Application of Enzymes in Regioselective and Stereoselective Organic Reactions


 ,
,  ,
, 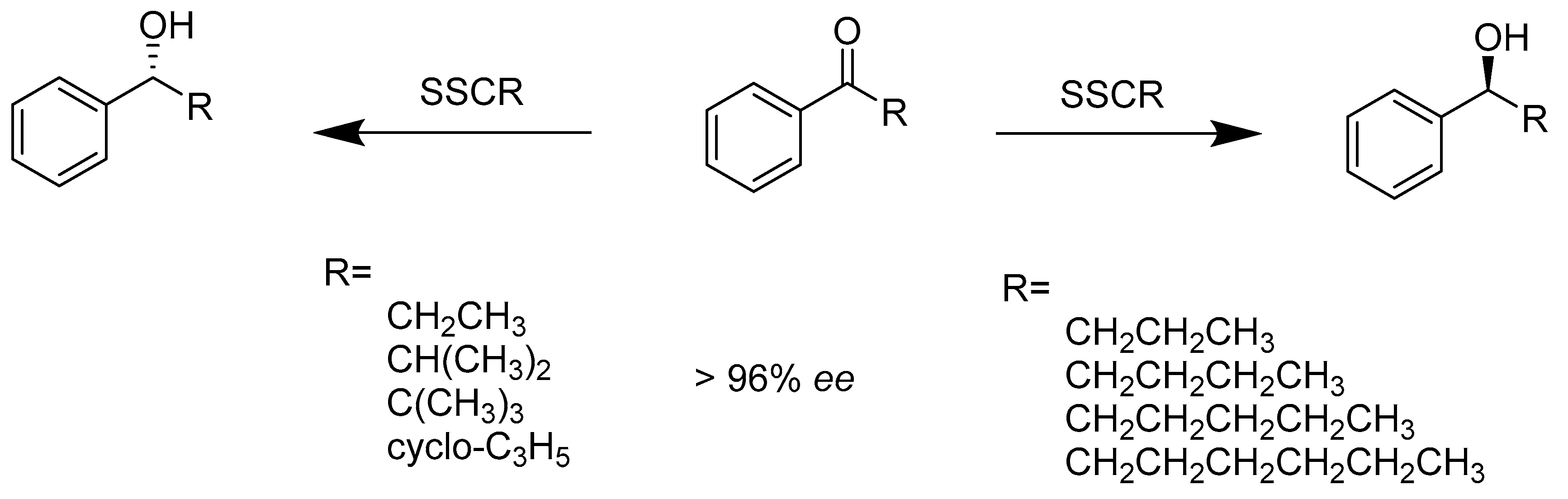{kind=link}
