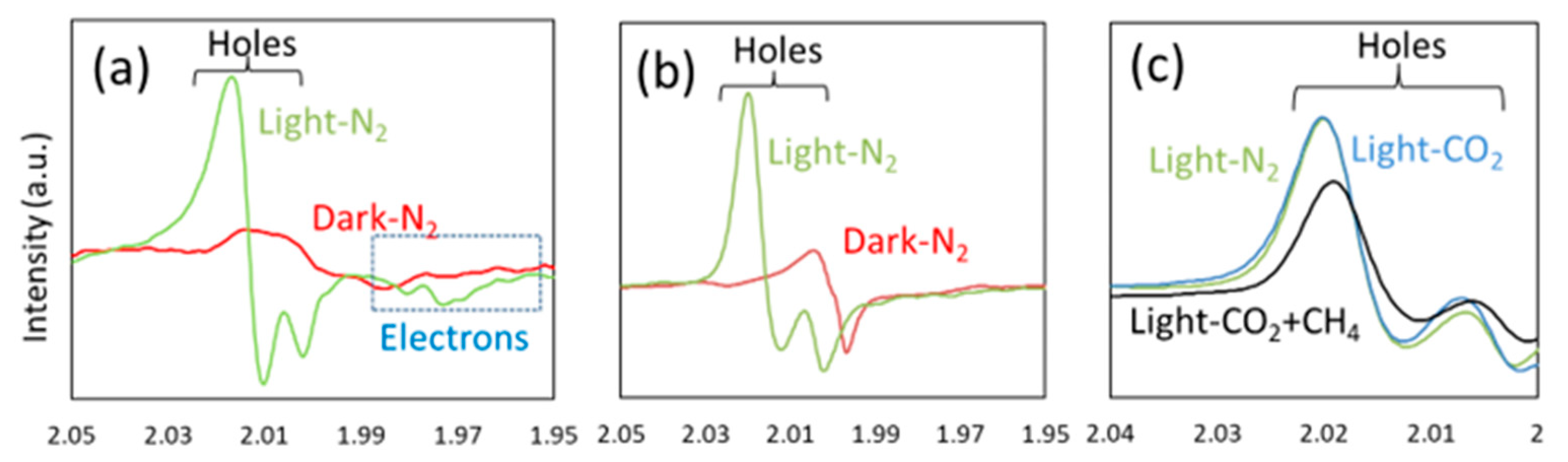
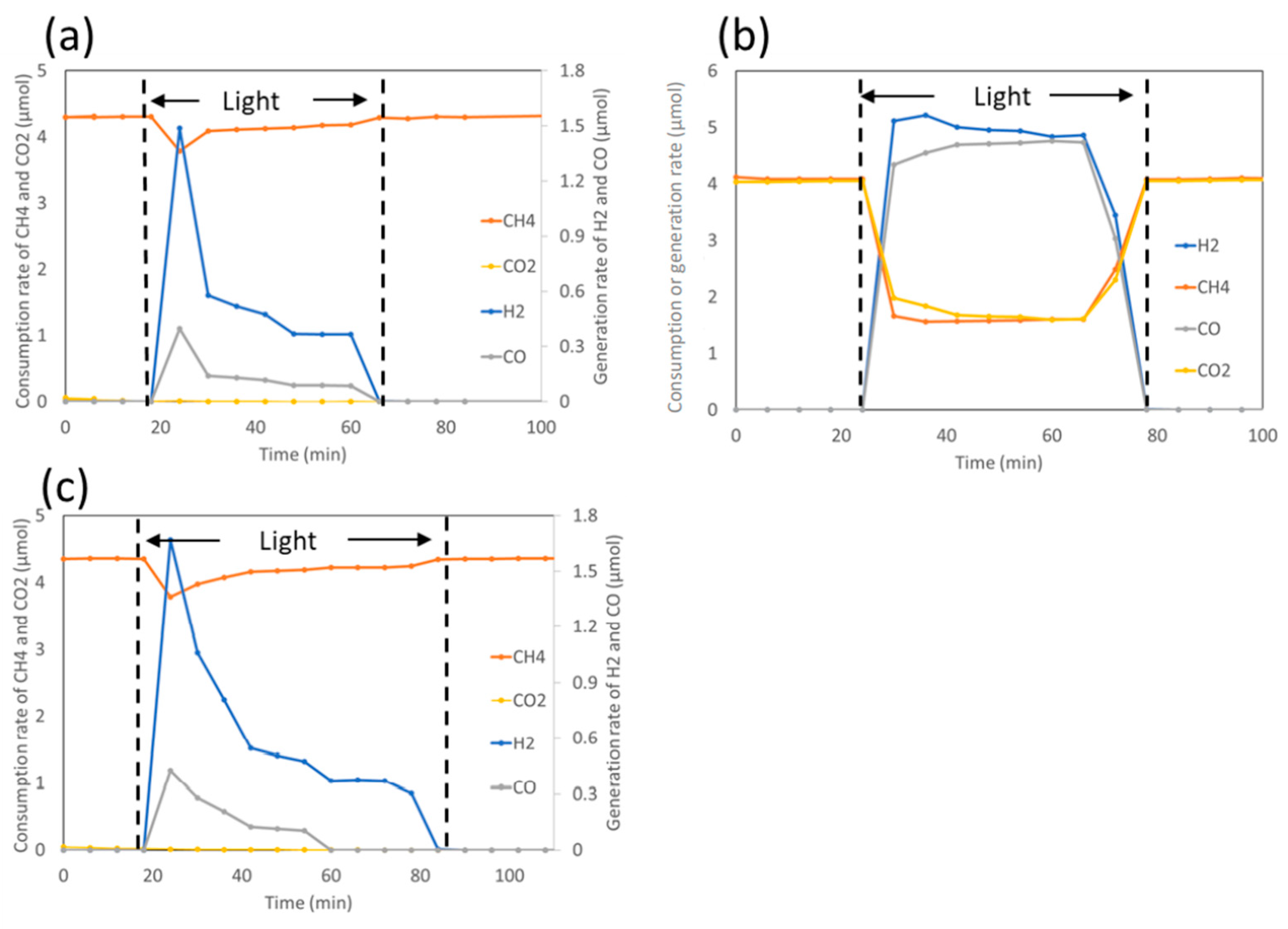
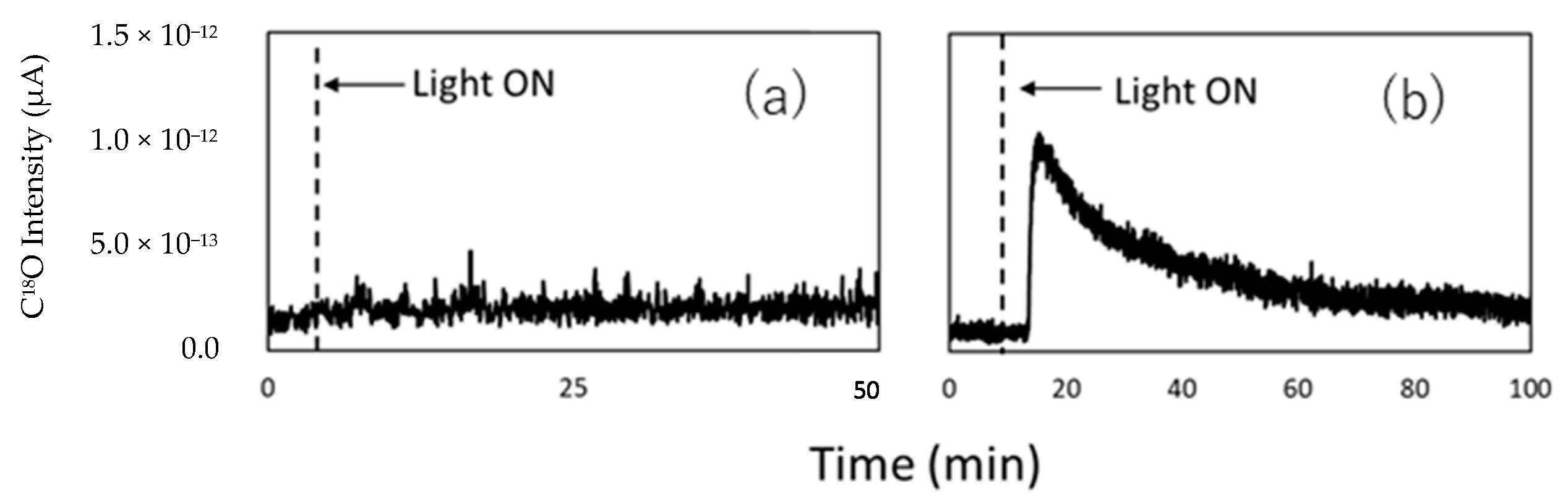
4.3. Mechanism of the Reaction
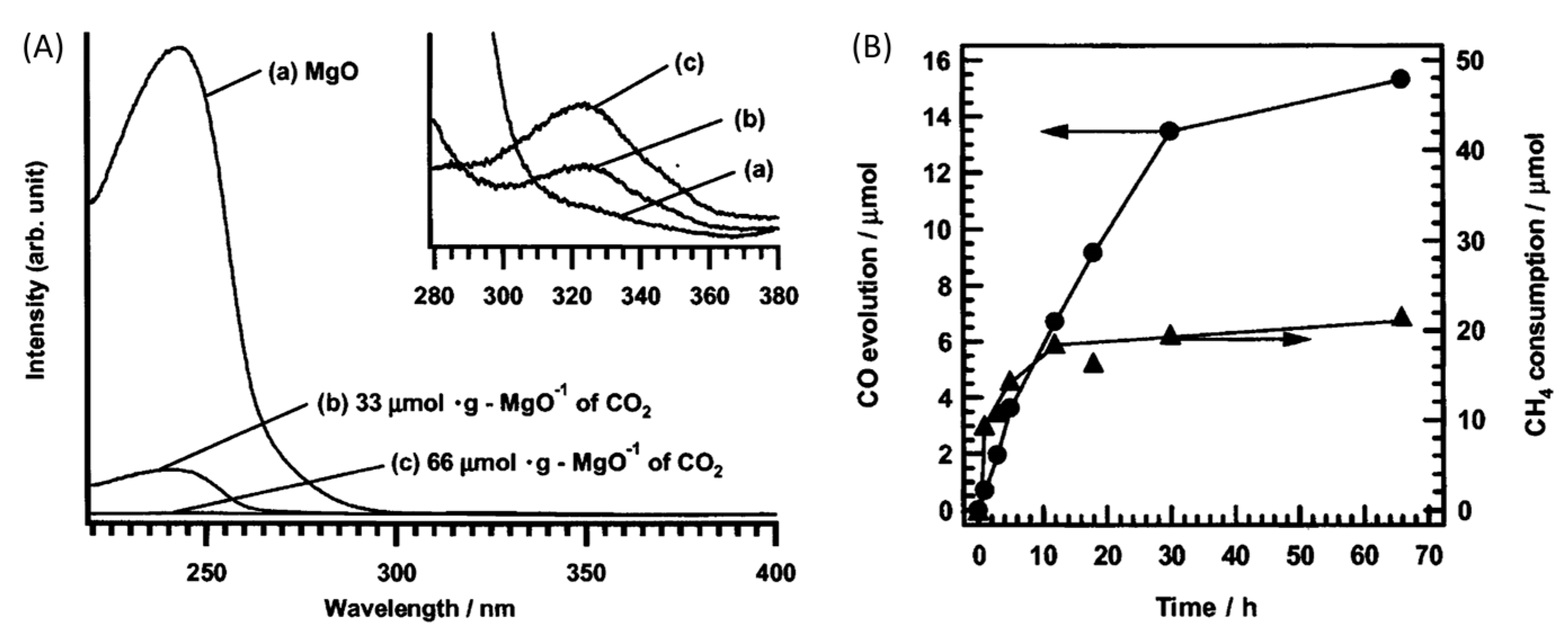
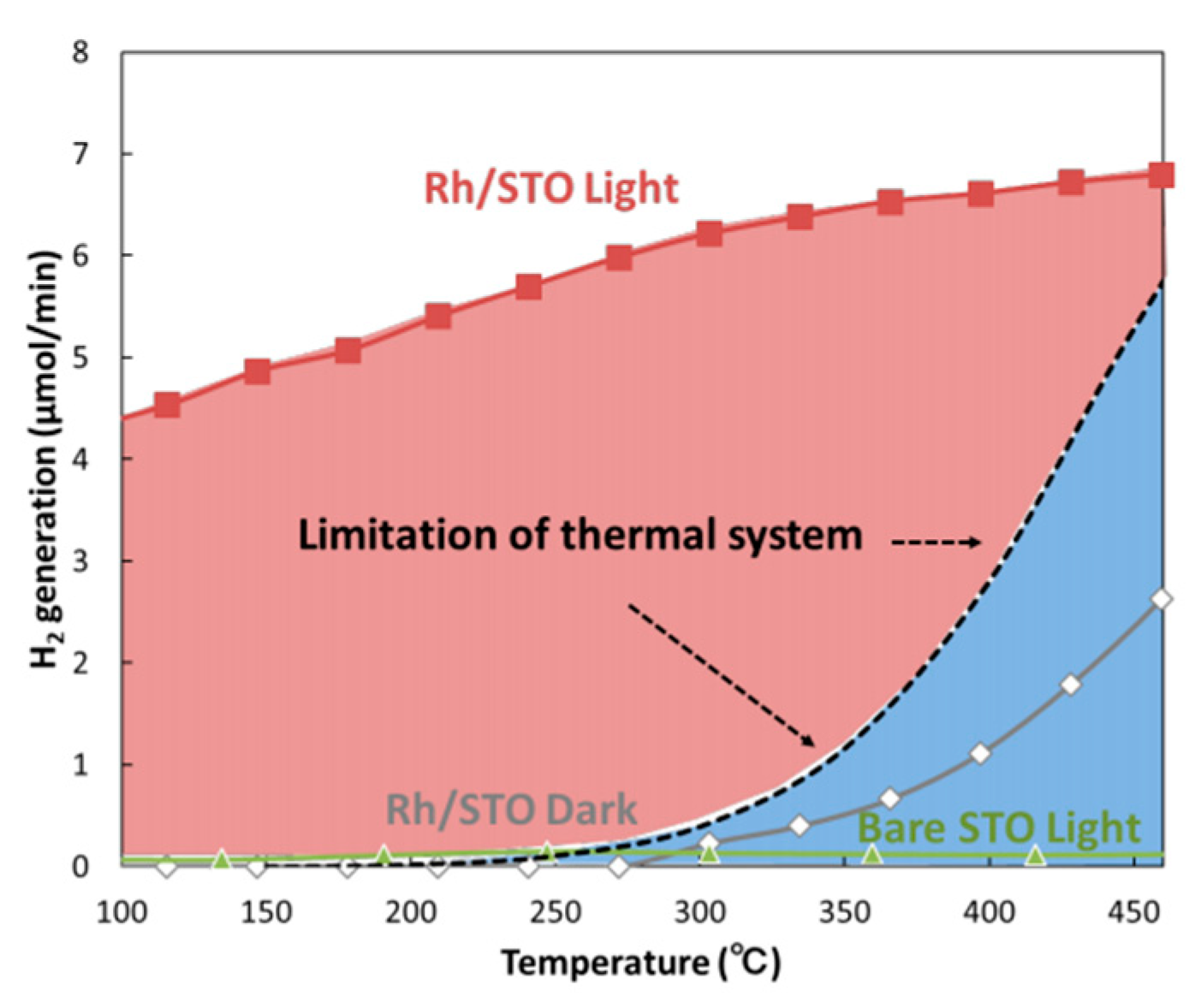
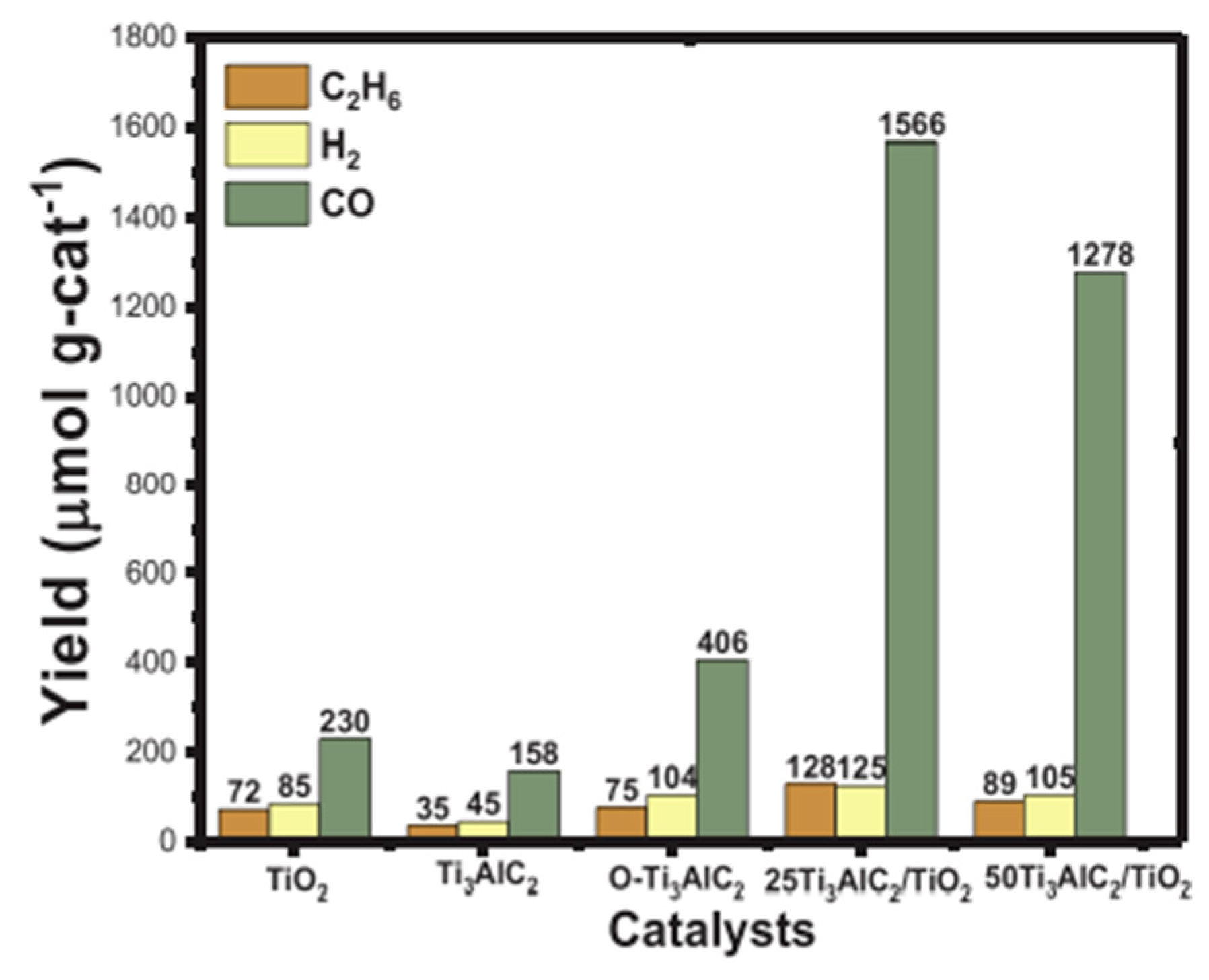
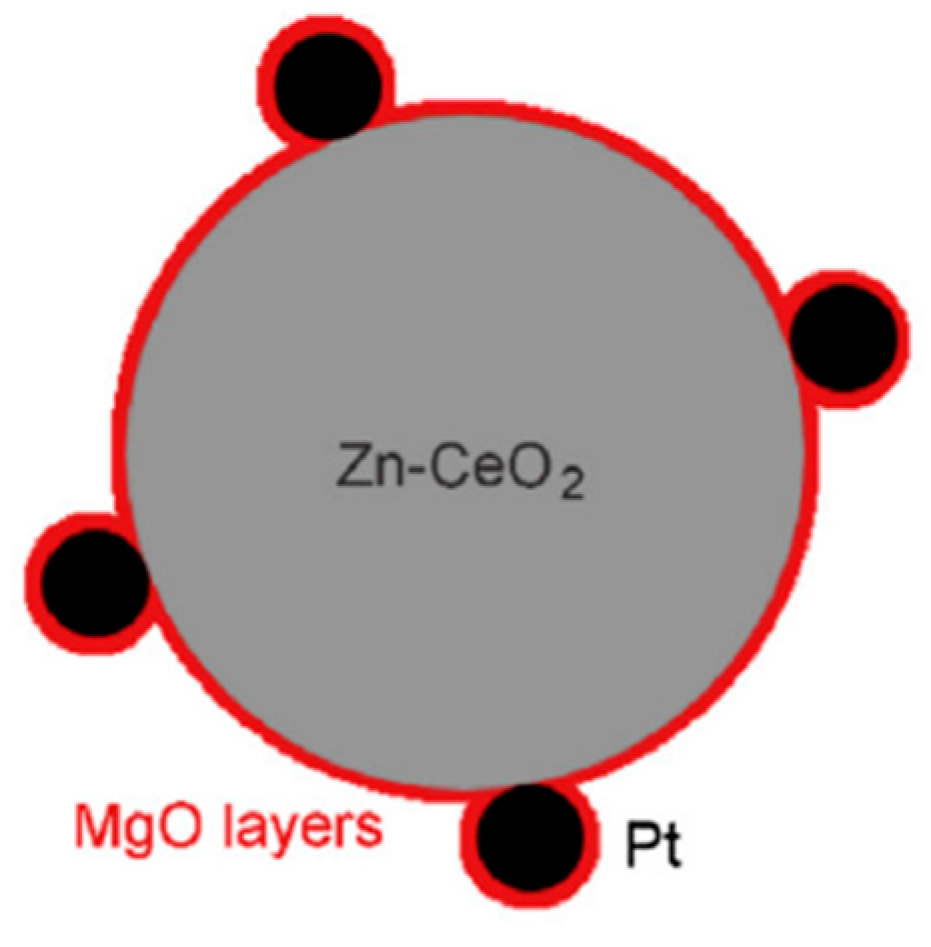
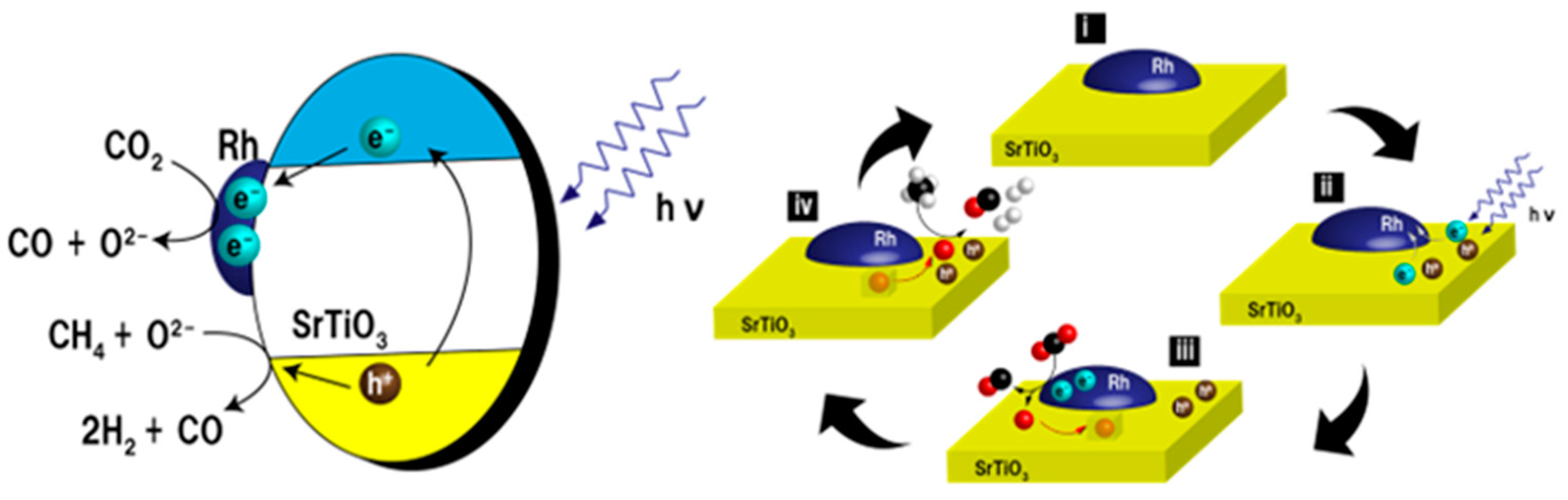
5. Photocatalytic Dry Reforming of Methane (DRM)
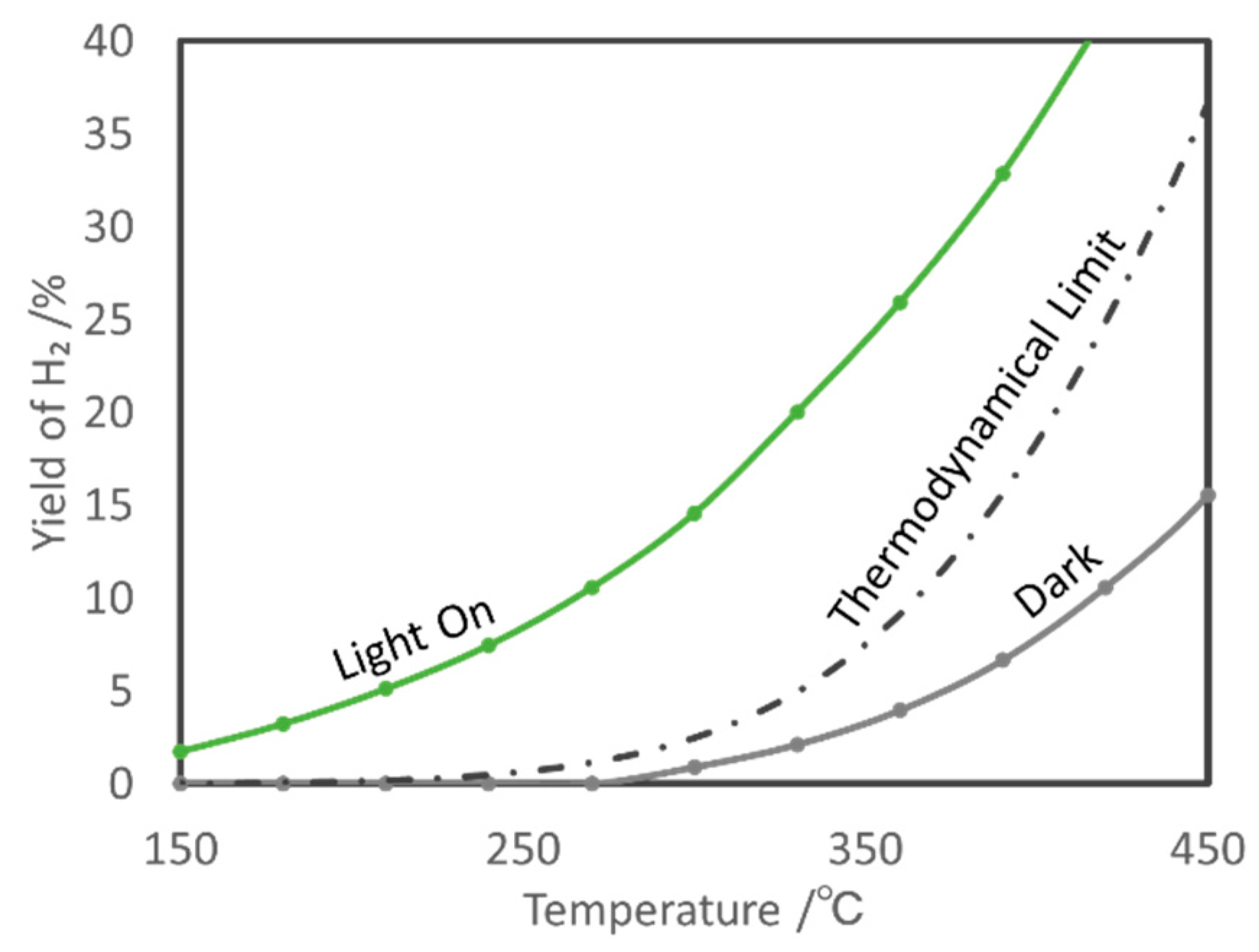
5.1. Overview of the Reaction
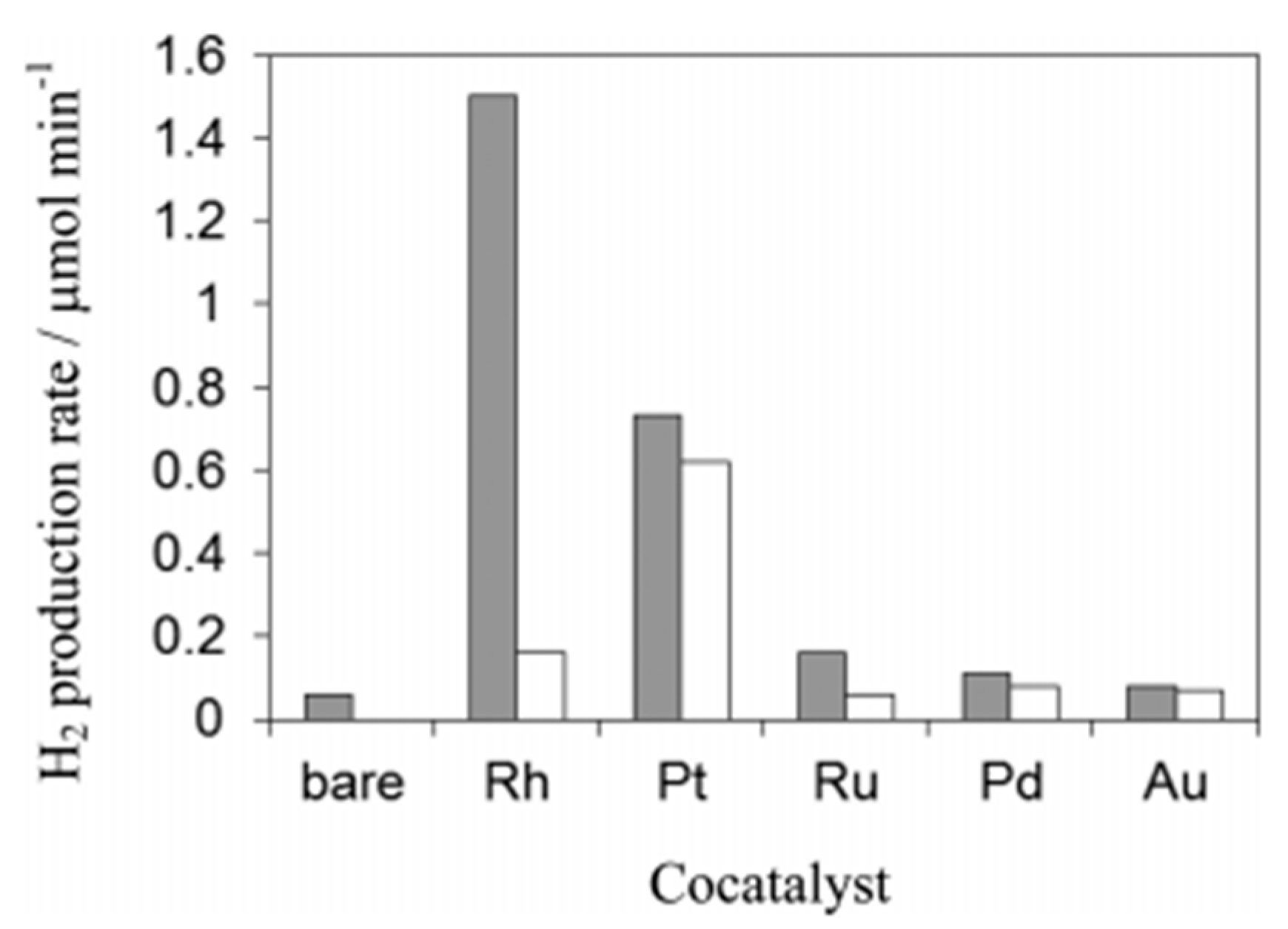
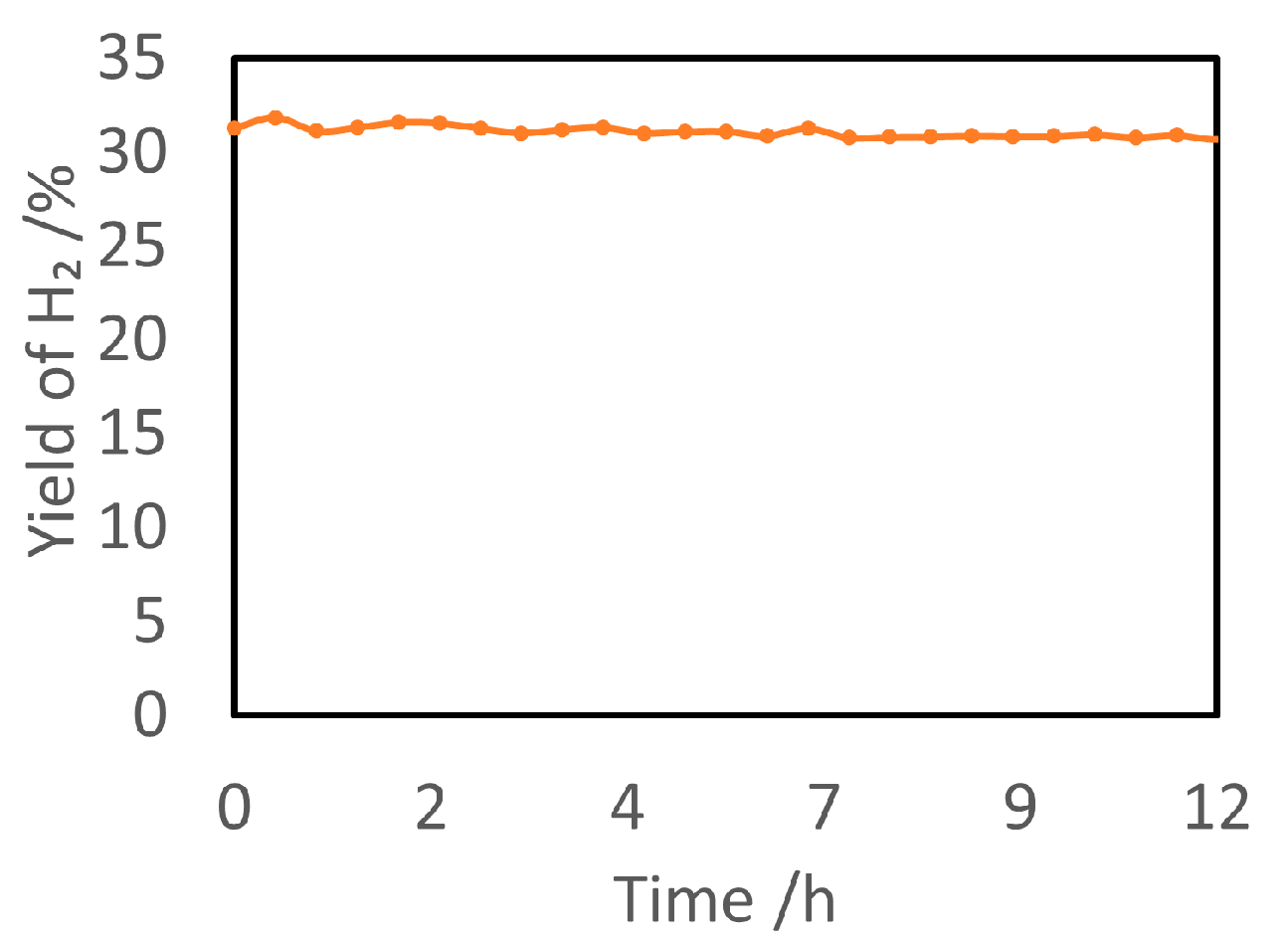
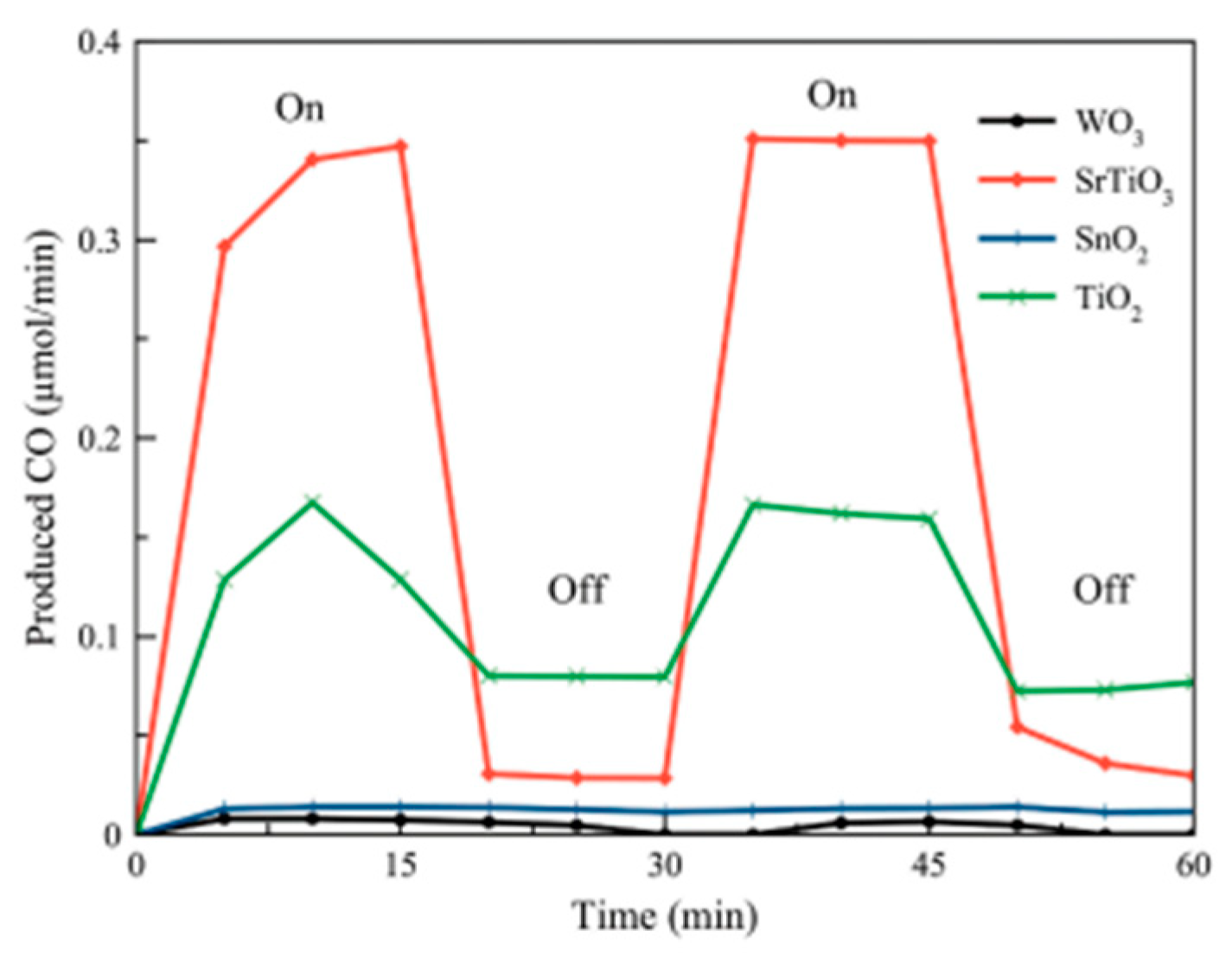
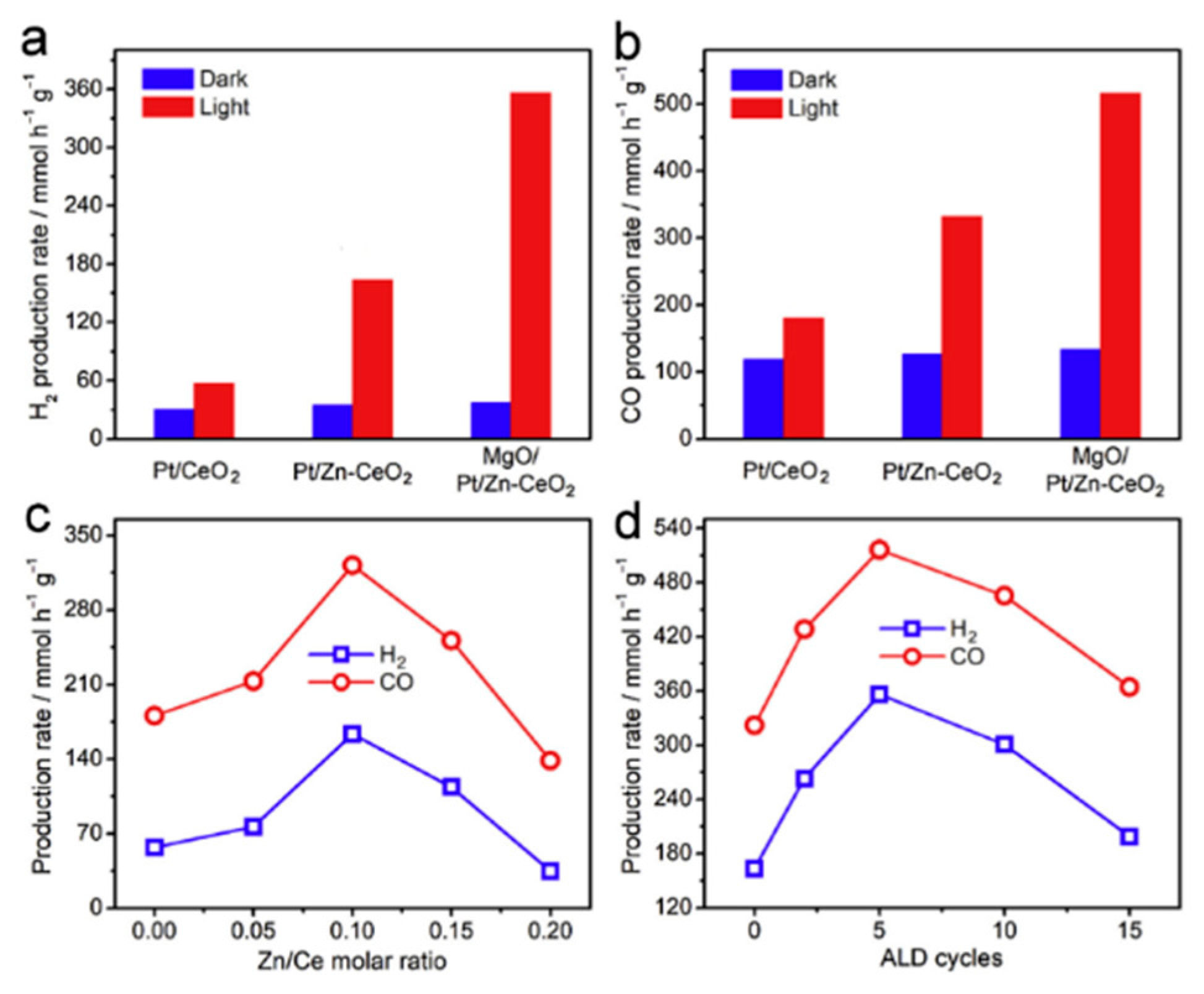
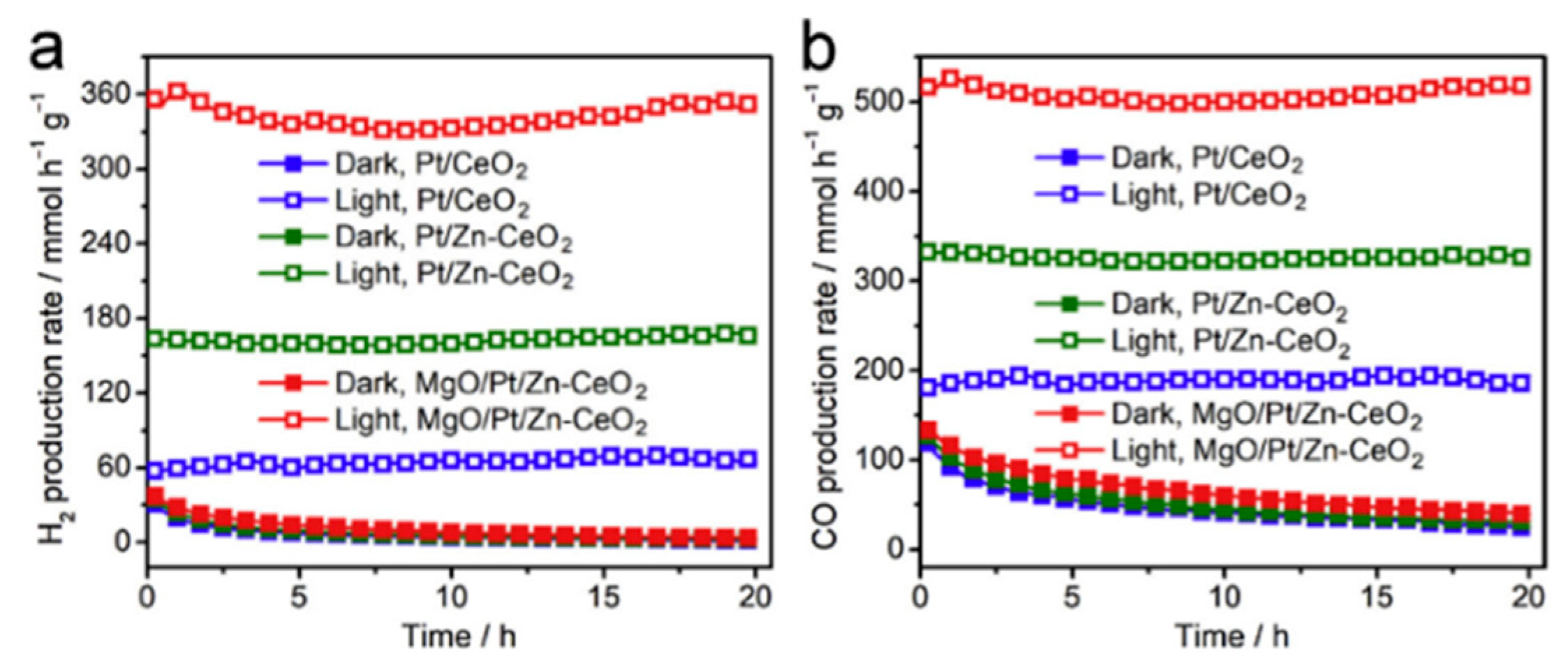
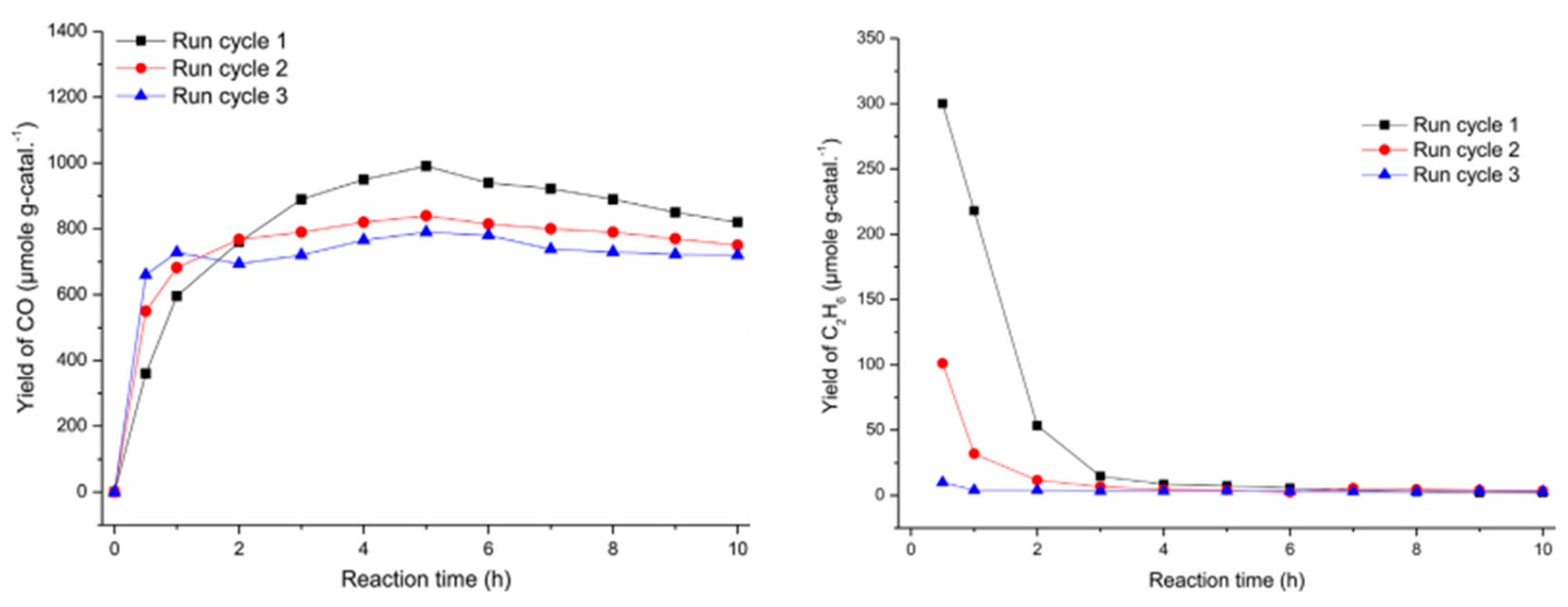
5.2. Material
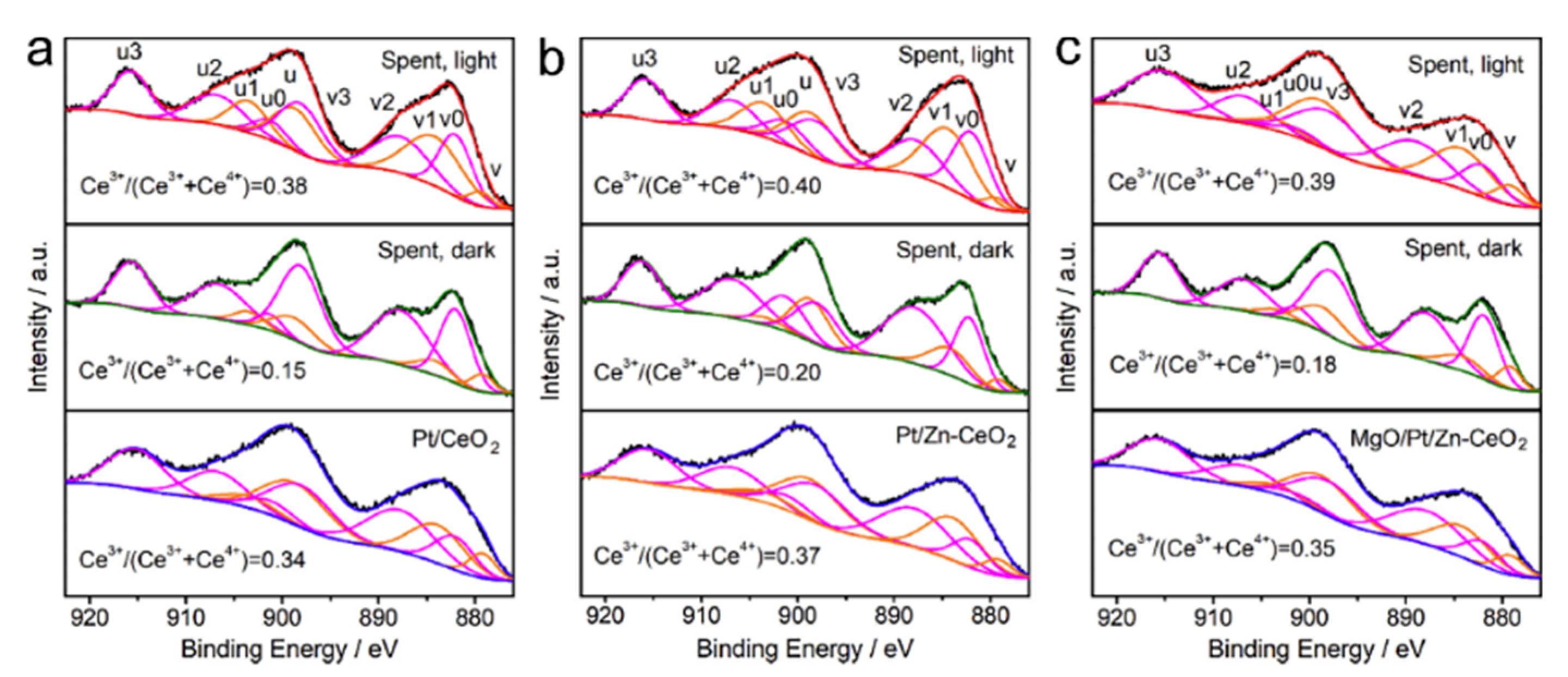
5.3. Mechanism of the Reaction
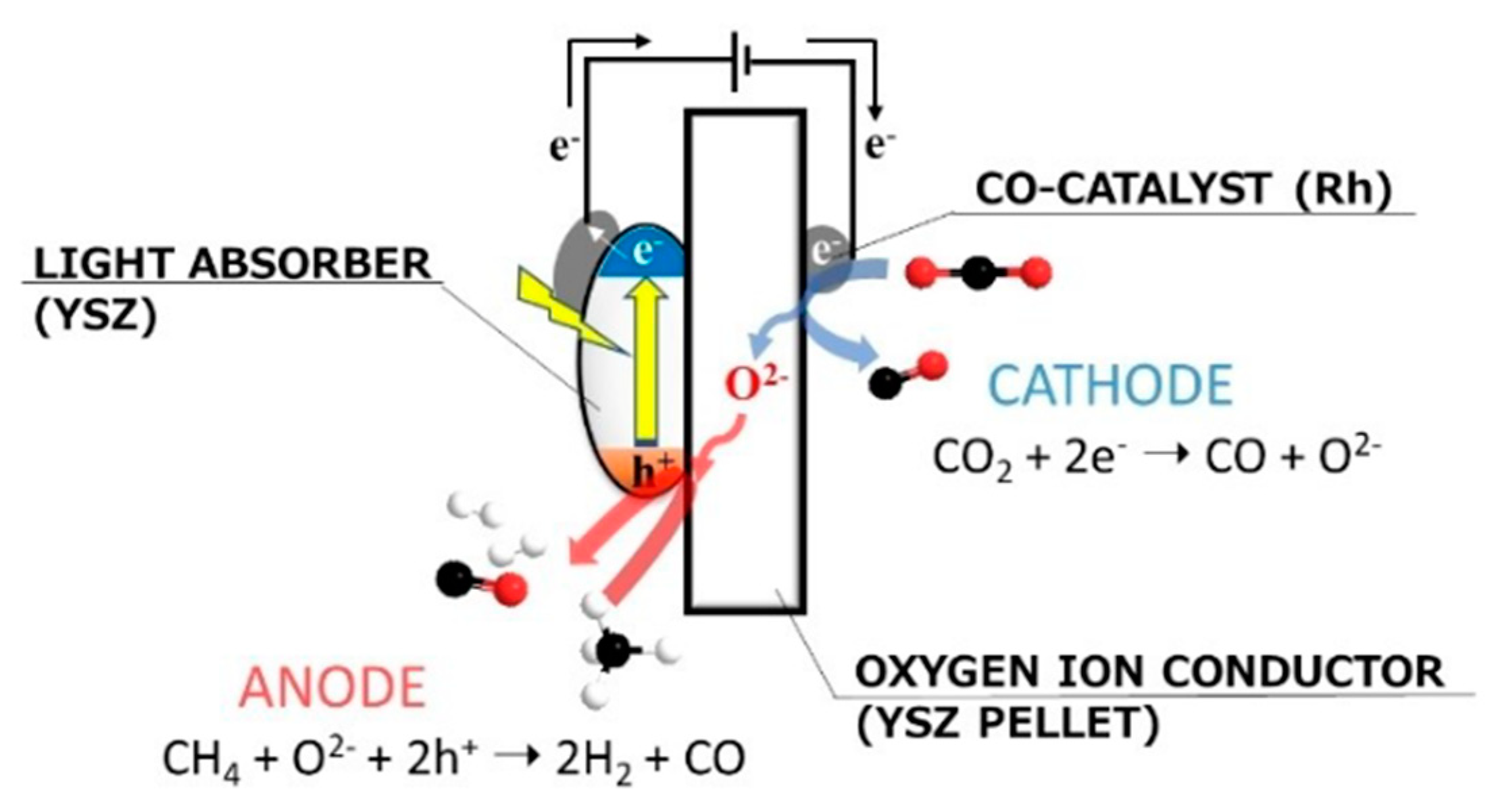
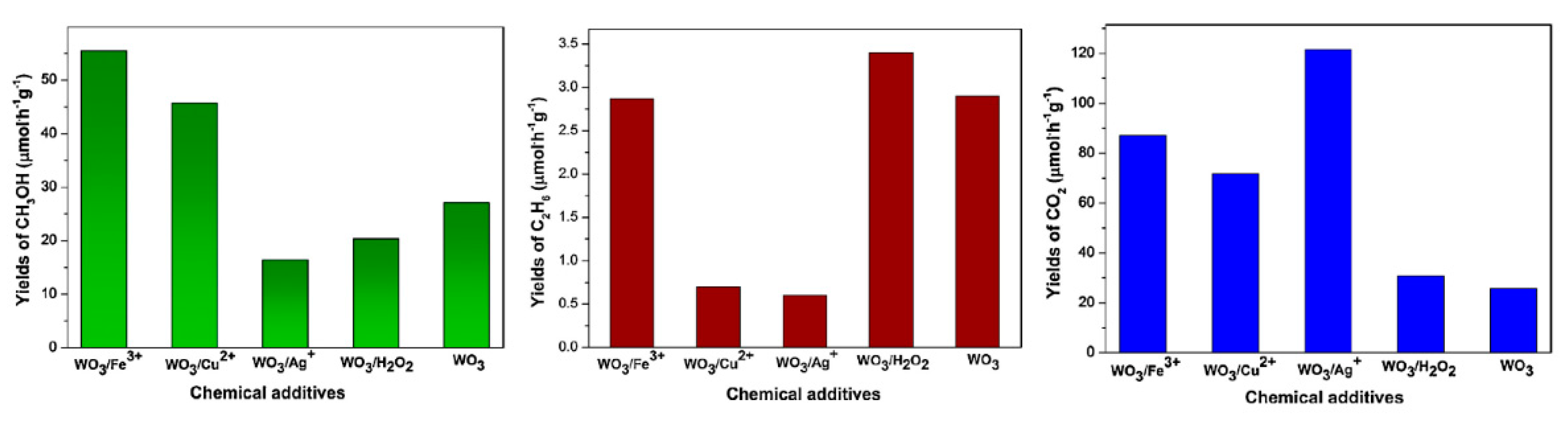
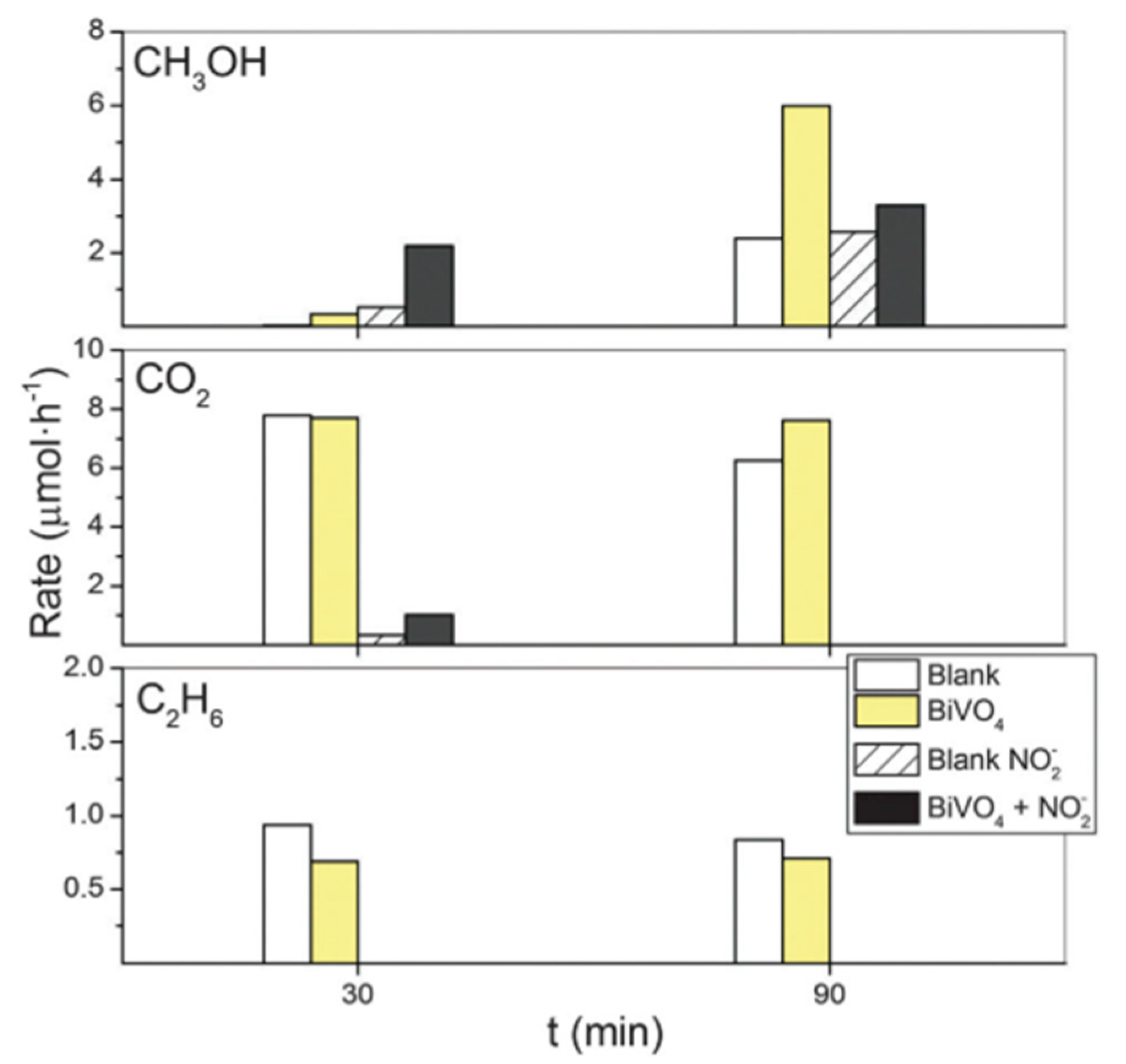
6. Photocatalytic Partial Oxidation of Methane (POM)
6.1. Overview of the Reaction
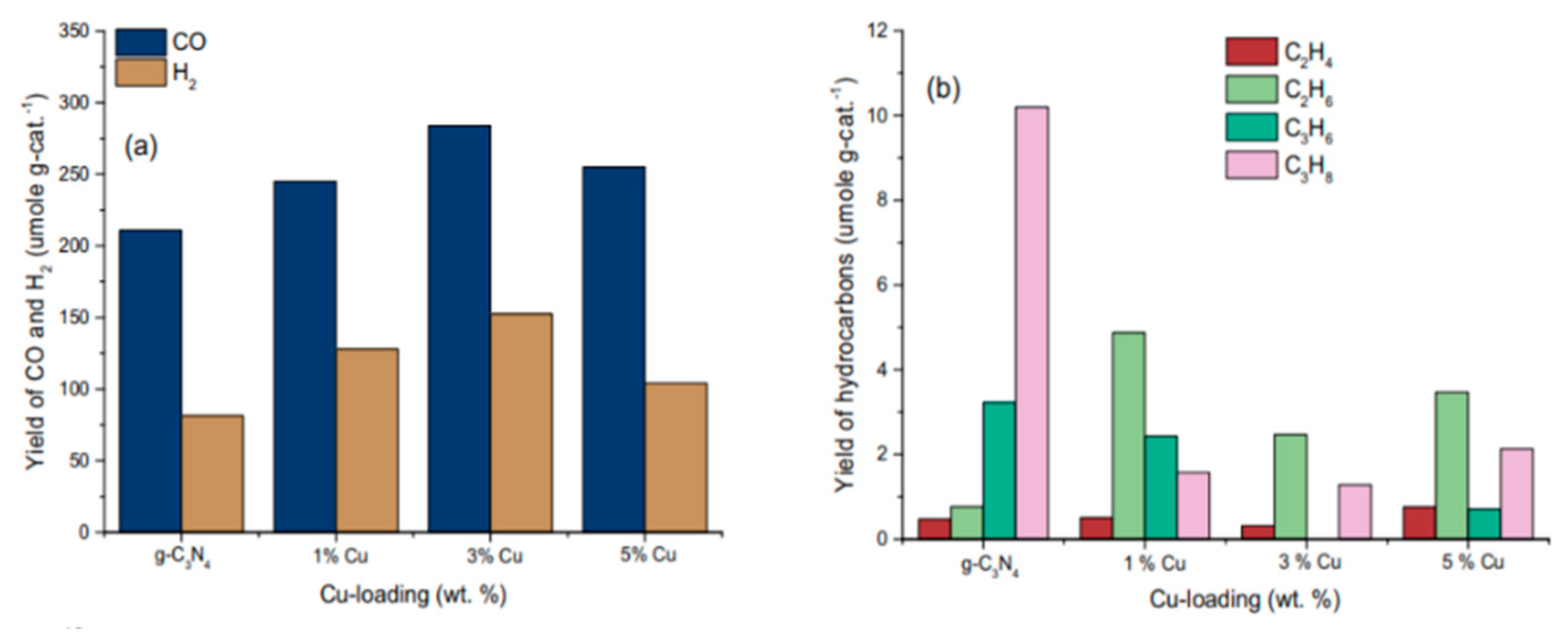
6.2. Material
6.3. Proposed Mechanism
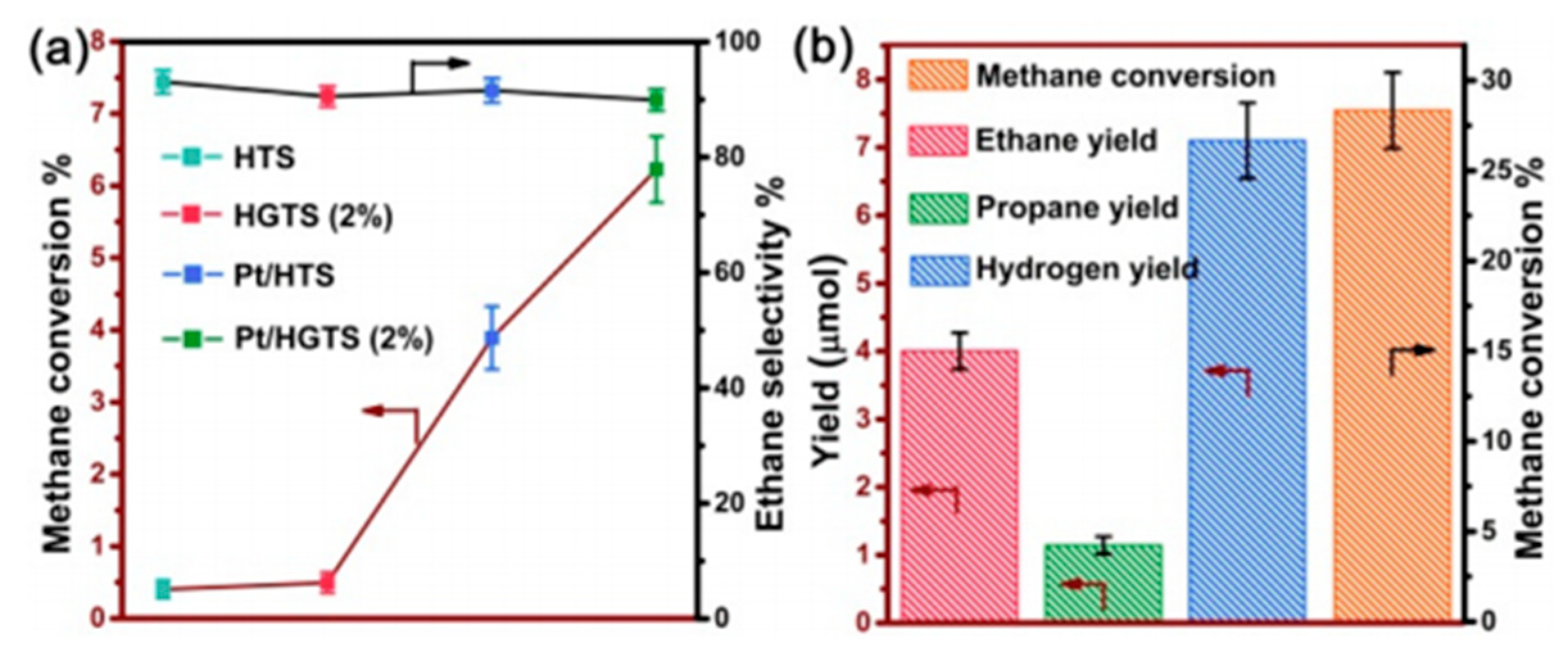
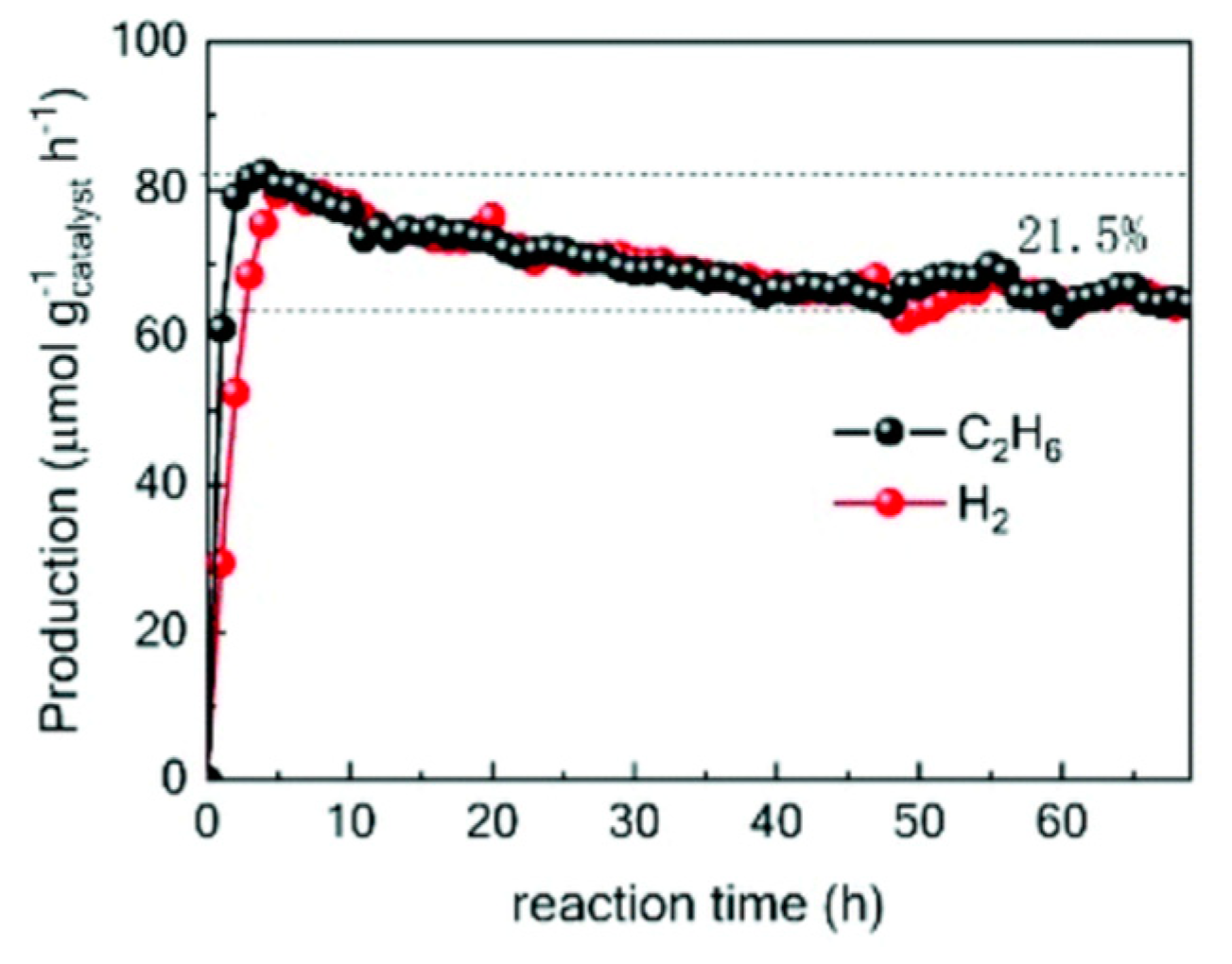
7. Photocatalytic Nonoxidative Coupling of Methane
7.1. Overview of the Reaction
7.2. Material
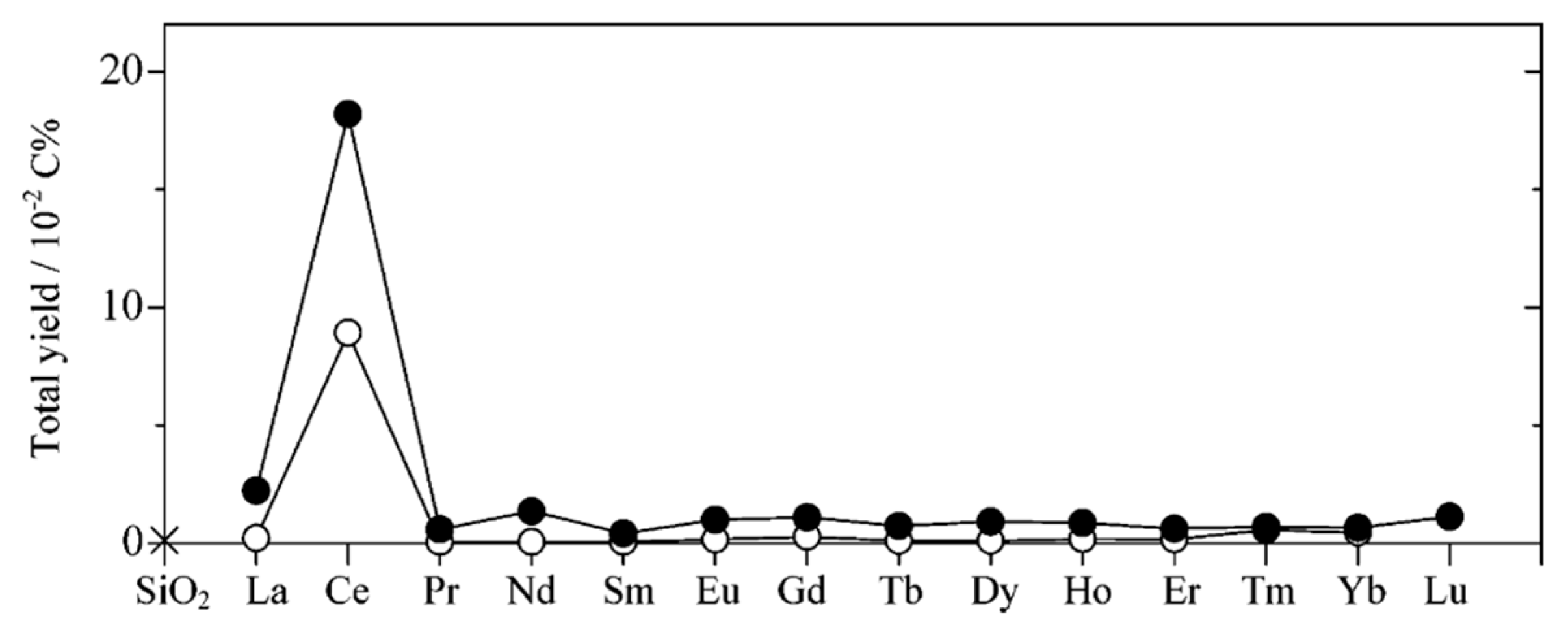
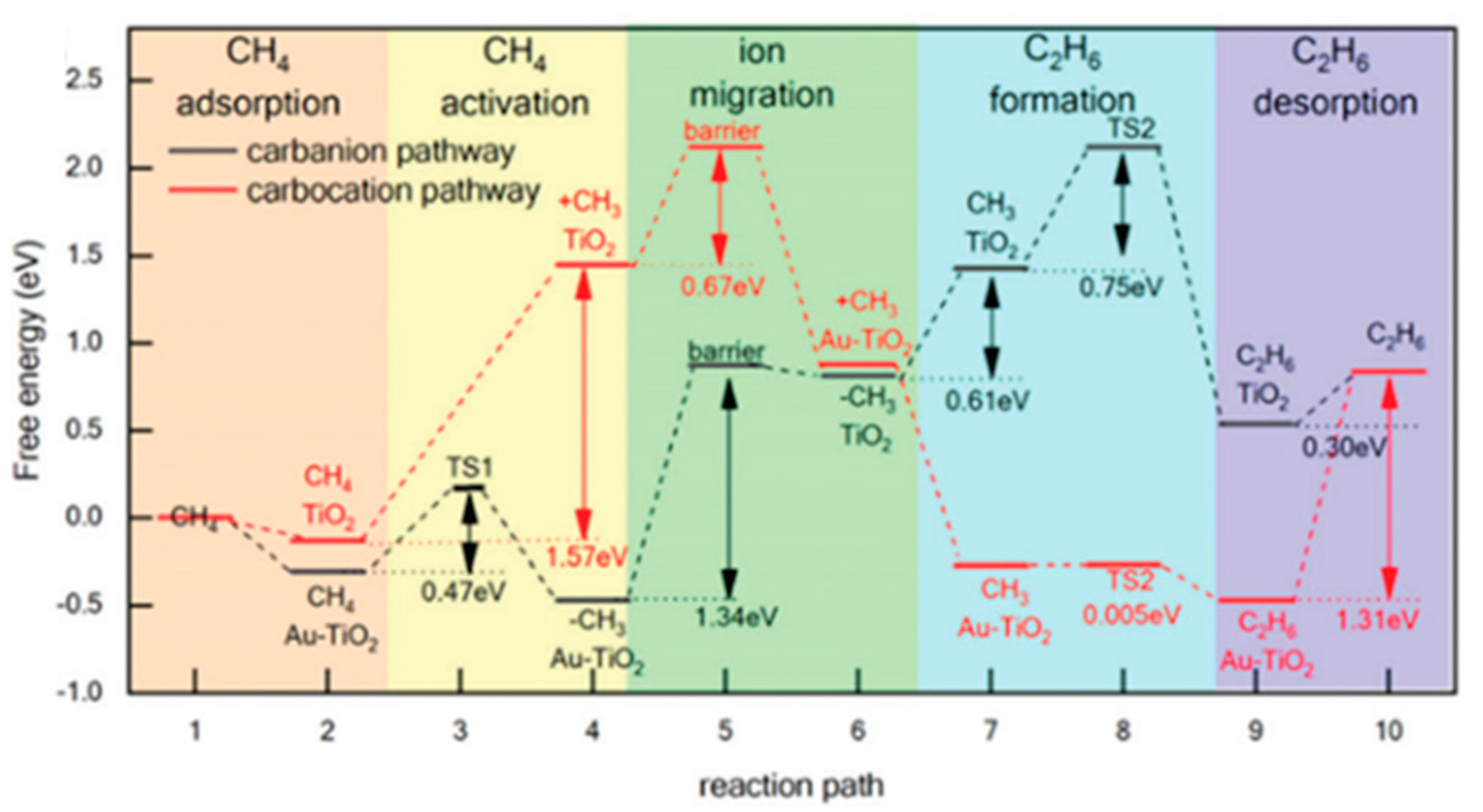
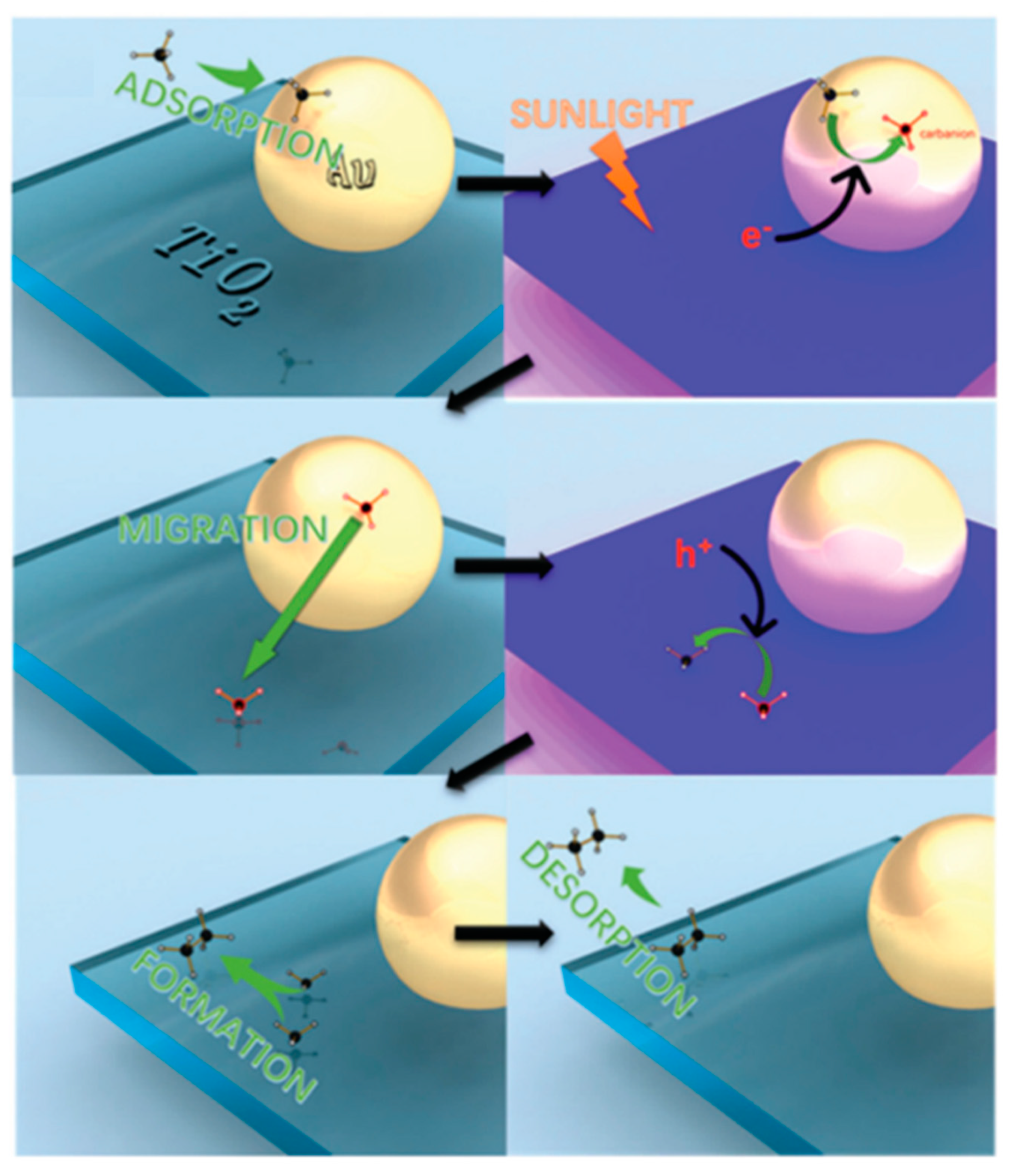
7.3. Proposed Mechanism
8. Other Important Phenomena that Must Be Considered
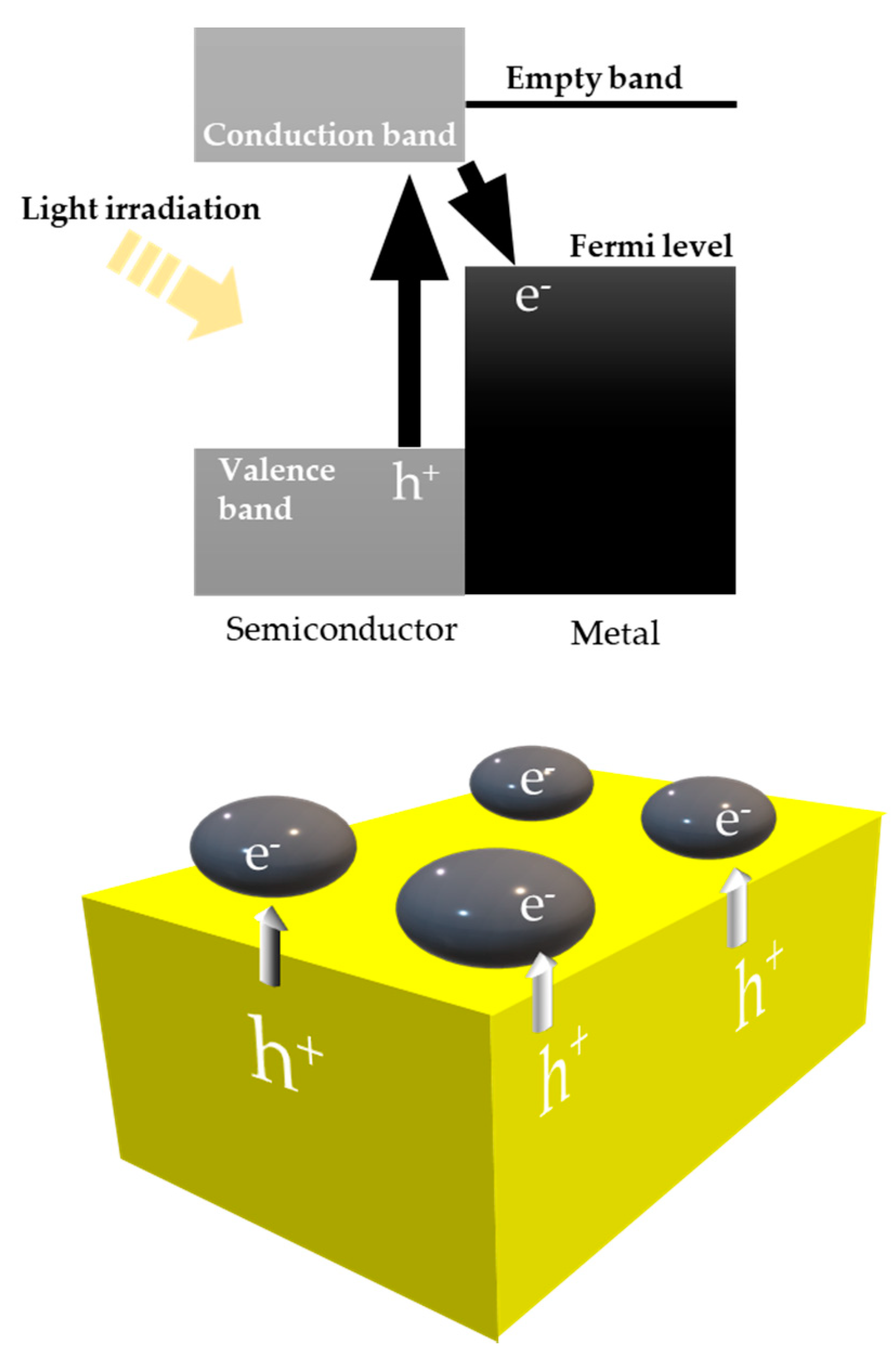
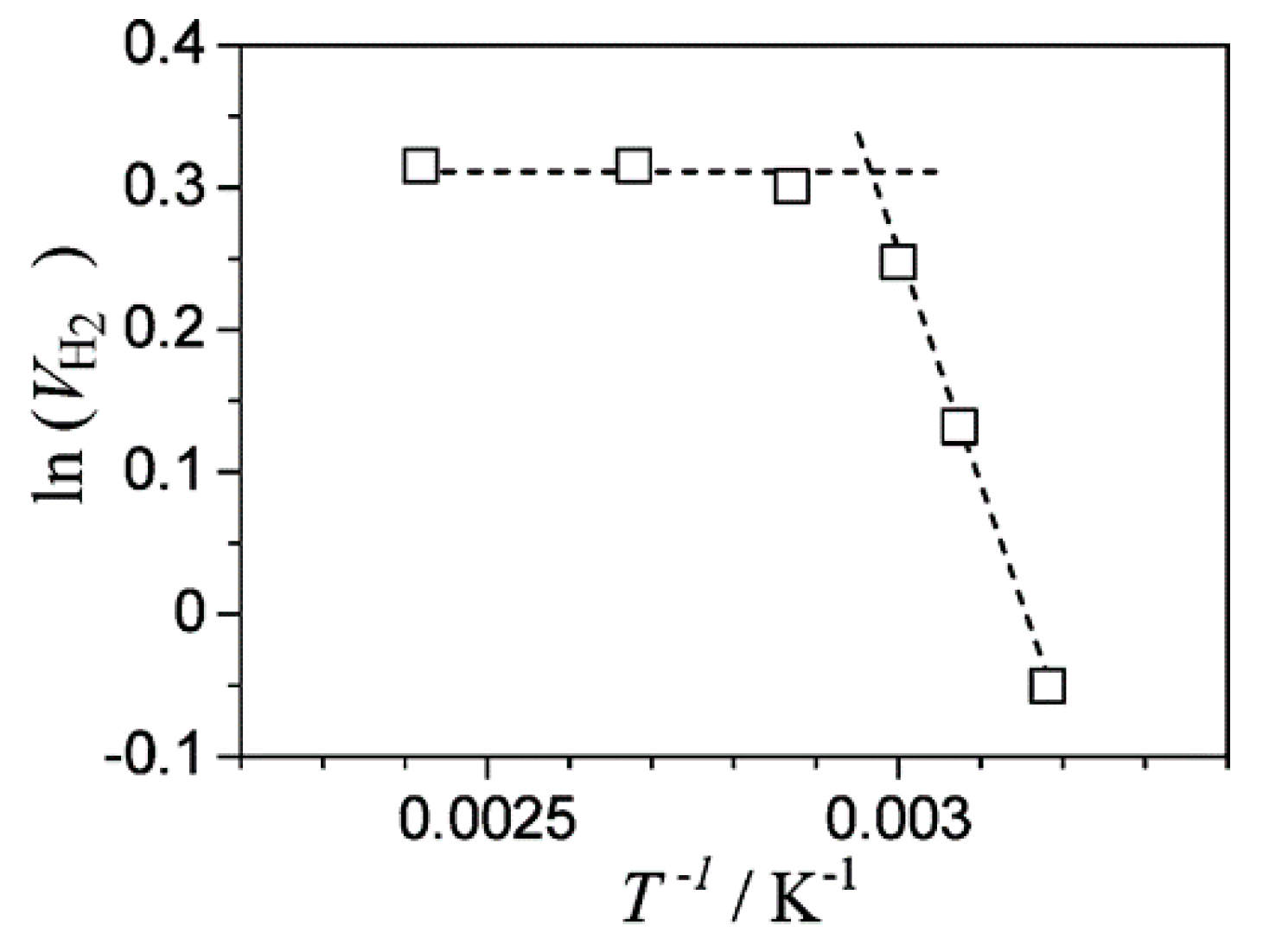
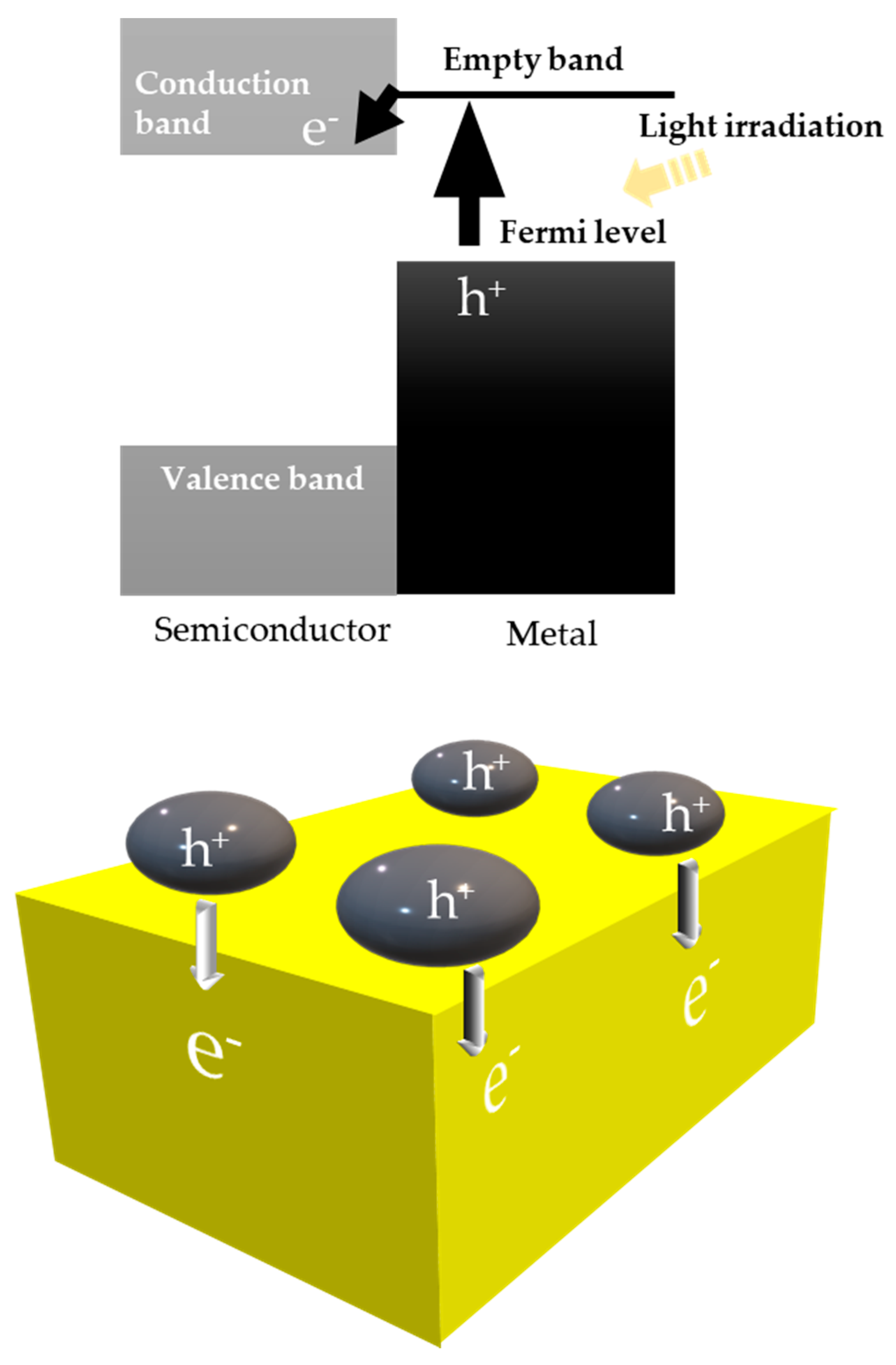
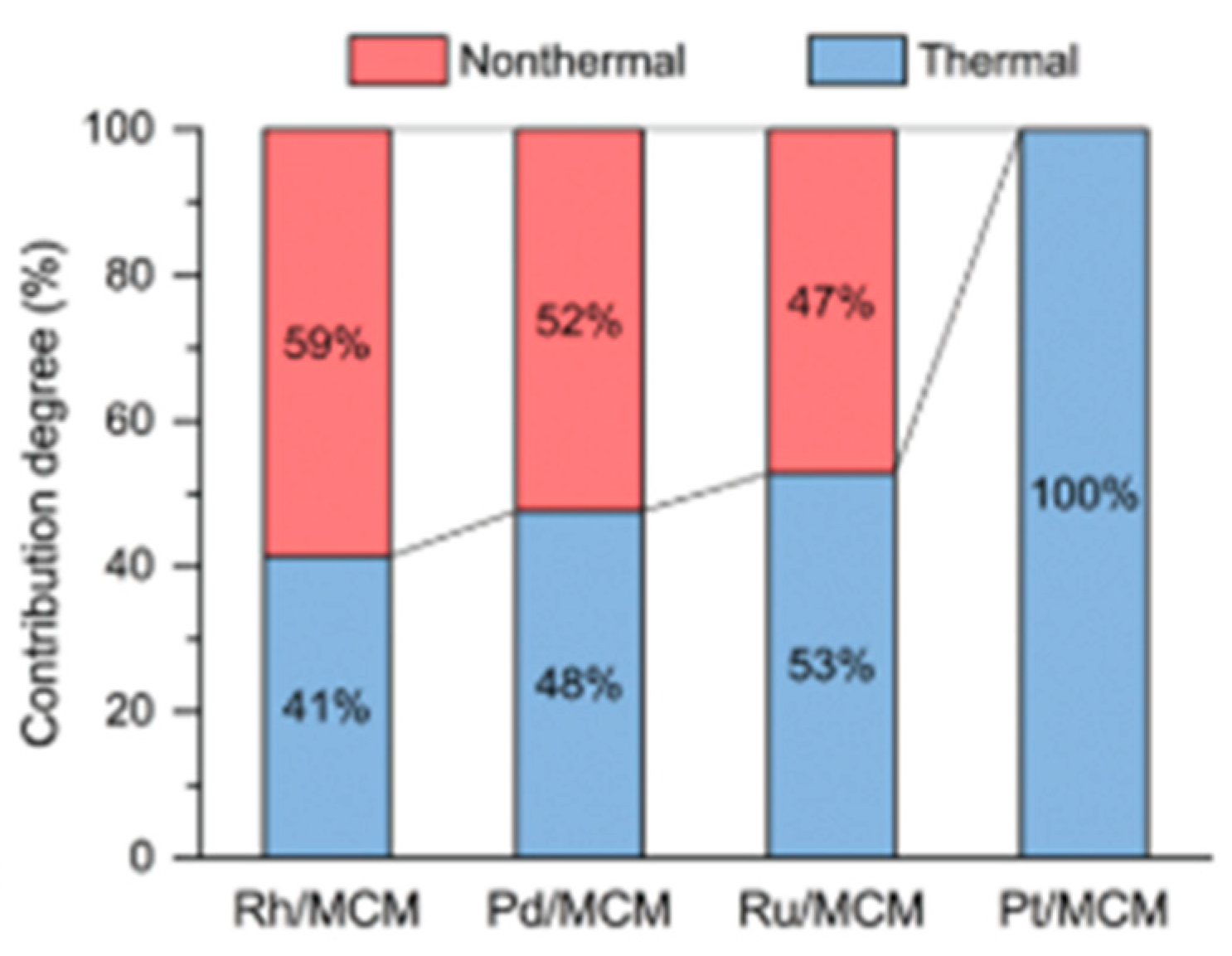
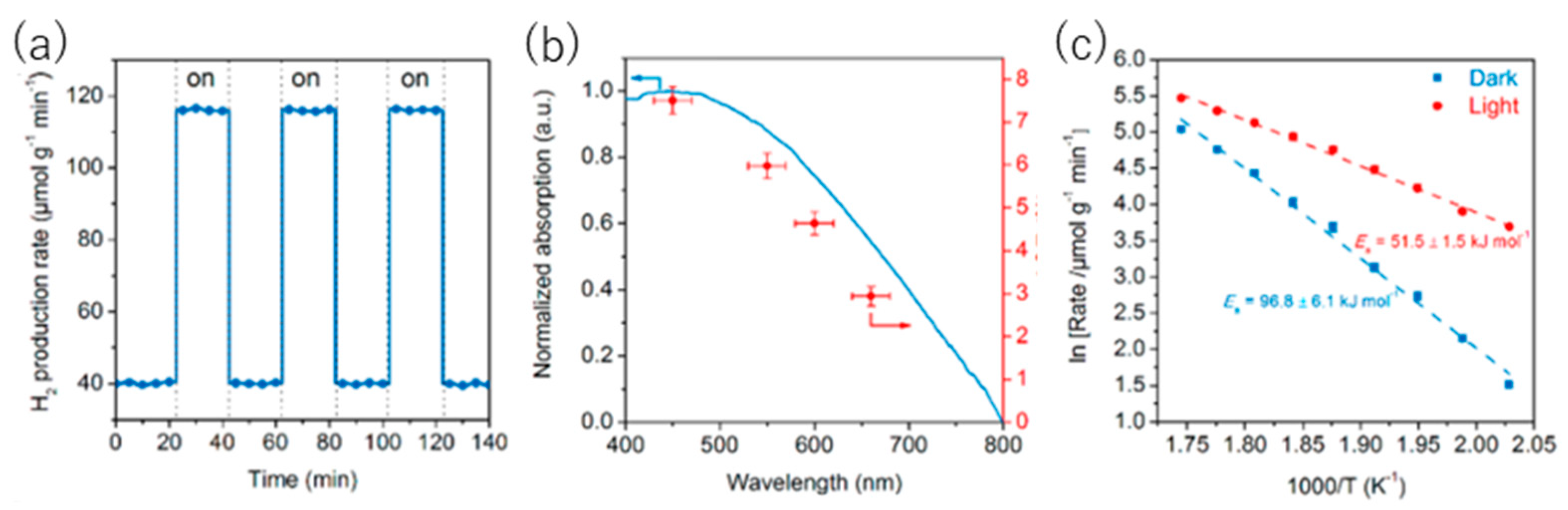
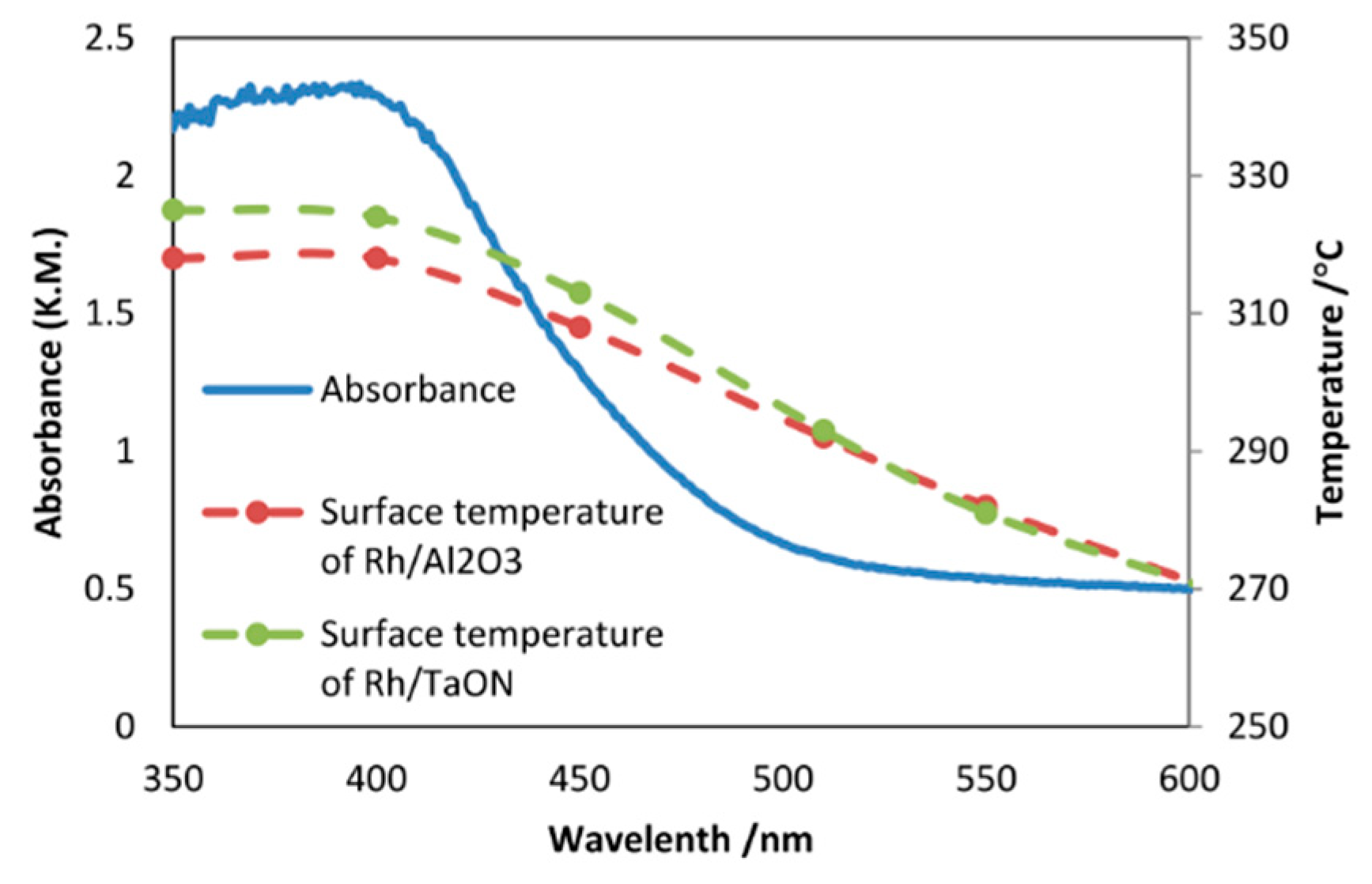
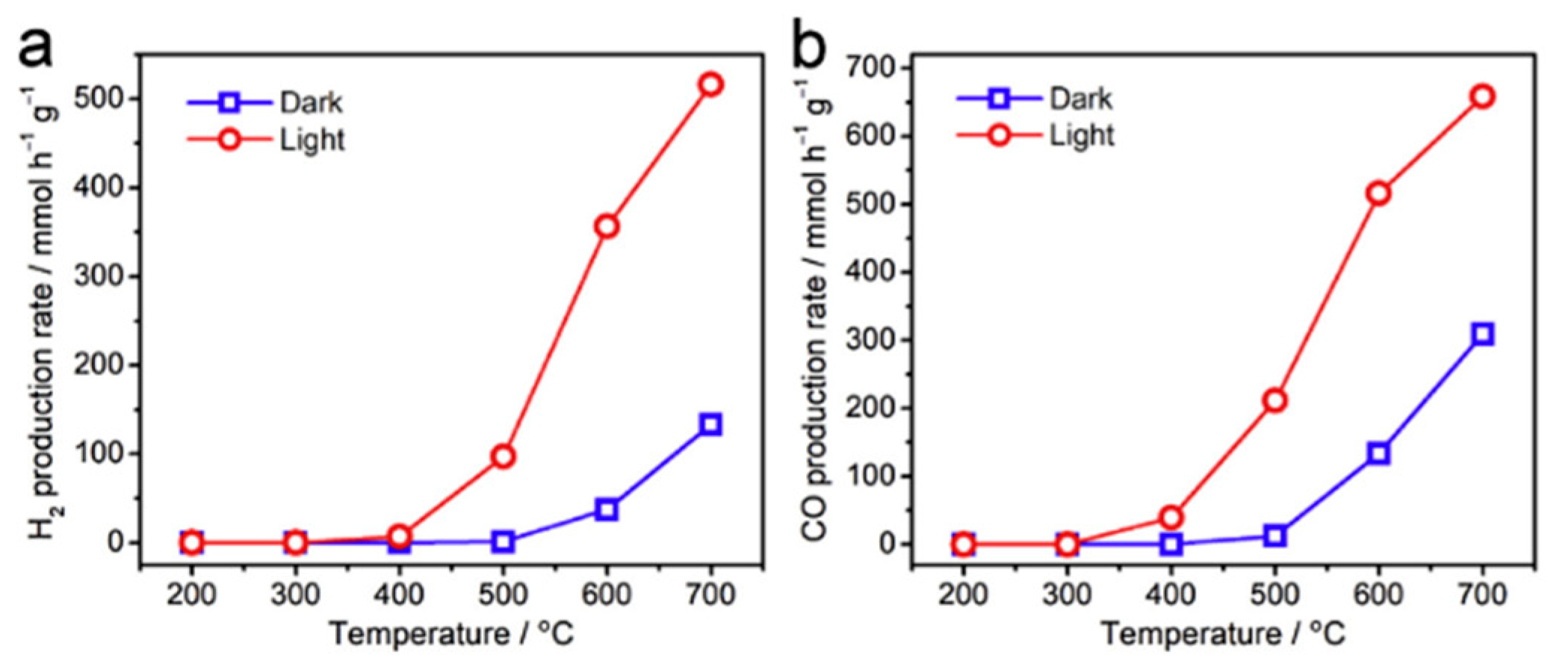
8.1. Another Excitation Mechanism: Hot Carriers
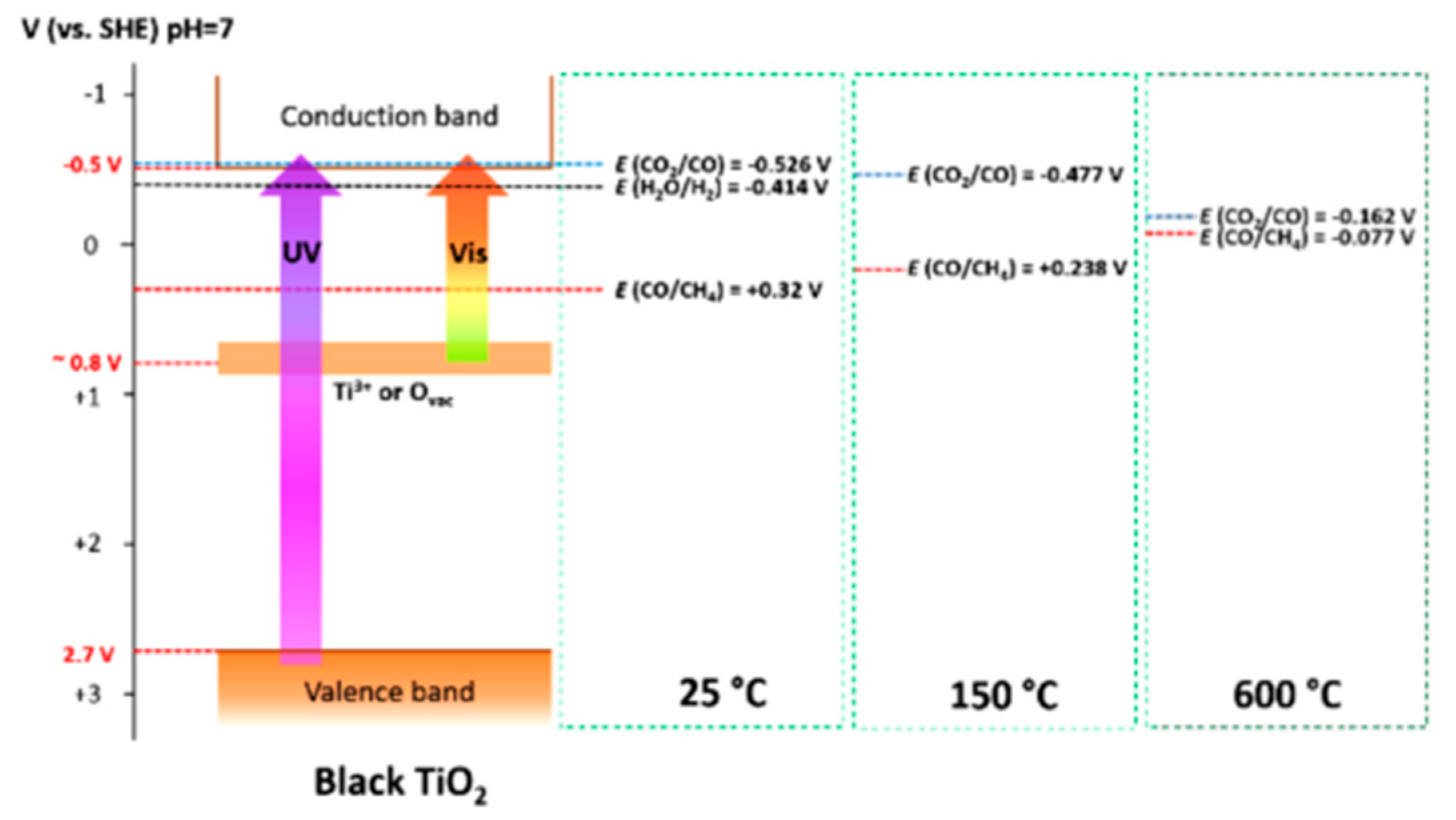
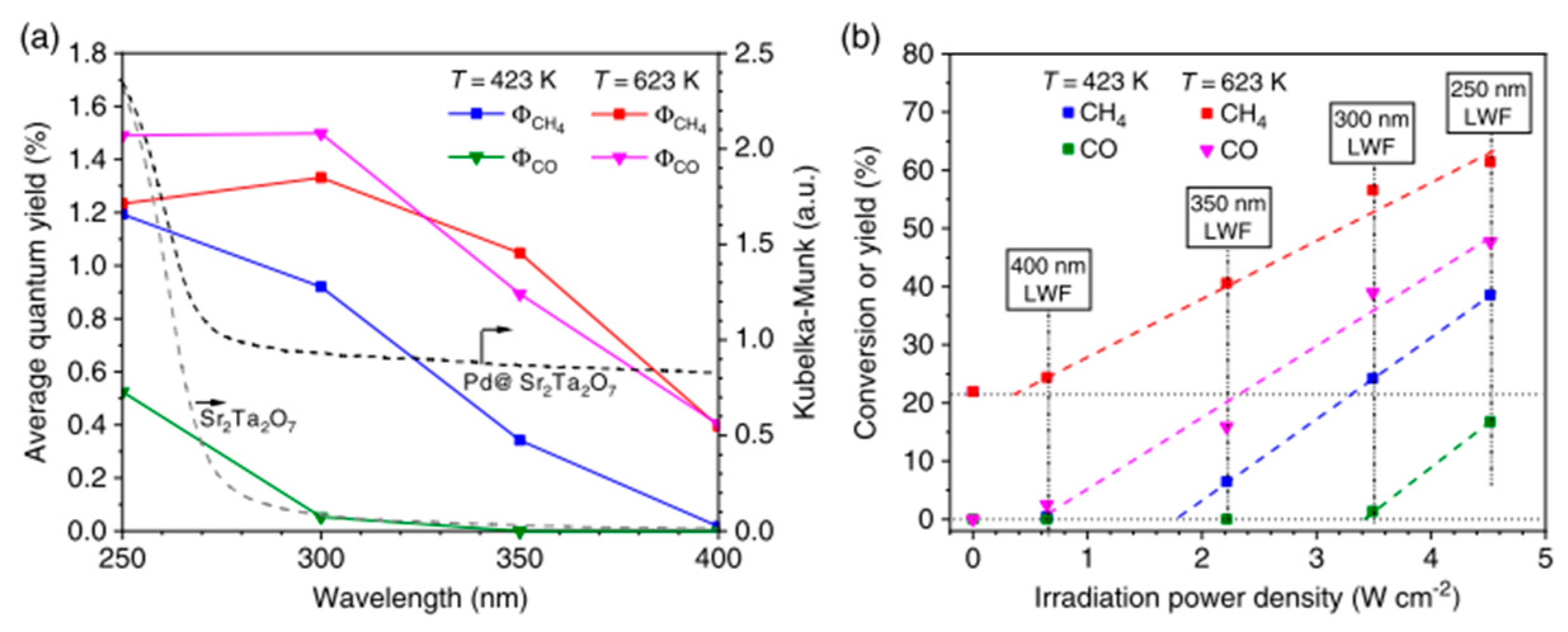
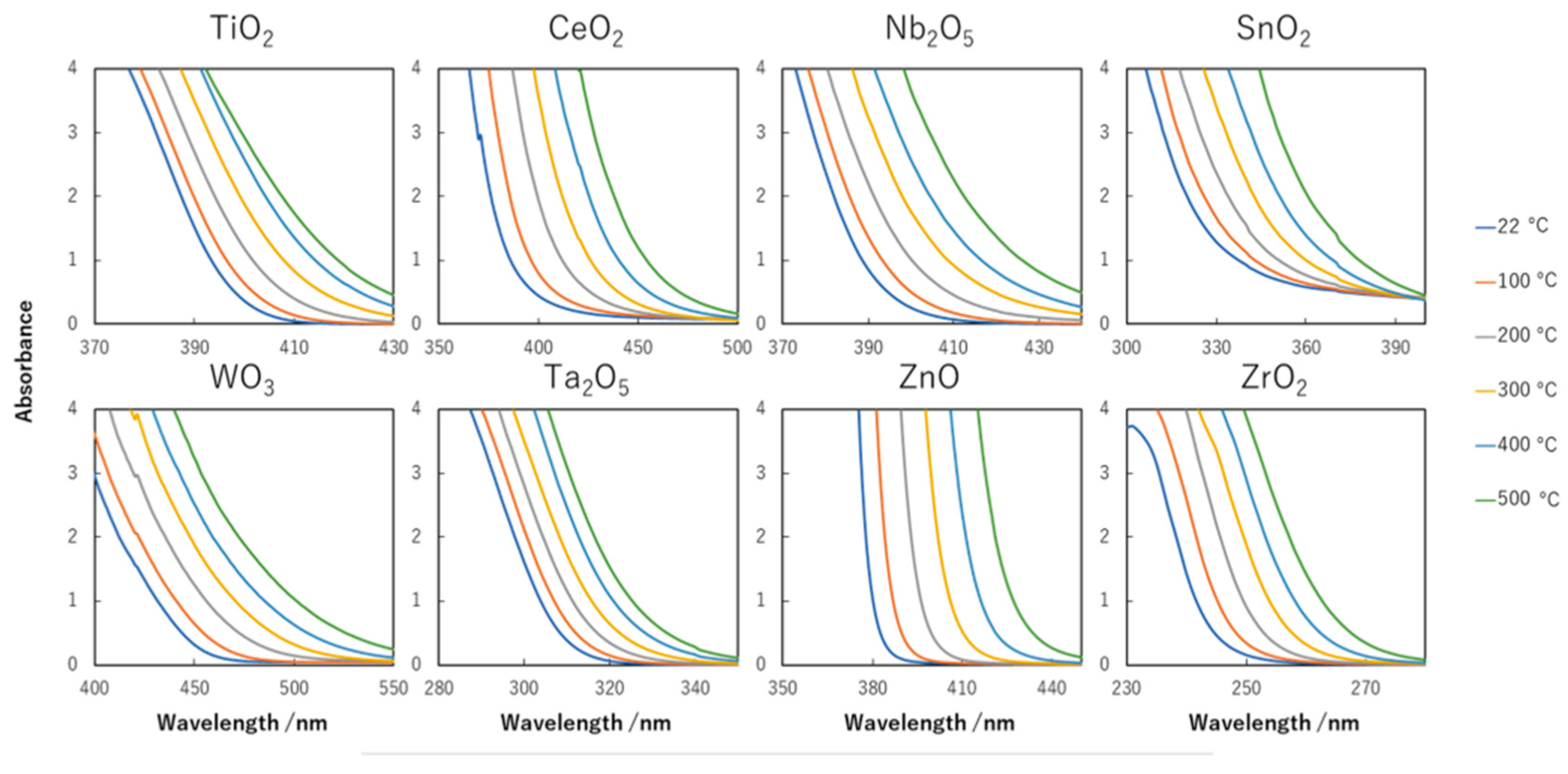
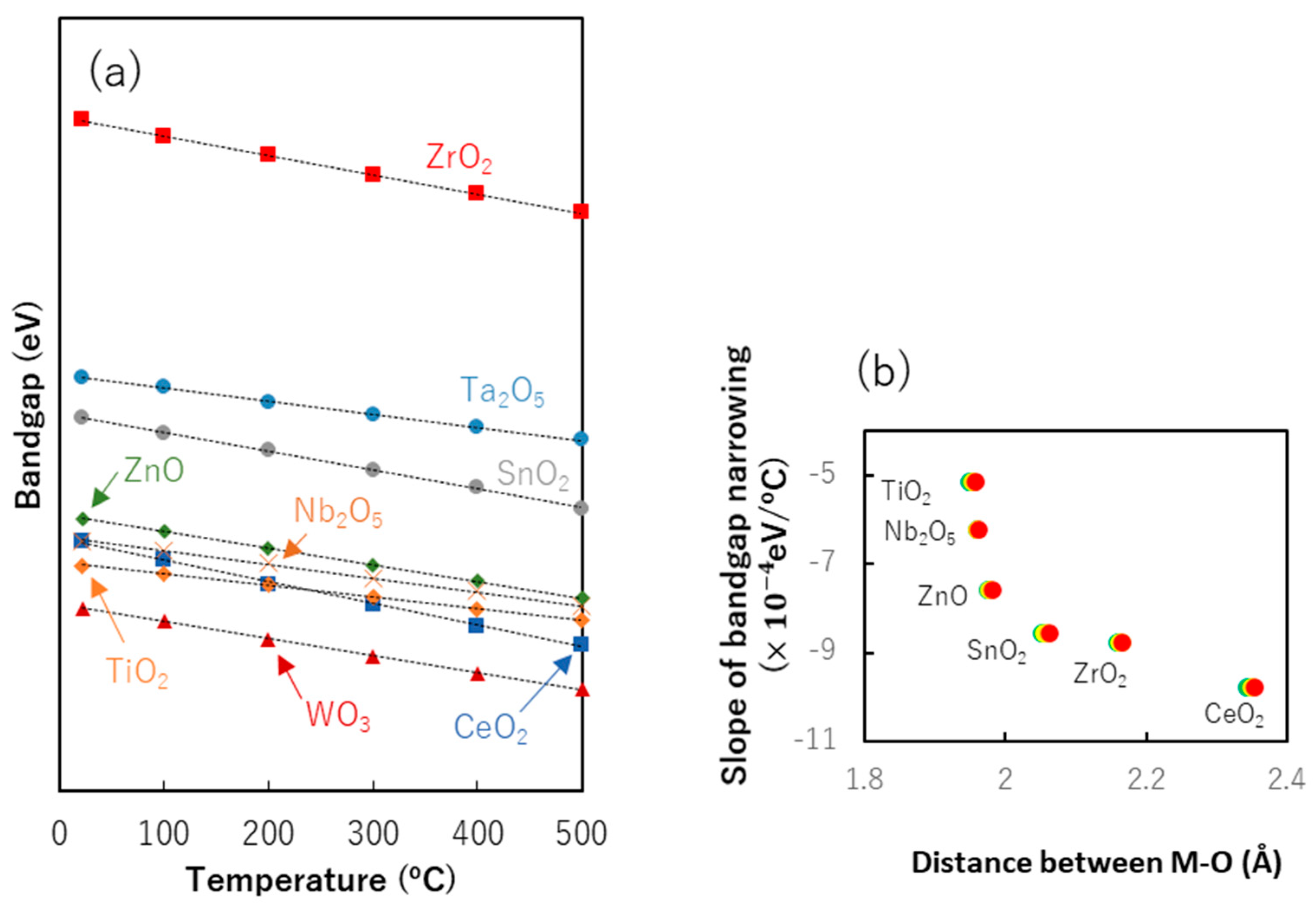
8.2. Bandgap Shift due to Thermal Energy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liu, C.; Ye, J.; Jiang, J.; Pan, Y. Progresses in the Preparation of Coke Resistant Ni-based Catalyst for Steam and CO2 Reforming of Methane. ChemCatChem 2011, 3, 529–541. [Google Scholar] [CrossRef]
