Mechanistic Study of Silane Alcoholysis Reactions with Self-Assembled Monolayer-Functionalized Gold Nanoparticle Catalysts



Abstract
1. Introduction
2. Results and Discussion
2.1. Fabrication of Alkanethiolate SAM-Functionalized AuNP-Arrays
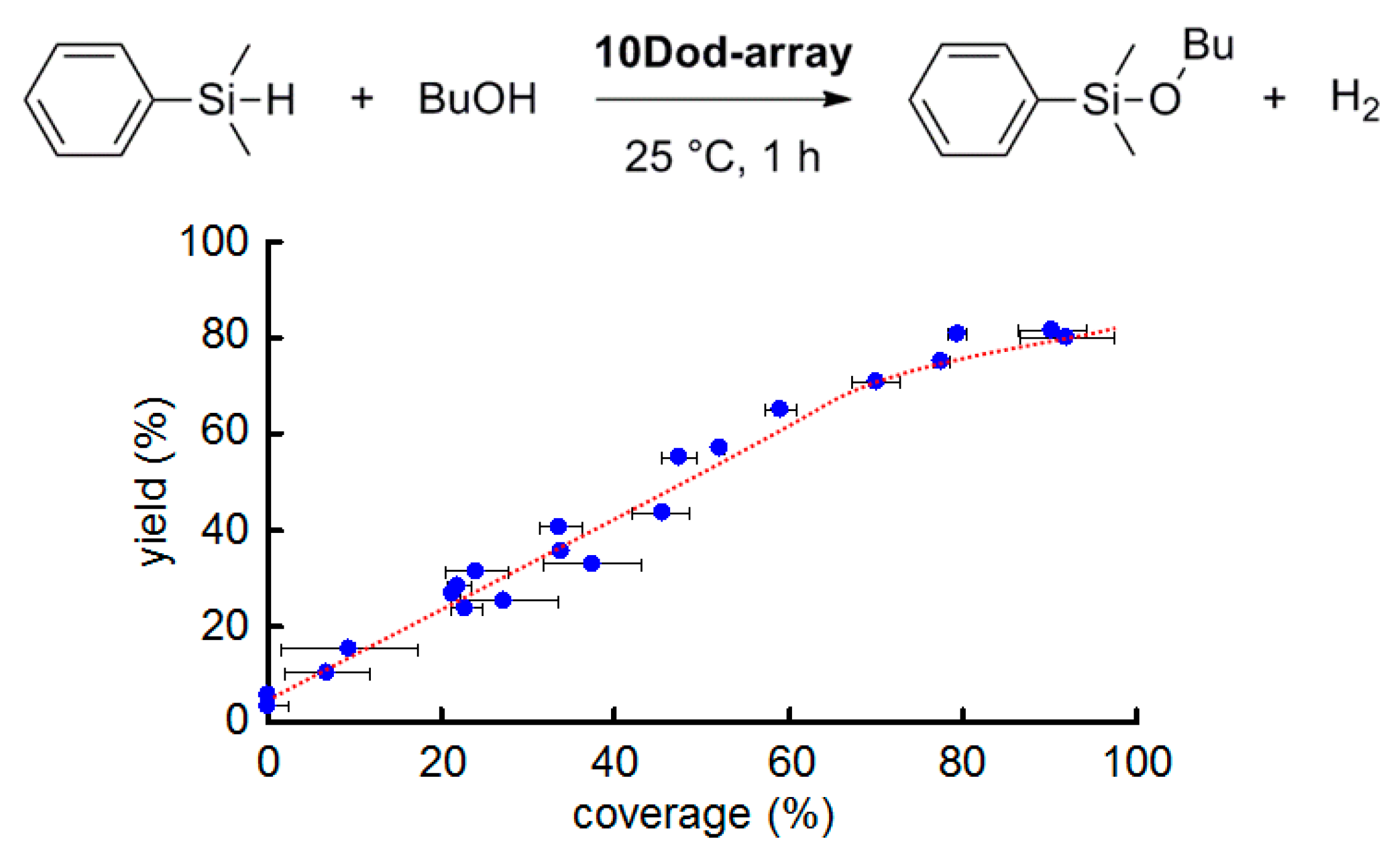
2.2. Effect of AuNP Coverage on the Support
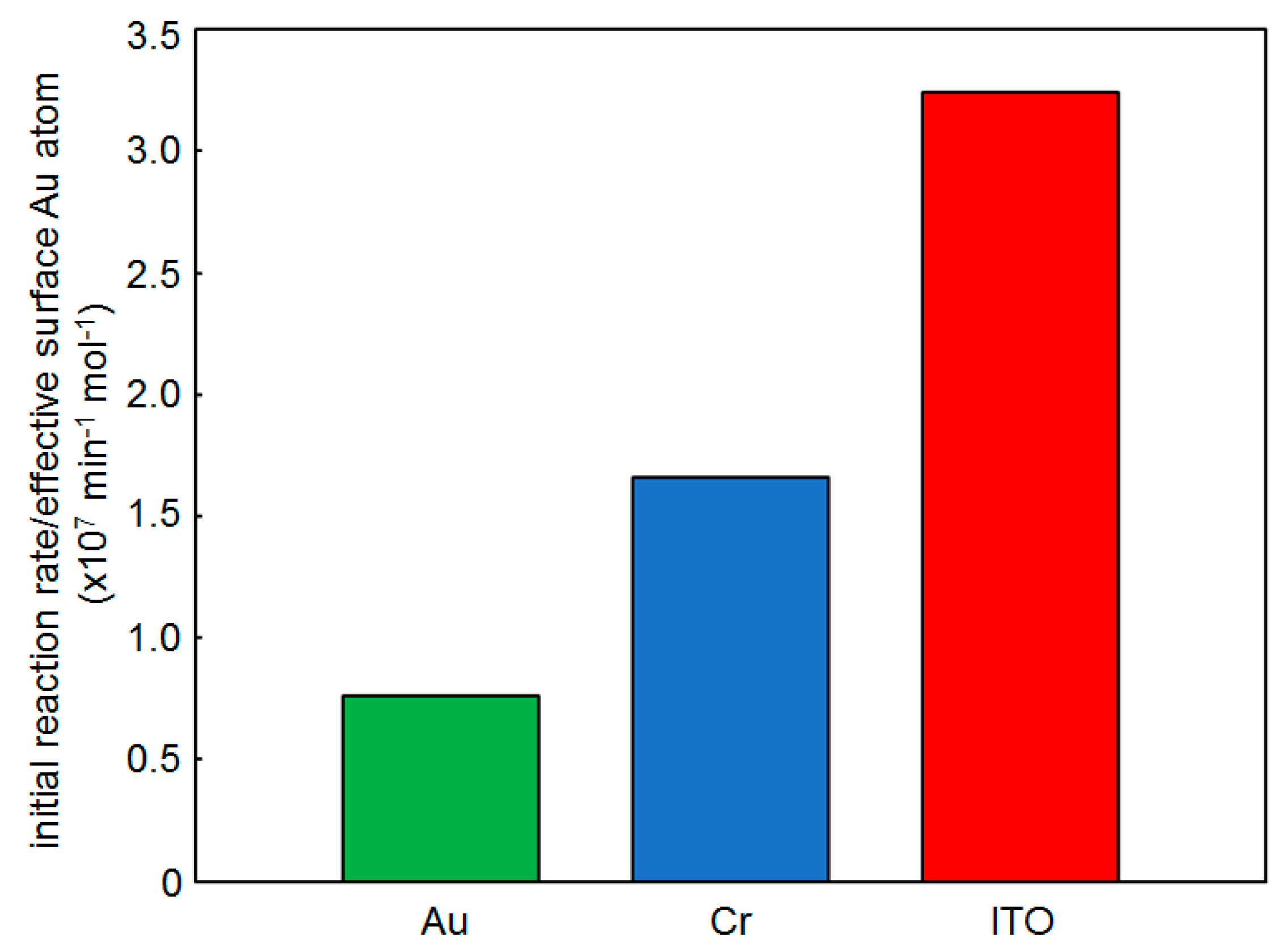
2.3. Effect of the Support
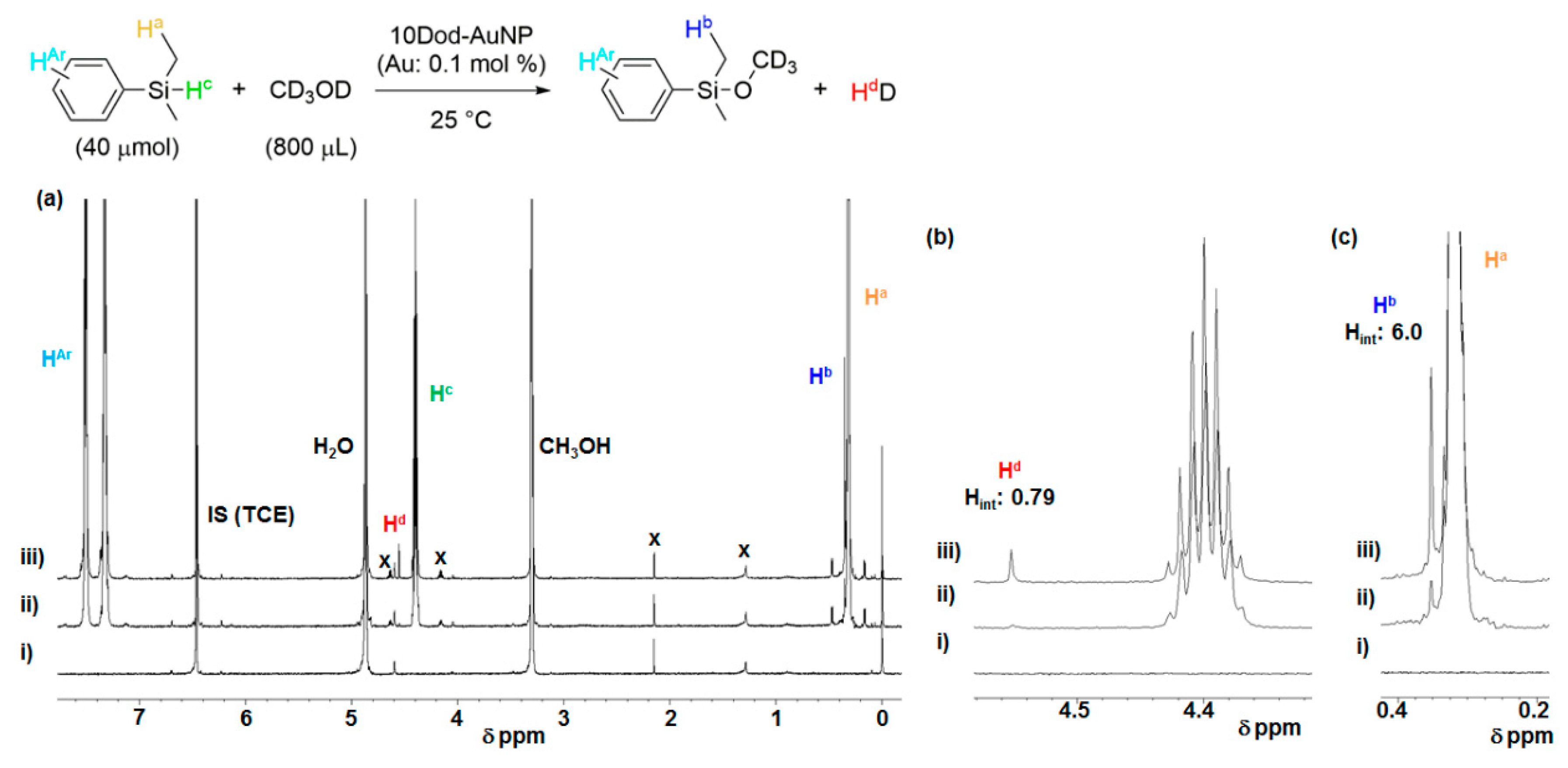
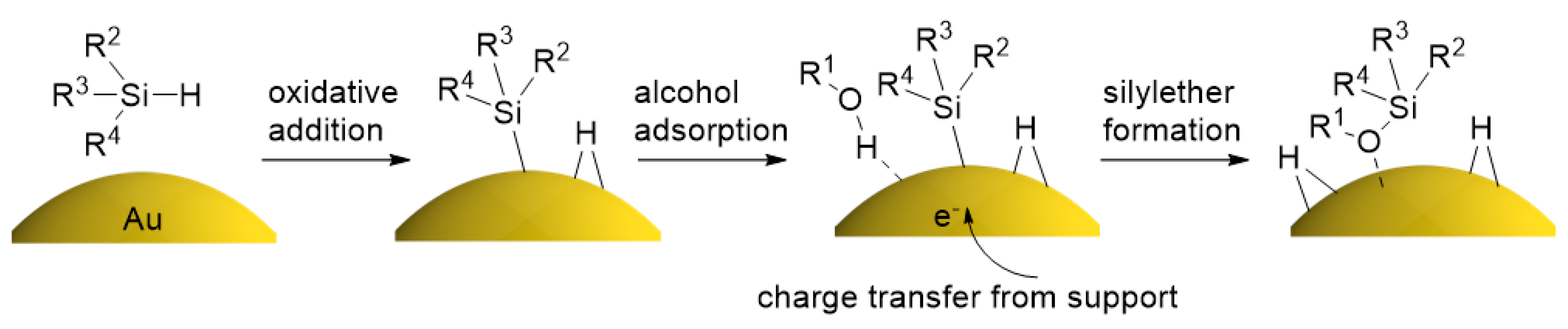
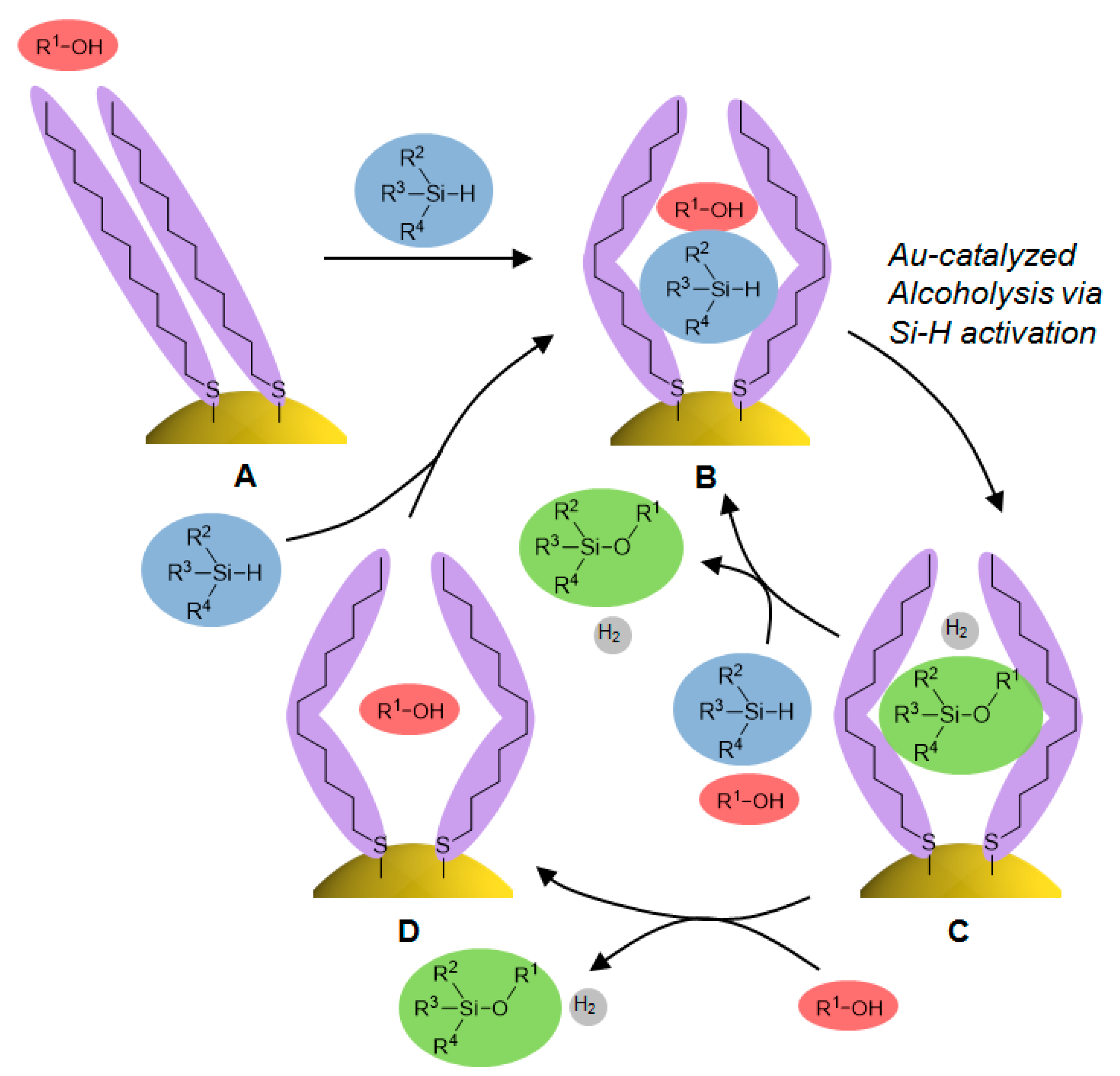
2.4. Investigation for Reactive Intermediates on the AuNP Surface


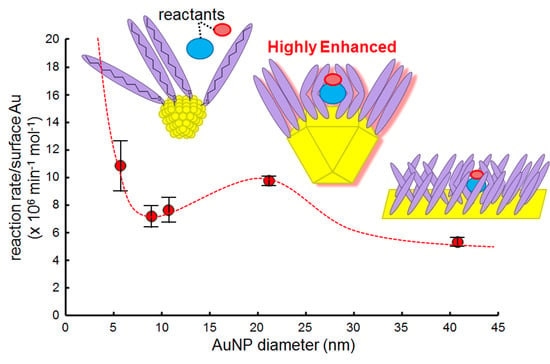
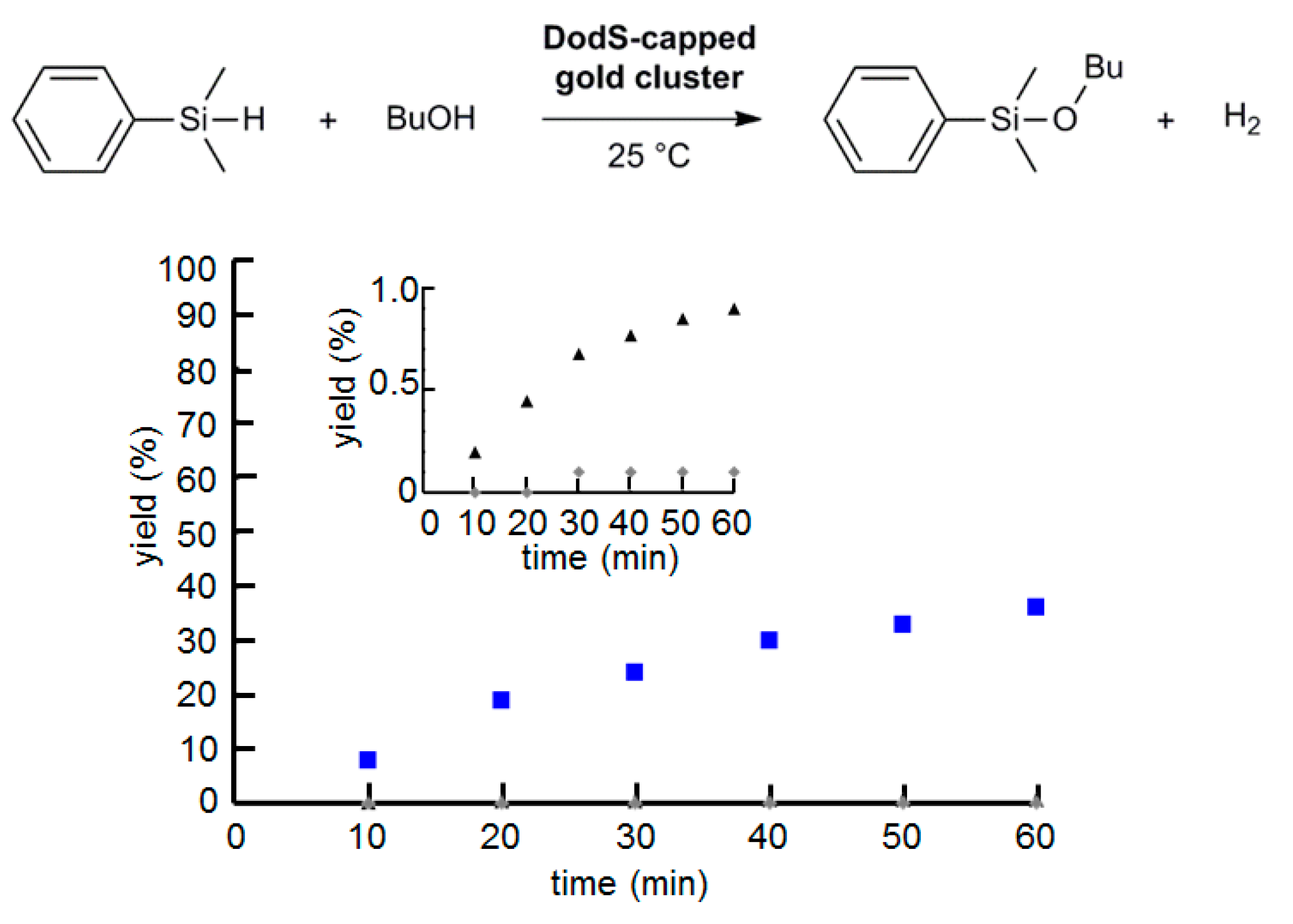
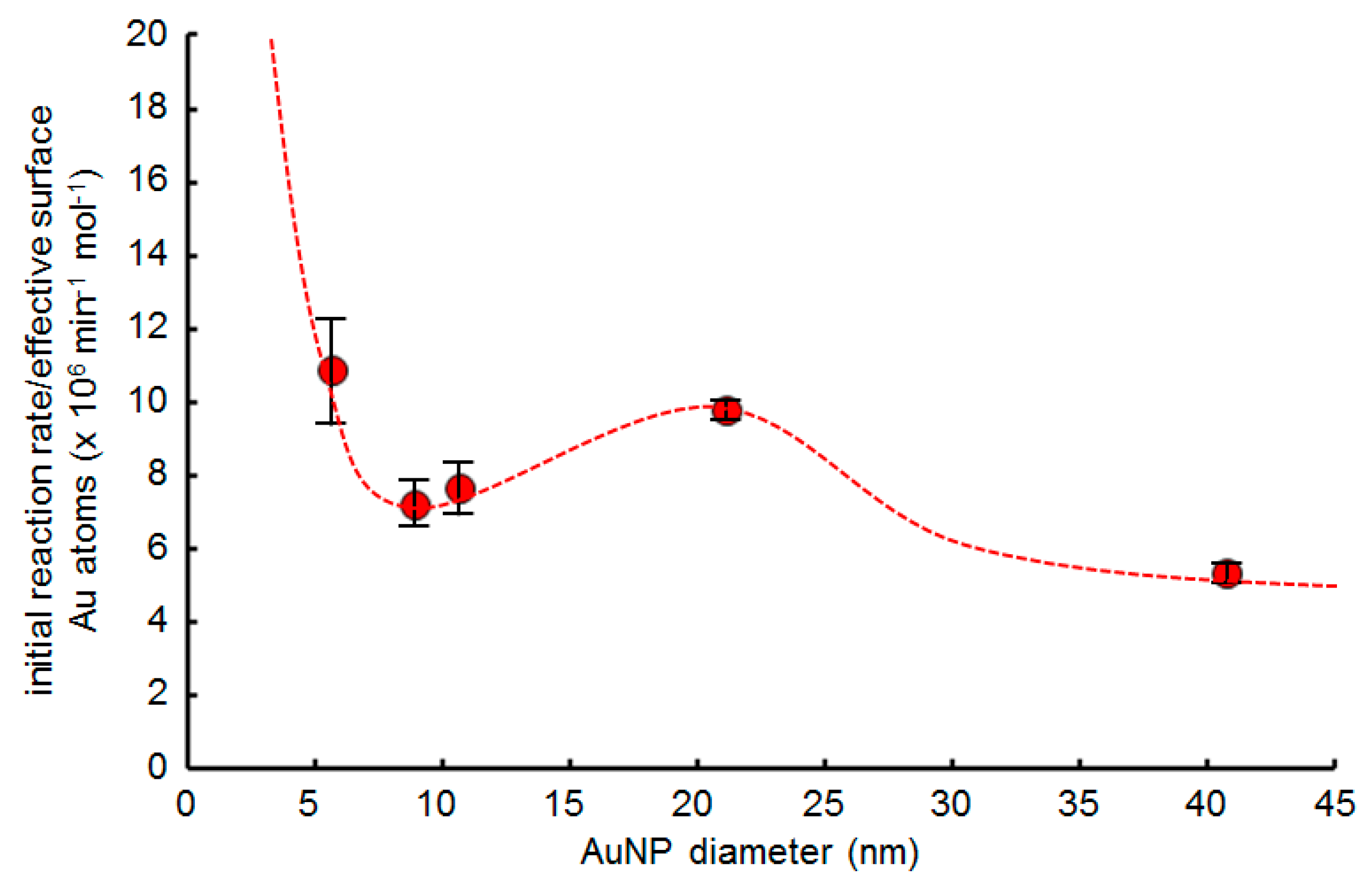
2.5. Effect of AuNP Size
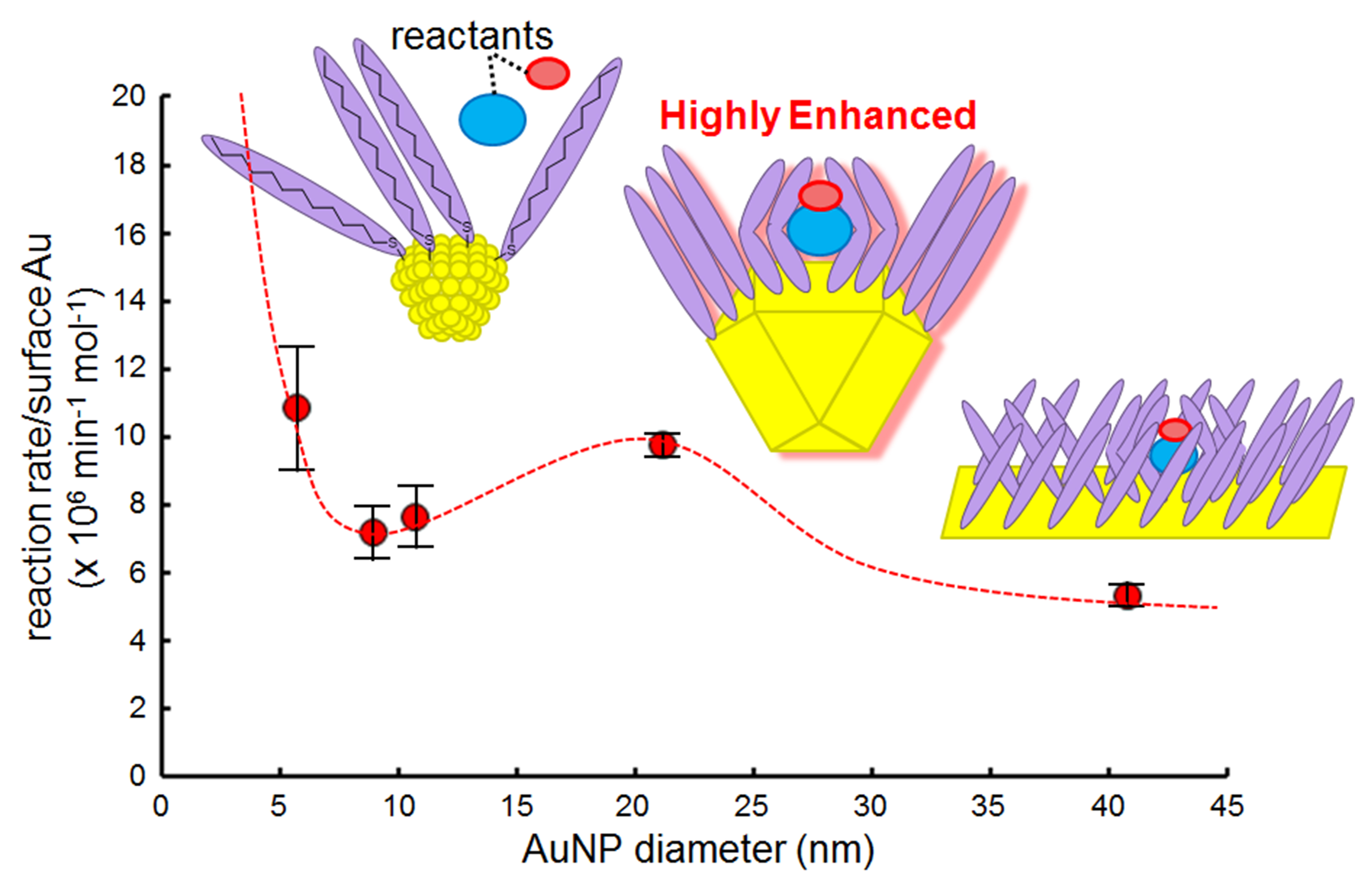
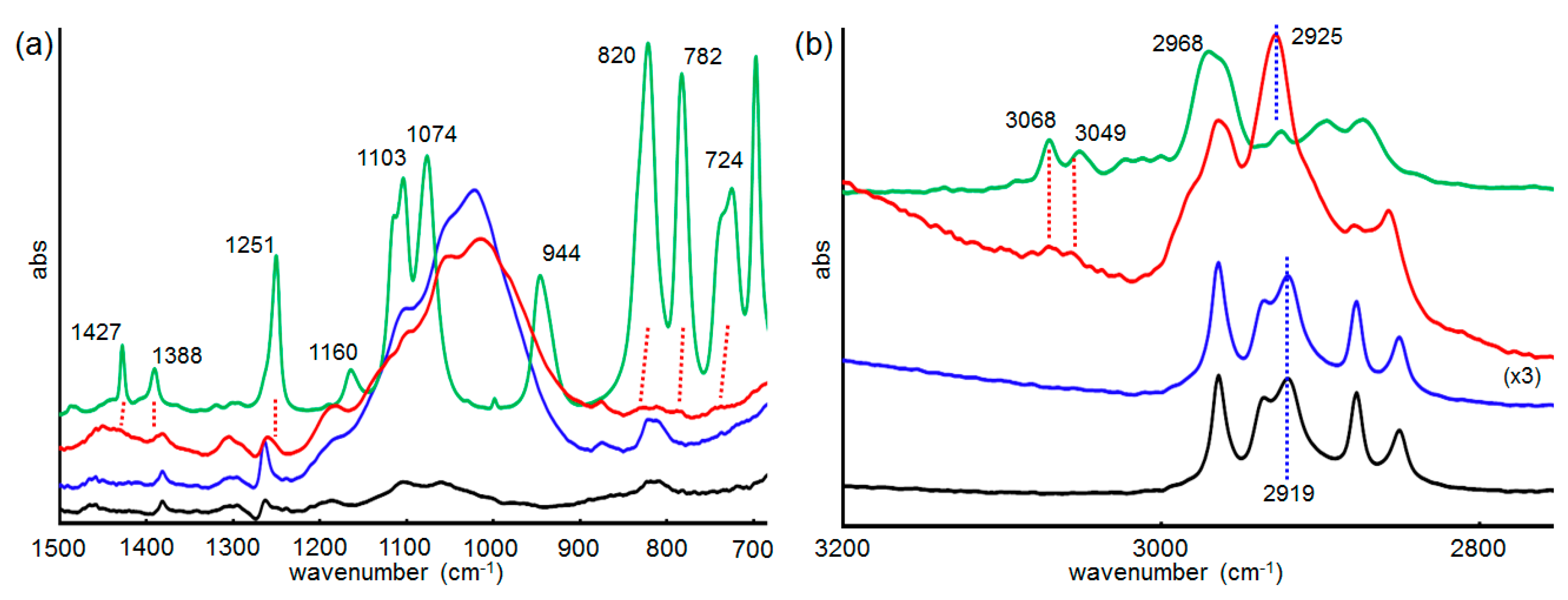
2.6. Infrared (IR) RA Analysis for the Conformational Change of SAM during the Reaction
3. Materials and Methods
3.1. General
3.2. Materials
3.3. Preparation of AuNP-Arrays
3.4. Typical Reaction Procedure for AuNP-Array Catalysts
3.5. Typical Reaction Procedure for Colloidal AuNP Catalysts
3.6. Preparation of Dodecanethiolate SAM-Functionalized Au Substrate (Dod-Au) for RA IR Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef]
- Cong, H.; Porco, J.A., Jr. Chemical synthesis of complex molecules using nanoparticle catalysis. ACS Catal. 2012, 2, 65–70. [Google Scholar] [CrossRef]
- Corma, A.; Garcia, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. [Google Scholar] [CrossRef]
- Yamazoe, S.; Koyasu, K.; Tsukuda, T. Nonscalable oxidation catalysis of gold clusters. Acc. Chem. Res. 2014, 47, 816–824. [Google Scholar] [CrossRef]
- Taketoshi, A.; Haruta, M. Size- and structure-specificity in catalysis by gold clusters. Chem. Lett. 2014, 43, 380–387. [Google Scholar] [CrossRef]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Chavda, N.; Trivedi, A.; Thakarda, J.; Agrawal, Y.K.; Maity, P. Size specific activity of polymer stabilized gold nanoparticles for transfer hydrogenation catalysis. Catal. Lett. 2016, 146, 1331–1339. [Google Scholar] [CrossRef]