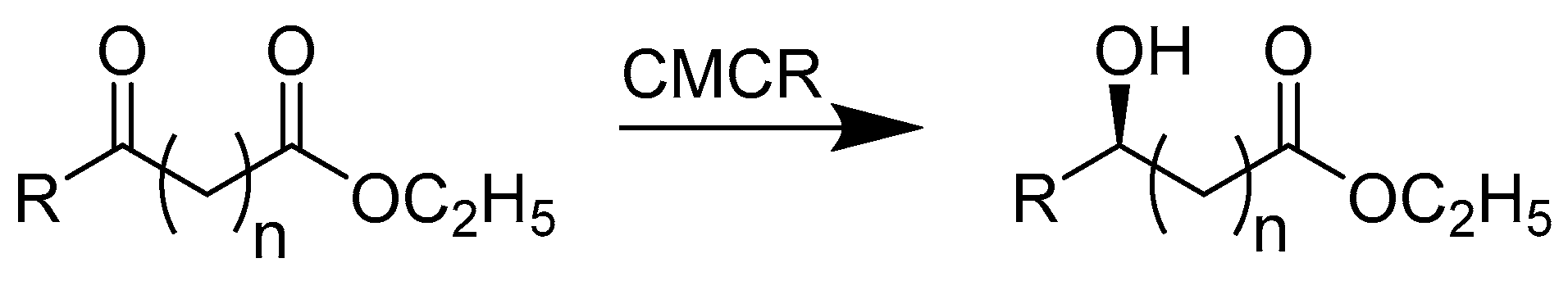{kind=link}
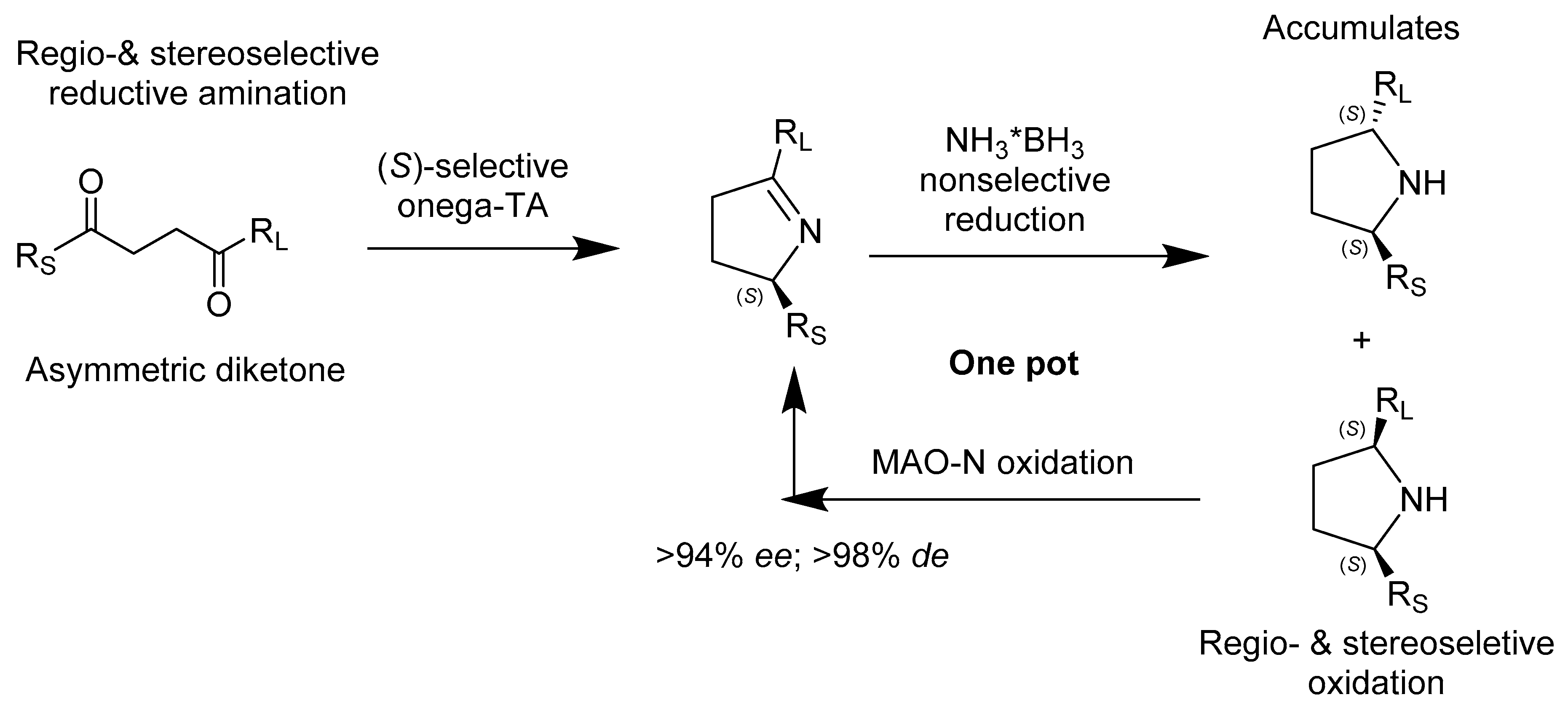{kind=link}
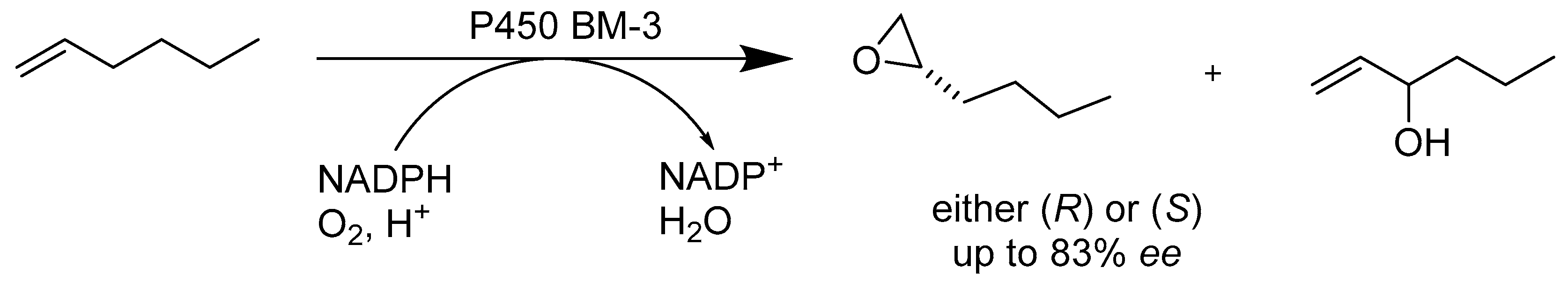{