- Tang, S.-B.; Qiu, F.-L.; Lu, S.-J. Effect of supports on the carbon deposition of nickel catalysts for methane reforming with CO2. Catal. Today 1995, 24, 253–255. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 81. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Pan, X.; Cortie, D.; Huang, X.; Yi, Z. Photocatalytic oxidation of methane over silver decorated zinc oxide nanocatalysts. Nat. Commun. 2016, 7, 12273. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Gordon, S.; Mcbride, B.J. Computer Program for Calculation of Complex Chemical Equilibrium Compositions and Application; NASA Reference Publication: Washington, DC, USA, 1994. [Google Scholar]
- Tomishige, K.; Himeno, Y.; Matsuo, Y.; Yoshinaga, Y.; Fujimoto, K. Catalytic Performance and Carbon Deposition Behavior of a NiO−MgO Solid Solution in Methane Reforming with Carbon Dioxide under Pressurized Conditions. Ind. Eng. Chem. Res. 2000, 39, 1891–1897. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, Non-oxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bao, X.; Lin, L. Direct conversion of methane under non-oxidative conditions. J. Catal. 2003, 216, 386–395. [Google Scholar] [CrossRef]
- Redondo, A.B.; Troussard, E.; van Bokhoven, J.A. Non-oxidative methane conversion assisted by corona discharge. Fuel Process. Technol. 2012, 104, 265–270. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Duan, X. Progress, challenge and perspective of heterogeneous photocatalysts. Chem. Soc. Rev. 2013, 42, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Web of Science. Analyze search Results for methane Reforming and methane Reforming Photo. Available online: https://apps.webofknowledge.com/WOS_GeneralSearch_input.do?product=WOS&search_mode=GeneralSearch&SID=F3OWgDOUyyDPjOy2WTg&preferencesSaved= (accessed on 11 November 2020).
- Maeda, K.; Teramura, K.; Lu, D.; Takata, T.; Saito, N.; Inoue, Y.; Domen, K. Photocatalyst releasing hydrogen from water. Nature 2006, 440, 295. [Google Scholar] [CrossRef] [PubMed]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic CO2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- Ye, S.; Wang, R.; Wu, M.-Z.; Yuan, Y.-P. A review on g-C3N4 for photocatalytic water splitting and CO2 reduction. Appl. Surf. Sci. 2015, 358, 15–27. [Google Scholar] [CrossRef]
- Wang, H.Y.; Ruckenstein, E. Carbon dioxide reforming of methane to synthesis gas over supported rhodium catalysts: The effect of support. Appl. Catal. A Gen. 2000, 204, 143–152. [Google Scholar] [CrossRef]
- Dębek, R.; Radlik, M.; Motak, M.; Galvez, M.E.; Turek, W.; Da Costa, P.; Grzybek, T. Ni-containing Ce-promoted hydrotalcite derived materials as catalysts for methane reforming with carbon dioxide at low temperature—On the effect of basicity. Catal. Today 2015, 257, 59–65. [Google Scholar] [CrossRef]