- Yao, Q.; Wang, C.; Wang, H.; Yan, H.; Lu, J. Revisiting the au particle size effect on TiO2-Coated Au/TiO2 catalysts in CO oxidation reaction. J. Phys. Chem. C 2016, 120, 9174–9183. [Google Scholar] [CrossRef]
- Hartadi, Y.; Widmann, D.; Behm, R.J. CO2 hydrogenation to methanol on supported au catalysts under moderate reaction conditions: Support and particle size effects. Chem. Sus. Chem. 2015, 8, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Donoeva, B.G.; Ovoshchnikov, D.S.; Golovko, V.B. Establishing a Au nanoparticle size effect in the Oxidation of Cyclohexene using gradually changing Au catalysts. ACS Catal. 2013, 3, 2986–2991. [Google Scholar] [CrossRef]
- Hartfelder, U.; Kartusch, C.; Makosch, M.; Rovezzi, M.; Sá, J.; van Bokhoven, J.A. Particle size and support effects in hydrogenation over supported gold catalysts. Catal. Sci. Technol. 2013, 3, 454–461. [Google Scholar] [CrossRef]
- Ousmane, M.; Liotta, L.F.; Pantaleo, G.; Venezia, A.M.; Carlo, G.D.; Aouine, M.; Retailleau, L.; Giroir-Fendler, A. Supported Au catalysts for propene total oxidation: Study of support morphology and gold particle size effects. Catal. Today 2011, 176, 7–13. [Google Scholar] [CrossRef]
- Laoufi, I.; Saint-Lager, M.-C.; Lazzari, R.; Jupille, J.; Robach, O.; Garaudée, S.; Gabailh, G.; Dolle, P.; Cruguel, H.; Bailly, A. Size and catalytic activity of supported gold nanoparticles: An in pperando study during CO oxidation. J. Phys. Chem. C 2011, 115, 4673–4679. [Google Scholar] [CrossRef]
- Baiker, A. Crucial aspects in the design of chirally modified noble metal catalysts for asymmetric hydrogenation of activated ketones. Chem. Soc. Rev. 2015, 44, 7449–7464. [Google Scholar] [CrossRef] [PubMed]