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
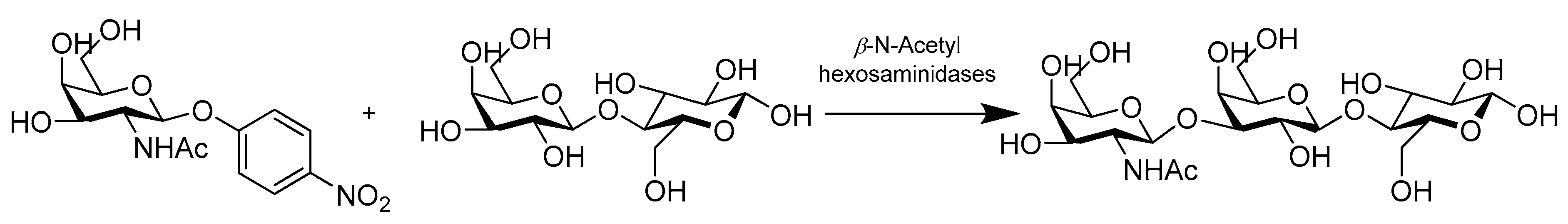{kind=link}
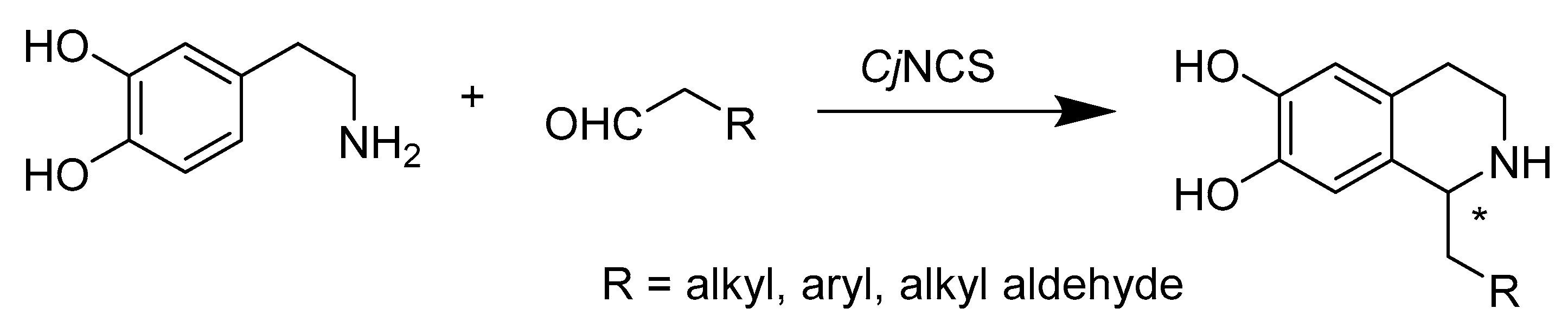{