- García, V.; Fernández, J.J.; Ruíz, W.; Mondragón, F.; Moreno, A. Effect of MgO addition on the basicity of Ni/ZrO2 and on its catalytic activity in carbon dioxide reforming of methane. Catal. Commun. 2009, 11, 240–246. [Google Scholar] [CrossRef]
- Cho, Y.; Shoji, S.; Yamaguchi, A.; Hoshina, T.; Fujita, T.; Abe, H.; Miyauchi, M. Visible-light-driven dry reforming of methane using a semiconductor-supported catalyst. Chem. Commun. 2020, 56. [Google Scholar] [CrossRef]
- Shimura, K.; Kato, S.; Yoshida, T.; Itoh, H.; Hattori, T.; Yoshida, H. Photocatalytic Steam Reforming of Methane over Sodium Tantalate. J. Phys. Chem. C 2010, 114, 3493–3503. [Google Scholar] [CrossRef]
- Shimura, K.; Kawai, H.; Yoshida, T.; Yoshida, H. Bifunctional Rhodium Cocatalysts for Photocatalytic Steam Reforming of Methane over Alkaline Titanate. ACS Catal. 2012, 2, 2126–2134. [Google Scholar] [CrossRef]
- Li, D.; Nakagawa, Y.; Tomishige, K. Methane reforming to synthesis gas over Ni catalysts modified with noble metals. Appl. Catal. A Gen. 2011, 408, 1–24. [Google Scholar] [CrossRef]
- Rezaei, M.; Alavi, S.M.; Sahebdelfar, S.; Yan, Z.-F. Syngas Production by Methane Reforming with Carbon Dioxide on Noble Metal Catalysts. J. Nat. Gas. Chem. 2006, 15, 327–334. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- László, B.; Baán, K.; Varga, E.; Oszkó, A.; Erdőhelyi, A.; Kónya, Z.; Kiss, J. Photo-induced reactions in the CO2-methane system on titanate nanotubes modified with Au and Rh nanoparticles. Appl. Catal. B Environ. 2016, 199, 473–484. [Google Scholar] [CrossRef]
- Shimura, K.; Yoshida, H. Semiconductor Photocatalysts for Non-oxidative Coupling, Dry Reforming and Steam Reforming of Methane. Catal. Surv. Asia 2014, 18, 24–33. [Google Scholar] [CrossRef]
- Shoji, S.; Peng, X.; Yamaguchi, A.; Watanabe, R.; Fukuhara, C.; Cho, Y.; Yamamoto, T.; Matsumura, S.; Yu, M.-W.; Ishii, S.; et al. Photocatalytic uphill conversion of natural gas beyond the limitation of thermal reaction systems. Nat. Catal. 2020, 3. [Google Scholar] [CrossRef]
- Tahir, M.; Tahir, B.; Amin, N.S. Photocatalytic CO2 reduction by CH4 over montmorillonite modified TiO2 nanocomposites in a continuous monolith photoreactor. Mater. Res. Bull. 2015, 63, 13–23. [Google Scholar] [CrossRef]