- Zaera, F. Regio-, Stereo-, and enantioselectivity in hydrocarbon conversion on metal surfaces. Acc. Chem. Res. 2009, 42, 1152–1160. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Studer, M. Cincona-modified platinum catalysts: From ligand acceleration to technical processes. Acc. Chem. Res. 2007, 40, 1348–1356. [Google Scholar] [CrossRef]
- Bürgi, T.; Baiker, A. Heterogeneous enantioselective hydrogenation over cincona alkaloid modified platinum: Mechanistic insights into a complex reaction. Acc. Chem. Res. 2004, 37, 909–917. [Google Scholar] [CrossRef]
- Hutchings, G.J. New approaches to rate enhancement in heterogeneous catalysis. Chem. Commun. 1999, 301–306. [Google Scholar] [CrossRef]
- Liu, P.; Qin, R.; Fu, G.; Zheng, N. Surface coordination chemistry of metal nanomaterials. J. Am. Chem. Soc. 2017, 139, 2122–2131. [Google Scholar] [CrossRef]
- Schoenbaum, C.A.; Schwartz, D.K.; Medlin, J.W. Controlling the surface environment of heterogeneous catalysts using self-assembled monolayers. Acc. Chem. Res. 2014, 47, 1438–1445. [Google Scholar] [CrossRef]
- Kumar, G.; Lien, C.-H.; Janik, M.J.; Medlin, J.W. Catalytic site selction via control over noncovalent interactions in self-assembled monolayers. ACS Catal. 2016, 6, 5086–5094. [Google Scholar] [CrossRef]
- Kahsar, K.R.; Schwartz, D.K.; Medlin, J.W. Control of metal catalyst selectivity through specific noncovalent molecular interactions. J. Am. Chem. Soc. 2014, 136, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.H.; Shoenbaum, C.A.; Schwartz, D.K.; Medlin, J.W. Directing reaction pathways by catalyst active-site selection using self-assembled monolayers. Nat. Commun. 2013, 4, 3448. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.T.; O’Brien, M.; Oetter, B.; Corpuz, A.; Richards, R.M.; Schwartz, D.K.; Medlin, J.W. Controlled selectivity for palladium catalysts using self-assembled monolayers. Nat. Mater. 2010, 9, 853–858. [Google Scholar] [CrossRef]