- Shi, D.; Feng, Y.; Zhong, S. Photocatalytic conversion of CH4 and CO2 to oxygenated compounds over Cu/CdS–TiO2/SiO2 catalyst. Catal. Today 2004, 98, 505–509. [Google Scholar] [CrossRef]
- Lang, J.; Ma, Y.; Wu, X.; Jiang, Y.; Hu, Y.H. Highly efficient light-driven methane coupling under ambient conditions based on an integrated design of a photocatalytic system. Green Chem. 2020, 22, 4669–4675. [Google Scholar] [CrossRef]
- Roy, S.; Bauer, T.; Al-Dahhan, M.; Lehner, P.; Turek, T. Monoliths as multiphase reactors: A review. AIChE J. 2004, 50, 2918–2938. [Google Scholar] [CrossRef]
- Tahir, M.; Tahir, B.; Zakaria, Z.Y.; Muhammad, A. Enhanced photocatalytic carbon dioxide reforming of methane to fuels over nickel and montmorillonite supported TiO2 nanocomposite under UV-light using monolith photoreactor. J. Clean. Prod. 2019, 213, 451–461. [Google Scholar] [CrossRef]
- Tahir, M. Enhanced photocatalytic CO2 reduction to fuels through bireforming of methane over structured 3D MAX Ti3AlC2/TiO2 heterojunction in a monolith photoreactor. J. CO2 Util. 2020, 38, 99–112. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Everitt, H.O.; Liu, J. Light-Induced Thermal Gradients in Ruthenium Catalysts Significantly Enhance Ammonia Production. Nano Lett. 2019, 19, 1706–1711. [Google Scholar] [CrossRef]
- Ohno, T.; Sarukawa, K.; Tokieda, K.; Matsumura, M. Morphology of a TiO2 Photocatalyst (Degussa, P-25) Consisting of Anatase and Rutile Crystalline Phases. J. Catal. 2001, 203, 82–86. [Google Scholar] [CrossRef]
- Park, H.; Park, Y.; Kim, W.; Choi, W. Surface modification of TiO2 photocatalyst for environmental applications. J. Photochem. Photobiol. C Photochem. Rev. 2013, 15, 1–20. [Google Scholar] [CrossRef]
- Yoshida, H.; Hirao, K.; Nishimoto, J.; Shimura, K.; Kato, S.; Itoh, H.; Hattori, T. Hydrogen Production from Methane and Water on Platinum Loaded Titanium Oxide Photocatalysts. J. Phys. Chem. C 2008, 112, 5542–5551. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Mondal, K.C. CO2 reforming of methane combined with steam reforming or partial oxidation of methane to syngas over NdCoO3 perovskite-type mixed metal-oxide catalyst. Appl. Energy 2006, 83, 1024–1032. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zhang, X.; Mi, Z. Thermodynamic analysis of autothermal steam and CO2 reforming of methane. Int. J. Hydrogen Energy 2008, 33, 2507–2514. [Google Scholar] [CrossRef]
- Wibowo, S.; Yamaguchi, A.; Shoji, S.; Fujita, T.; Abe, H.; Miyauchi, M. Photo-assisted Dry Reforming of Methane over Strontium Titanate. Chem. Lett. 2018, 47, 935–937. [Google Scholar] [CrossRef]