- Kwon, S.G.; Krylova, G.; Sumer, A.; Schwartz, M.M.; Bunel, E.E.; Marshall, C.L.; Chattopadhyay, S.; Lee, B.; Jellinek, J.; Shevchenko, E.V. Capping ligands as selectivity switchers in hydrogenation reactions. Nano Lett. 2012, 12, 5382–5388. [Google Scholar] [CrossRef]
- Moreno, M.; Kissell, L.N.; Jasinski, J.B.; Zamborini, F.P. Selectivity and reactivity of alkylamine- and alkanethiolate-stabilized Pd and PdAg nanoparticles for hydrogenation and isomerization of allyl alcohol. ACS Catal. 2012, 2, 2602–2613. [Google Scholar] [CrossRef]
- Sadeghmoghaddam, E.; Gu, H.; Shon, Y.-S. Pd nanoparticle-catalyzed isomerization vs hydrogenation of allyl alcohol: Solvent-dependent regioselectivity. ACS Catal. 2012, 2, 1838–1845. [Google Scholar] [CrossRef]
- Wu, B.; Huang, H.; Yang, J.; Zheng, N.; Fu, G. Selective hydrogenation of α,β-unsaturated aldehydes catalyzed by amine-capped platinum-cobalt nanocrystals. Angew. Chem. Int. Ed. 2012, 51, 344–3443. [Google Scholar] [CrossRef]
- Taguchi, T.; Isozaki, K.; Miki, K. Enhanced catalytic activity of self-assembled-monolayer-capped gold nanoparticles. Adv. Mater. 2012, 24, 6462–6467. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Seeding growth for size control of 5–40 nm diameter gold nanoparticles. Langmuir 2001, 17, 6782–6786. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Ochiai, T.; Isozaki, K.; Nishiyama, S.; Miki, K. Enhancement of self-assembly of large (>10 nm) gold nanoparticles on an ITO substrate. Appl. Phys. Express 2014, 7, 065001. [Google Scholar] [CrossRef]
- Ochiai, T.; Isozaki, K.; Pincella, F.; Taguchi, T.; Nittoh, K.; Miki, K. Plasmon-resonant optics on an indium–tin-oxide film for exciting a two-pthoton photochromic reaction. Appl. Phys. Express 2013, 6, 102001. [Google Scholar] [CrossRef]
- Isozaki, K.; Ochiai, T.; Taguchi, T.; Nittoh, K.; Miki, K. Chemical coating of large-area Au nanoparticle two-dimensional arrays as plasmon-resonant optics. Appl. Phys. Lett. 2010, 97, 221101. [Google Scholar] [CrossRef]