- Teramura, K.; Tanaka, T.; Ishikawa, H.; Kohno, Y.; Funabiki, T. Photocatalytic Reduction of CO2 to CO in the Presence of H2 or CH4 as a Reductant over MgO. J. Phys. Chem. B 2003, 108, 346–354. [Google Scholar] [CrossRef]
- Chang, S.; Li, M.; Hua, Q.; Zhang, L.; Ma, Y.; Ye, B.; Huang, W. Shape-dependent interplay between oxygen vacancies and Ag–CeO2 interaction in Ag/CeO2 catalysts and their influence on the catalytic activity. J. Catal. 2012, 293, 195–204. [Google Scholar] [CrossRef]
- Pan, F.; Xiang, X.; Du, Z.; Sarnello, E.; Li, T.; Li, Y. Integrating photocatalysis and thermocatalysis to enable efficient CO2 reforming of methane on Pt supported CeO2 with Zn doping and atomic layer deposited MgO overcoating. Appl. Catal. B Environ. 2020, 260, 118189. [Google Scholar] [CrossRef]
- Lei, J.; Chen, Y.; Shen, F.; Wang, L.; Liu, Y.; Zhang, J. Surface modification of TiO2 with g-C3N4 for enhanced UV and visible photocatalytic activity. J. Alloys Compd. 2015, 631, 328–334. [Google Scholar] [CrossRef]
- Tahir, B.; Tahir, M.; Amin, N.A.S. Photo-induced CO2 reduction by CH4/H2O to fuels over Cu-modified g-C3N4 nanorods under simulated solar energy. Appl. Surf. Sci. 2017, 419, 875–885. [Google Scholar] [CrossRef]
- Tahir, B.; Tahir, M.; Amin, N.A.S. Silver loaded protonated graphitic carbon nitride (Ag/pg-C3N4) nanosheets for stimulating CO2 reduction to fuels via photocatalytic bi-reforming of methane. Appl. Surf. Sci. 2019, 493, 18–31. [Google Scholar] [CrossRef]
- Han, B.; Wei, W.; Chang, L.; Cheng, P.; Hu, Y.H. Efficient Visible Light Photocatalytic CO2 Reforming of CH4. ACS Catal. 2015, 6, 494–497. [Google Scholar] [CrossRef]
- Han, B.; Wei, W.; Li, M.; Sun, K.; Hu, Y.H. A thermo-photo hybrid process for steam reforming of methane: Highly efficient visible light photocatalysis. Chem. Commun. 2019, 55, 7816–7819. [Google Scholar] [CrossRef]
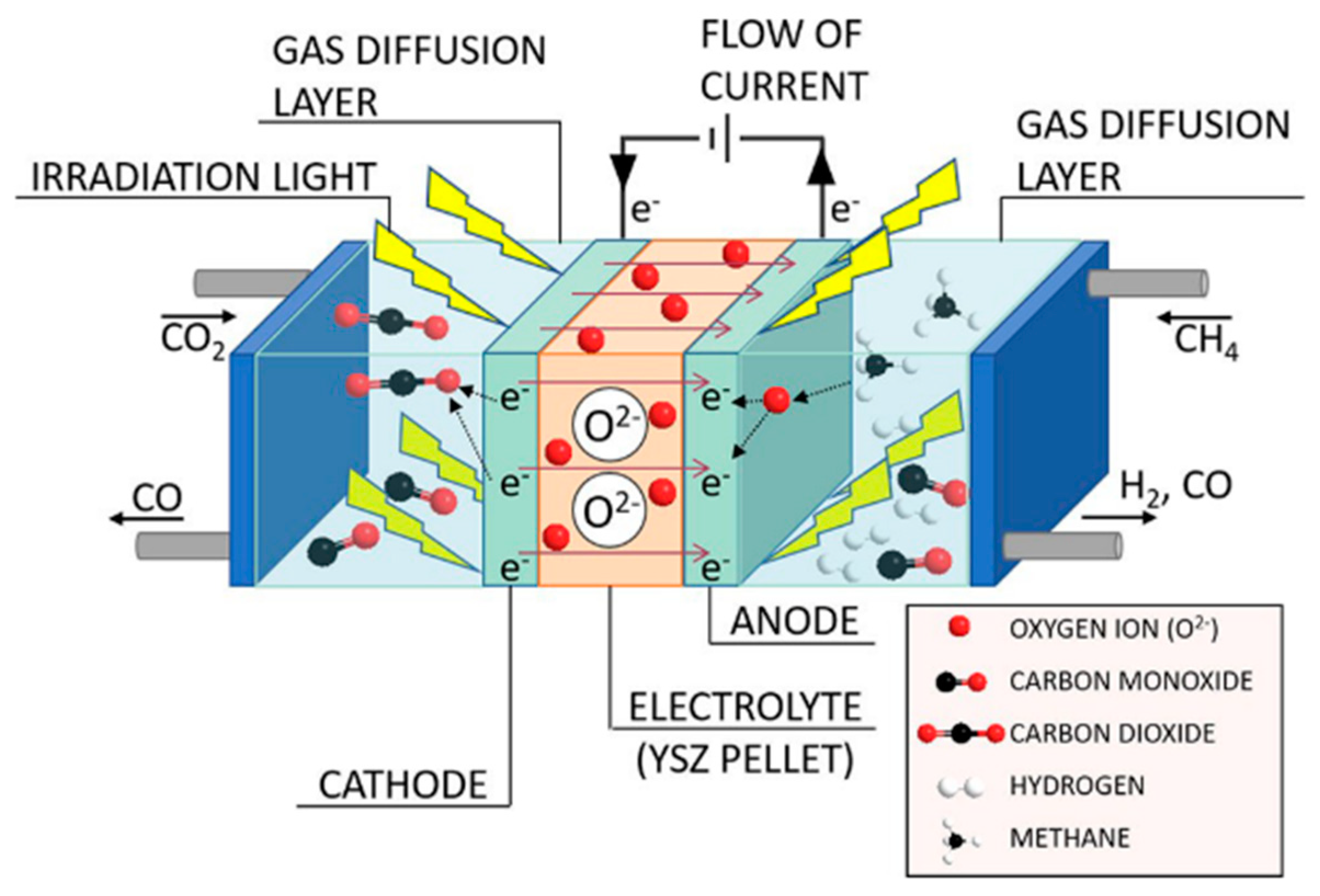
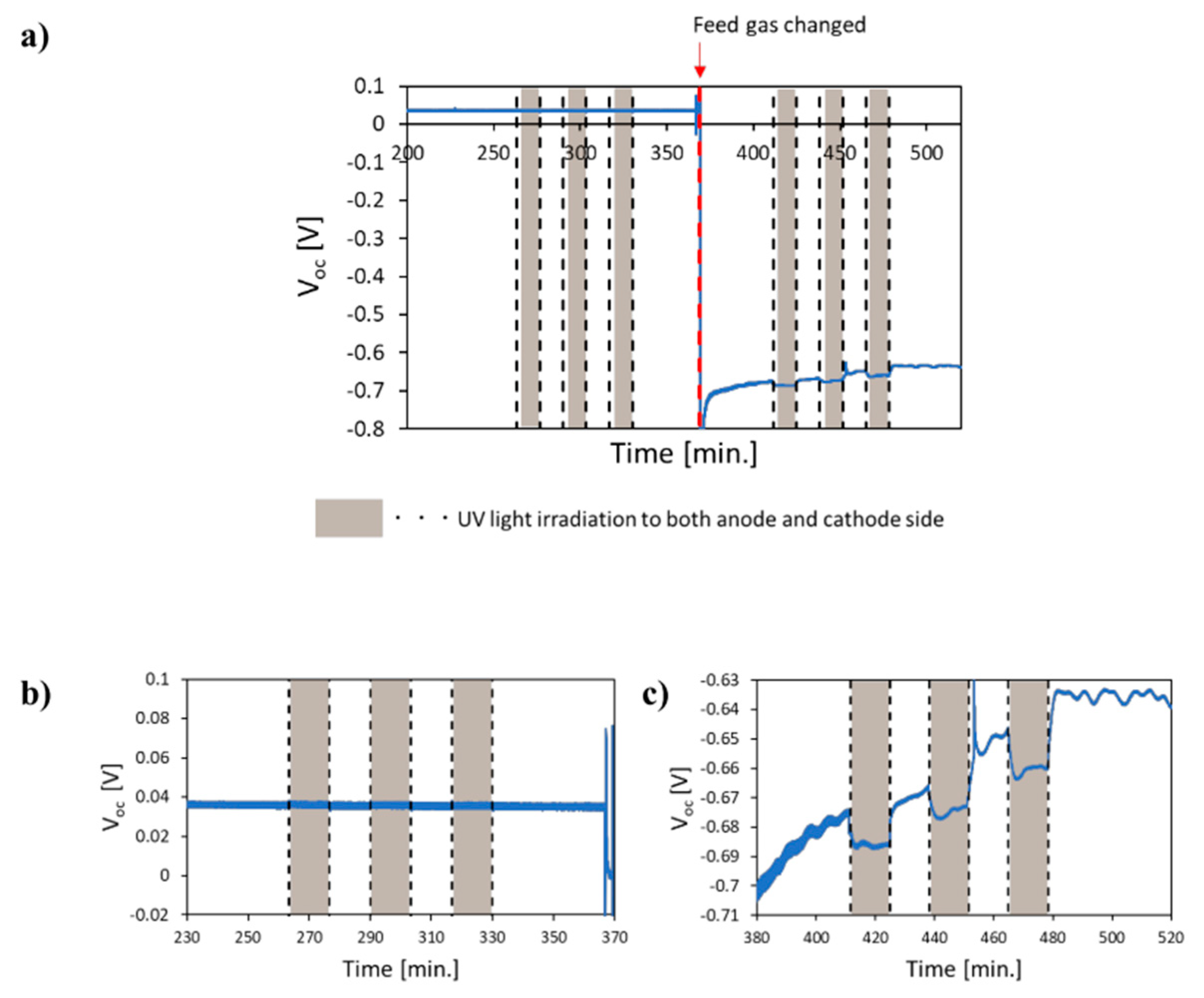
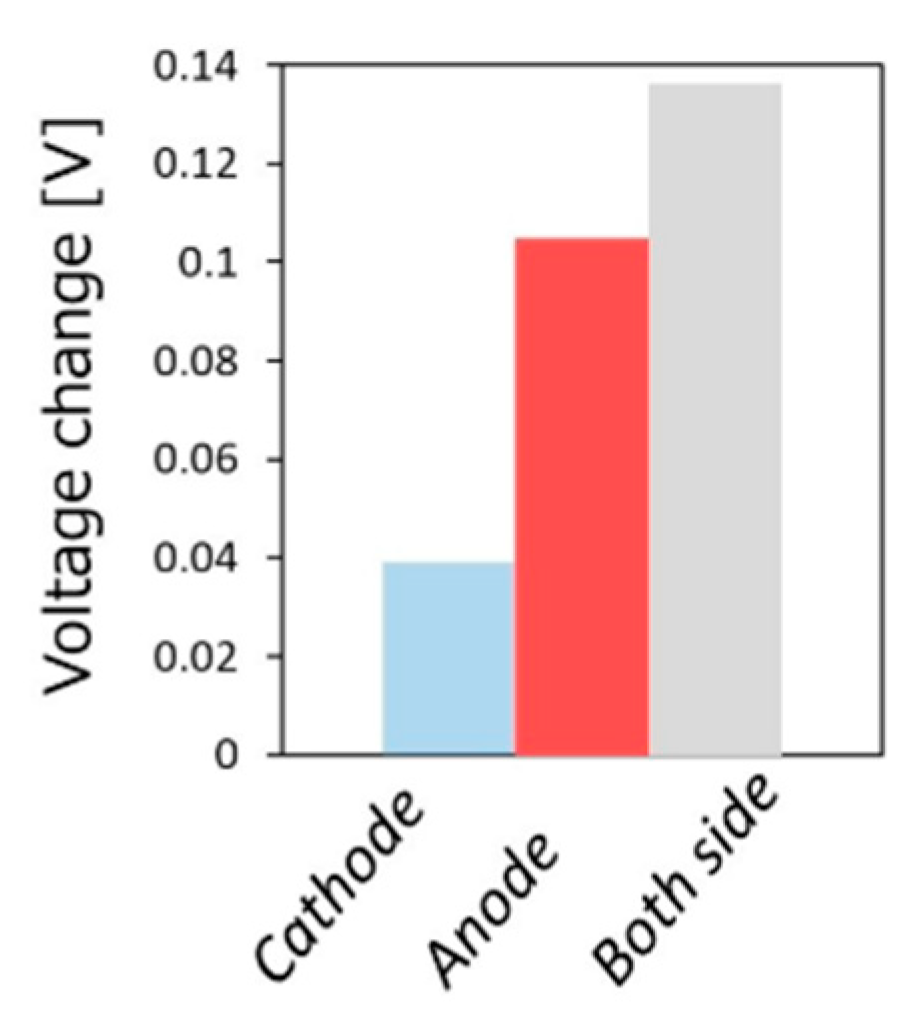
- Kushida, M.; Yamaguchi, A.; Cho, Y.; Fujita, T.; Abe, H.; Miyauchi, M. Gas-Phase Photoelectrocatalysis Mediated by Oxygen Ions for Uphill Conversion of Greenhouse Gases. ChemPhotoChem 2020. [Google Scholar] [CrossRef]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in methane conversion processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef]