- Complete exchange of surface ligands on AuNPs (citrate or ascorbate) with dodecanethiol was confirmed by ATR IR measurement (Figure S10).
- Michaelson, H.B. The work function of the elements and its periodicity. J. Appl. Phys. 1977, 48, 4729–4733. [Google Scholar] [CrossRef]
- Schlaf, R.; Murata, H.; Kafafi, Z.H. Work function measurements on indium tin oxide films. J. Electron Spectrosc. Relat. Phenom. 2001, 120, 149–154. [Google Scholar] [CrossRef]
- The yields of 1,6-hexanethiolate SAM-capped Au, Cr, and ITO substrates after 1 h reaction were 9, 6, and 4%, respectively. The lower yields with the support films clarify that the catalytic activity of supported AuNPs originates from AuNPs.
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Dirk, K.; Ulf, H.; Joop, A.P. Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 585–630. [Google Scholar]
- Mitsudome, T.; Urayama, T.; Maeno, Z.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Highly efficient dehydrogenative coupling of hydrosilanes with amines or amides using supported gold nanoparticles. Chem. Eur. J. 2015, 21, 3202–3205. [Google Scholar] [CrossRef]
- Mitsudome, T.; Yamamoto, Y.; Noujima, A.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Highly efficient etherification of silanes by using a gold nanoparticle catalyst: Remarkable effect of O2. Chem. Eur. J. 2013, 19, 14398–14402. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Datta, A. Role of carbon support for subnanometer gold-cluster-catalyzed disiloxane synthesis from hydrosilane and water. J. Phys. Chem. C 2017, 121, 20101–20112. [Google Scholar] [CrossRef]
- Li, M.-B.; Tian, S.-K.; Wu, Z.; Jin, R. Cu2+ induced formation of Au44(SC2H4Ph)32 and its high catalytic activity for the reduction of 4-nitrophenol at low temperature. Chem. Commun. 2015, 51, 4433–4436. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zeng, C.; Jin, R. Thermally robust Au99(SPh)42 nanoclusters for chemoselective hydrogenation of nitrobenzaldehyde derivatives in water. J. Am. Chem. Soc. 2014, 136, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jiang, D.-E.; Kumar, S.; Chen, Y.; Jin, R. Size dependence of atomically precise gold nanoclusters in chemoselective hydrogenation and active site structure. ACS Catal. 2014, 4, 2463–2469. [Google Scholar] [CrossRef]
- Liu, Y.; Tsunoyama, H.; Akita, T.; Xie, S.; Tsukuda, T. Aerobic oxidation of cyclohexane catalyzed by size-controlled Au clusters on hydroxyapatite: Size effect in the Sub-2 nm regime. ACS Catal. 2011, 1, 2–6. [Google Scholar] [CrossRef]
- Negishi, Y.; Nakazaki, T.; Malola, S.; Takano, S.; Niihori, Y.; Kurashige, W.; Yamazoe, S.; Tsukuda, T.; Häkkinen, H. A critical size for emergence of nonbulk electronic and geometric structures in dodecanethiolate-protected Au clusters. J. Am. Chem. Soc. 2015, 137, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Aikens, C.M.; Hollander, F.J.; Schatz, G.C.; Jin, R. Correlating the crystal structure of a thiol-protected Au25 cluster and optical properties. J. Am. Chem. Soc. 2008, 130, 5883–5885. [Google Scholar] [CrossRef]
- Heaven, M.W.; Dass, A.; White, P.S.; Holt, K.M.; Murray, R.W. Crystal structure of the gold nanoparticle [N(C8H17)4][Au25(SCH2CH2Ph)18]. J. Am. Chem. Soc. 2008, 130, 3754–3755. [Google Scholar] [CrossRef]
- Negishi, Y.; Chaki, N.K.; Shichibu, Y.; Whetten, R.L.; Tsukuda, T. Origin of Magic stability of thiolated gold clusters: A case study on Au25(SC6H13)18. J. Am. Chem. Soc. 2007, 129, 11322–11323. [Google Scholar] [CrossRef]
- Dolaminc, I.; Knoppe, S.; Dass, A.; Bürgi, T. First enantioseparation and circular dichroism spectra of Au38 clusters protected by achiral ligands. Nat. Commun. 2012, 3, 1802. [Google Scholar]
- Qian, H.; Zhu, M.; Anderson, U.N.; Jin, R. Facile, large-scale synthesis of dodecanethiol-stabilized Au38 clusters. J. Phys. Chem. A 2009, 113, 4281–4284. [Google Scholar] [CrossRef]
- Chaki, N.K.; Negishi, Y.; Tsunoyama, H.; Shichibu, Y.; Tsukuda, T. Ubiquitous 8 and 29 kDa Gold: Alkanethiolate cluster compounds: Mass-Spectrometric determination of molecular formulas and structural implications. J. Am. Chem. Soc. 2008, 130, 8608–8610. [Google Scholar] [CrossRef] [PubMed]
- Donkers, R.L.; Lee, D.; Murray, R.W. Synthesis and isolation of the molecule-like cluster Au38(PhCH2CH2S)24. Langmuir 2004, 20, 1945–1952. [Google Scholar] [CrossRef]
- Shichibu, Y.; Negishi, Y.; Tsukuda, T.; Teranishi, T. Large-Scale synthesis of thiolated Au25 clusters via ligand exchange reactions of phosphine-stabilized Au11 clusters. J. Am. Chem. Soc. 2005, 127, 13464–13465. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Takasugi, Y.; Sato, S.; Yao, H.; Kimura, K.; Tsukuda, T. Magic-numbered Aun clusters protected by glutathione monolayers (n = 18, 21, 25, 28, 32, 39): Isolation and spectroscopic characterization. J. Am. Chem. Soc. 2004, 126, 6518–6519. [Google Scholar] [CrossRef]
- The respective core size of [Au25SR18]− and Au38SR24 clusters is ca. 1.0 [47] and 1.1 nm [55] from TEM.
- Reaction yield: 6.7% (Au thin film) and 8.7% (Dod-Au thin film) [29]. The slightly increased yield by SAM functionalization clarifies the reaction acceleration effect of SAMs even on the flat Au surface.
- Ghorai, P.K.; Glotzer, S.C. Molecular dynamics simulation study of self-assembled monolayers of alkanethiol surfactants on spherical gold nanoparticles. J. Phys. Chem. C 2007, 111, 15857–15862. [Google Scholar] [CrossRef]
- Hasegawa, T. Quantitative Infrared Spectroscopy for Understanding of a Condensed Matter; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Hostetler, M.J.; Stokes, J.J.; Murray, R.W. Infrared spectroscopy of three-dimensional self-assembled monolayers: N-alkanethiolate monolayers on gold cluster compounds. Langmuir 1996, 12, 3604–3612. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Coverage (%) | Yield (%) b | Initial Reaction Rate (×10−4 min−1) | Effective Surface Au Atom (nmol) | Initial Reaction Rate/Effective Surface Au Atom (×106 min−1 mol−1) |