- Sari, F.N.I.; Lu, S.-H.; Ting, J.-M. Wide-bandgap HfO2-V2O5 nanowires heterostructure for visible light-driven photocatalytic degradation. J. Am. Ceram. Soc. 2020, 103, 2252–2261. [Google Scholar] [CrossRef]
- Murcia-López, S.; Villa, K.; Andreu, T.; Morante, J.R. Improved selectivity for partial oxidation of methane to methanol in the presence of nitrite ions and BiVO4 photocatalyst. Chem. Commun. 2015, 51, 7249–7252. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Nagai, Y.; Rahmawaty, D.; Wei, C.; Anpo, M. Characteristics of the Photocatalytic Oxidation of Methane into Methanol on V-Containing MCM-41 Catalysts. Catal. Lett. 2008, 124, 80. [Google Scholar] [CrossRef]
- Villa, K.; Murcia-López, S.; Andreu, T.; Morante, J.R. Mesoporous WO3 photocatalyst for the partial oxidation of methane to methanol using electron scavengers. Appl. Catal. B Environ. 2015, 163, 150–155. [Google Scholar] [CrossRef]
- Murcia-López, S.; Villa, K.; Andreu, T.; Morante, J.R. Partial Oxidation of Methane to Methanol Using Bismuth-Based Photocatalysts. ACS Catal. 2014, 4, 3013–3019. [Google Scholar] [CrossRef]
- Murcia-López, S.; Bacariza, M.C.; Villa, K.; Lopes, J.M.; Henriques, C.; Morante, J.R.; Andreu, T. Controlled Photocatalytic Oxidation of Methane to Methanol through Surface Modification of Beta Zeolites. ACS Catal. 2017, 7, 2878–2885. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Aksoylu, E.; Wayne Goodman, D. Nonoxidative Activation of Methane. Catal. Rev. 2003, 45, 151–203. [Google Scholar] [CrossRef]
- Wu, S.; Tan, X.; Lei, J.; Chen, H.; Wang, L.; Zhang, J. Ga-Doped and Pt-Loaded Porous TiO2–SiO2 for Photocatalytic Nonoxidative Coupling of Methane. J. Am. Chem. Soc. 2019, 141, 6592–6600. [Google Scholar] [CrossRef]
- Yuliati, L.; Hamajima, T.; Hattori, T.; Yoshida, H. Nonoxidative Coupling of Methane over Supported Ceria Photocatalysts. J. Phys. Chem. C 2008, 112, 7223–7232. [Google Scholar] [CrossRef]
- Yuliati, L.; Hattori, T.; Itoh, H.; Yoshida, H. Photocatalytic non-oxidative coupling of methane on gallium oxide and silica-supported gallium oxide. J. Catal. 2008, 257, 396–402. [Google Scholar] [CrossRef]
- Singh, S.P.; Anzai, A.; Kawaharasaki, S.; Yamamoto, A.; Yoshida, H. Non-oxidative coupling of methane over Pd-loaded gallium oxide photocatalysts in a flow reactor. Catal. Today 2020. [Google Scholar] [CrossRef]
- Clavero, C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photonics 2014, 8, 95–103. [Google Scholar] [CrossRef]