|---|---|---|---|---|---|---|
| 1 | 10Dod-array | 38 | 38 | 78.0 | 1.02 | 7.60 |
| 2 | 10Dod-array/Cr | 21 | 44 | 94.0 | 0.56 | 16.6 |
| 3 | 10Dod-array/ITO | 24 | 76 | 210 | 0.65 | 32.4 |
| 4 | 5Dod-array | 84 | 61 | 176 | 1.63 | 10.8 |
| 5 c | 9Dod-array | 90 | 83 | 147 | 2.07 | 7.15 |
| 6 | 20Dod-array | 47 | 50 | 152 | 1.56 | 9.72 |
| 7 | 40Dod-array | 74 | 58 | 145 | 2.75 | 5.27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isozaki, K.; Taguchi, T.; Ishibashi, K.; Shimoaka, T.; Kurashige, W.; Negishi, Y.; Hasegawa, T.; Nakamura, M.; Miki, K. Mechanistic Study of Silane Alcoholysis Reactions with Self-Assembled Monolayer-Functionalized Gold Nanoparticle Catalysts. Catalysts 2020, 10, 908. https://doi.org/10.3390/catal10080908
Isozaki K, Taguchi T, Ishibashi K, Shimoaka T, Kurashige W, Negishi Y, Hasegawa T, Nakamura M, Miki K. Mechanistic Study of Silane Alcoholysis Reactions with Self-Assembled Monolayer-Functionalized Gold Nanoparticle Catalysts. Catalysts. 2020; 10(8):908. https://doi.org/10.3390/catal10080908
Chicago/Turabian StyleIsozaki, Katsuhiro, Tomoya Taguchi, Kosuke Ishibashi, Takafumi Shimoaka, Wataru Kurashige, Yuichi Negishi, Takeshi Hasegawa, Masaharu Nakamura, and Kazushi Miki. 2020. "Mechanistic Study of Silane Alcoholysis Reactions with Self-Assembled Monolayer-Functionalized Gold Nanoparticle Catalysts" Catalysts 10, no. 8: 908. https://doi.org/10.3390/catal10080908
APA StyleIsozaki, K., Taguchi, T., Ishibashi, K., Shimoaka, T., Kurashige, W., Negishi, Y., Hasegawa, T., Nakamura, M., & Miki, K. (2020). Mechanistic Study of Silane Alcoholysis Reactions with Self-Assembled Monolayer-Functionalized Gold Nanoparticle Catalysts. Catalysts, 10(8), 908. https://doi.org/10.3390/catal10080908