- Cui, J.; Li, Y.; Liu, L.; Chen, L.; Xu, J.; Ma, J.; Fang, G.; Zhu, E.; Wu, H.; Zhao, L.; et al. Near-Infrared Plasmonic-Enhanced Solar Energy Harvest for Highly Efficient Photocatalytic Reactions. Nano Lett. 2015, 15, 6295–6301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Swearer, D.F.; Zhang, C.; Robatjazi, H.; Zhao, H.; Henderson, L.; Dong, L.; Christopher, P.; Carter, E.A.; Nordlander, P.; et al. Quantifying hot carrier and thermal contributions in plasmonic photocatalysis. Science 2018, 362, 69–72. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Reish, M.E.; Zhang, D.; Su, N.Q.; Gutiérrez, Y.; Moreno, F.; Yang, W.; Everitt, H.O.; Liu, J. Plasmon-Enhanced Catalysis: Distinguishing Thermal and Nonthermal Effects. Nano Lett. 2018, 18, 1714–1723. [Google Scholar] [CrossRef]
- Tian, Y.; Tatsuma, T. Mechanisms and Applications of Plasmon-Induced Charge Separation at TiO2 Films Loaded with Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 7632–7637. [Google Scholar] [CrossRef]
- Jiang, H.; Peng, X.; Yamaguchi, A.; Ueda, S.; Fujita, T.; Abe, H.; Miyauchi, M. Photocatalytic Partial Oxidation of Methane on Palladium-Loaded Strontium Tantalate. Sol. RRL 2019, 3, 1900076. [Google Scholar] [CrossRef]
- Jiang, H.; Peng, X.; Yamaguchi, A.; Fujita, T.; Abe, H.; Miyauchi, M. Synergistic photo-thermal and photochemical partial oxidation of methane over noble metals incorporated in mesoporous silica. Chem. Commun. 2019, 55, 13765–13768. [Google Scholar] [CrossRef]
- Song, H.; Meng, X.; Wang, Z.; Wang, Z.; Chen, H.; Weng, Y.; Ichihara, F.; Oshikiri, M.; Kako, T.; Ye, J. Visible-Light-Mediated Methane Activation for Steam Methane Reforming under Mild Conditions: A Case Study of Rh/TiO2 Catalysts. ACS Catal. 2018, 8, 7556–7565. [Google Scholar] [CrossRef]
- Cho, Y.; Yamaguchi, A.; Uehara, R.; Yasuhara, S.; Hoshina, T.; Miyauchi, M. Temperature dependence on bandgap of semiconductor photocatalysts. J. Chem. Phys. 2020, 152, 231101. [Google Scholar] [CrossRef] [PubMed]



























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Light | CdS | Bi2S3 | TaON | Ta3N5 | Rh-Dope SrTiO3 | g-C3N4 | Bi2WO6 | MgFe2O4 | ZnFe2O4 | CaFe2O4 |
| OFF | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 1.6 | 0.1 | N.D. | N.D. |
| ON | 1.2 | 0.2 | N.D. | N.D. | N.D. | 0.2 | N.D. | N.D. | N.D. | N.D. |
| Light | Cu2O | Rh/Bi2WO6 | Ru/ZnFe2O4 | Ru/CaFe2O4 | Rh/Cu2O | Rh/g-C3N4 | Rh/TaON | Rh/Ta3N5 | Thermal Catalyst Limit | |
| OFF | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 15.6 | 22.8 | 36.3 | |
| ON | 0.6 | N.D. | 2.2 | 2.4 | N.D. | 0.1 | 50.3 | 33.6 | - | |
| Entry | Catalyst | Feed Gas a | H2 Production Rate b (μmol min−1) |
|---|---|---|---|
| 1 | TiO2 | H2O | n.d. |
| 2 | - | CH4 | n.d. |
| 3 | - | H2O+CH4 | 0.004 |
| 4 | PtM(0.1)/TiO2 | H2O | n.d. |
| 5 | - | CH4 | 0.063 c |
| 6 | - | H2O+CH4 | 0.76 d |
| Entry | Photocatalyst | SBET a (m2 g−1) | D b (nm) | Production Rate (μmol min−1) | |||
|---|---|---|---|---|---|---|---|
| Feed Gas | H2O | H2O + CH4 | |||||
| Product | H2 | H2 | CO2 | ||||
| 1 | NaTaO3:LaC(2%) c | 3.6 | 50 | - | 0.08 | 0.21 | - d |
| 2 | NaTaO3:LaBM(2%) c | 1.9 | 62 | - | 0.12 | 0.29 | - d |
| 3 | Pt(0.1)/NaTaO3:LaC(2%) c,e | - | - | - | 0.00 | 1.5 | 0.39 |
| 4 | Pt(0.05)/NaTaO3:LaBM(2%) c,e | - | - | - | - | 1.8 | 0.46 |
| 5 | NiO(0.2)/NaTaO3:LaC(2%) c | - | - | - | 0.16 | 0.18 | - f |
| 6 | Pt(0.1)/TiO2 c,g | 335 | - | - | 0.00 | 0.76 | 0.18 |
| 7 | NaTaO3BM c | 0.67 | 58 | - | - | 0.06 | - f |
| 8 | KTaO3BM c | 1.5 | 58 | - | - | 0.04 | - f |
| 9 | LiTaO3BM c | 0.48 | 57 | - | - | 0.02 | - f |
| 10 | Pt(0.01)KTaO3:LaBM(2%) c,h | 5.1 | 58 | - | - | 0.28 | - d |
| 11 | Pt(0.03)/NaTaO3:LaBM(2%) e,i | - | - | - | 0.00 | 4.5 | 1.2 |
| Temperature (°C) | Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|
| CO2 | CH4 | CH3COOH | CH3CH3 | CH3COCH3 | CO | |
| 30 | 0 | 0 | - | - | - | - |
| 80 | 0.07 | 0.11 | - | 46.7 | 0 | 53.3 |
| 120 | 0.74 | 1.47 | Trace | 3.1 | 92.3 | 4.6 |
| 150 | 0.79 | 1.54 | Trace | 7.5 | 87.6 | 4.9 |
| Catalyst | Yield Rate a (μmol g-cat−1 h−1) | |
|---|---|---|
| CO | CH4 | |
| TiO2 | 19 | 0.74 |
| 5% MMT/TiO2 | 26 | 1.01 |
| 10% MMT/TiO2 | 51 | 1.06 |
| 15% MMT/TiO2 | 42 | 1.06 |
| 1% Ni-10% MMT/TiO2 | 2236 | 3.69 |
| 3% Ni-10% MMT/TiO2 | 3813 | 5.58 |
| 5% Ni-10% MMT/TiO2 | 2958 | 4.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.; Yamaguchi, A.; Miyauchi, M. Photocatalytic Methane Reforming: Recent Advances. Catalysts 2021, 11, 18. https://doi.org/10.3390/catal11010018
Cho Y, Yamaguchi A, Miyauchi M. Photocatalytic Methane Reforming: Recent Advances. Catalysts. 2021; 11(1):18. https://doi.org/10.3390/catal11010018
Chicago/Turabian StyleCho, Yohei, Akira Yamaguchi, and Masahiro Miyauchi. 2021. "Photocatalytic Methane Reforming: Recent Advances" Catalysts 11, no. 1: 18. https://doi.org/10.3390/catal11010018
APA StyleCho, Y., Yamaguchi, A., & Miyauchi, M. (2021). Photocatalytic Methane Reforming: Recent Advances. Catalysts, 11(1), 18. https://doi.org/10.3390/catal11010018