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
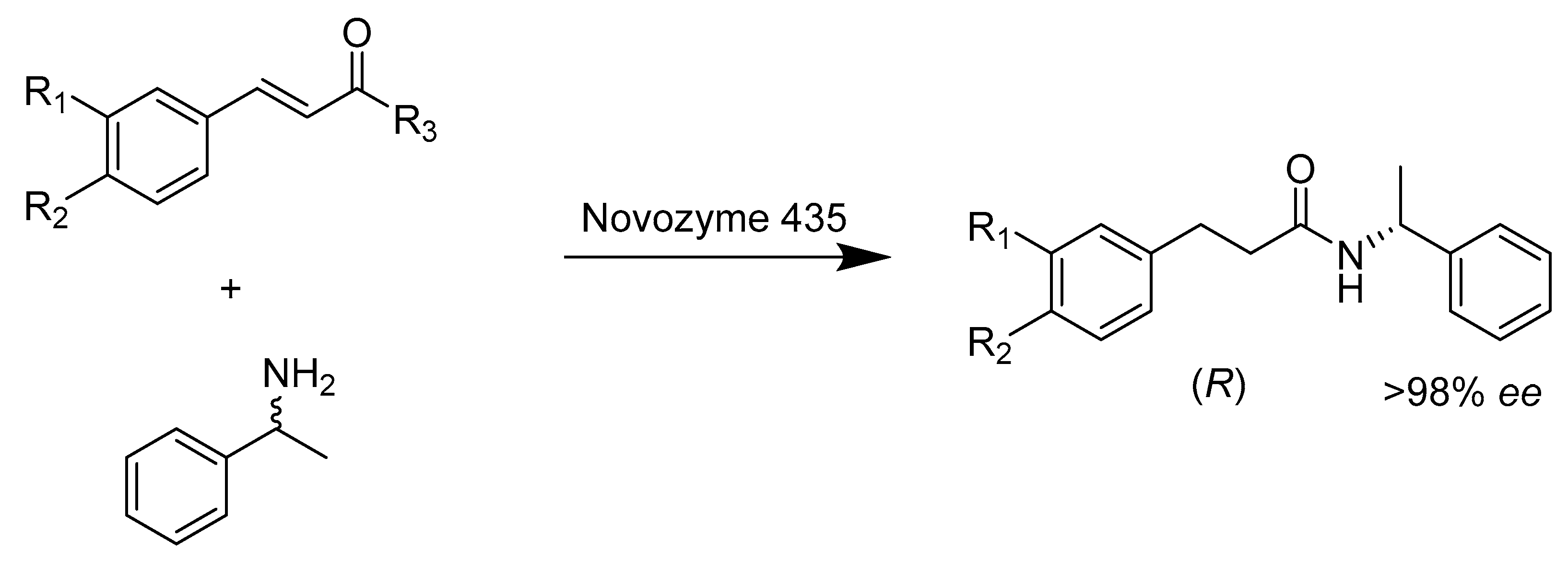{kind=link}
{kind=link}
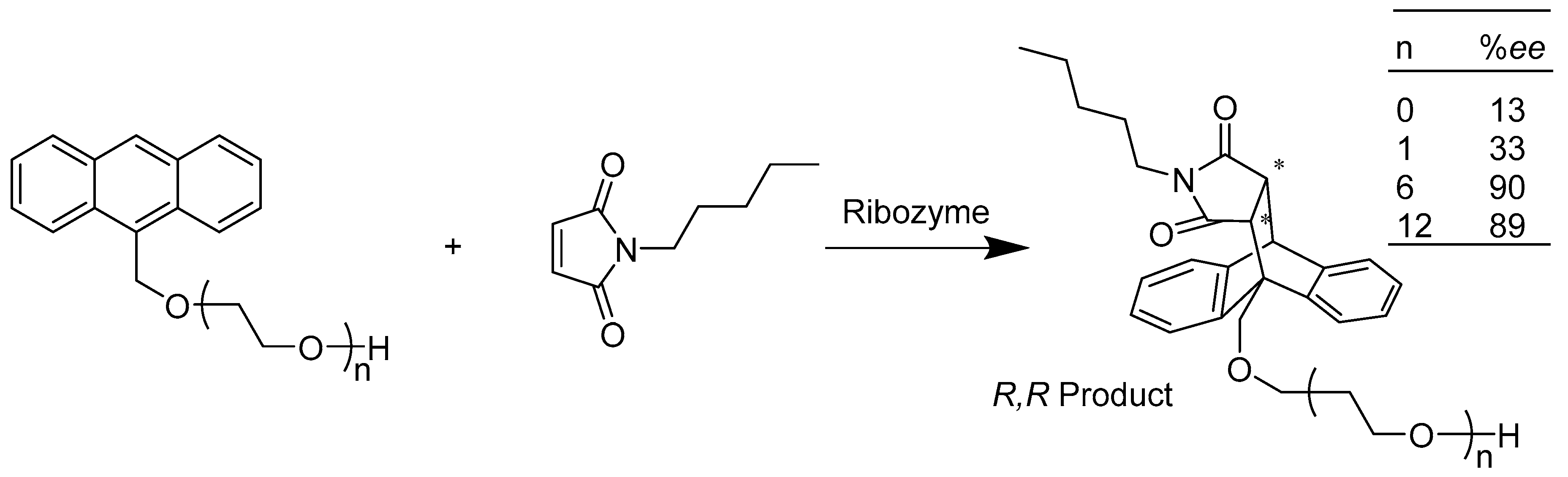{kind=link}
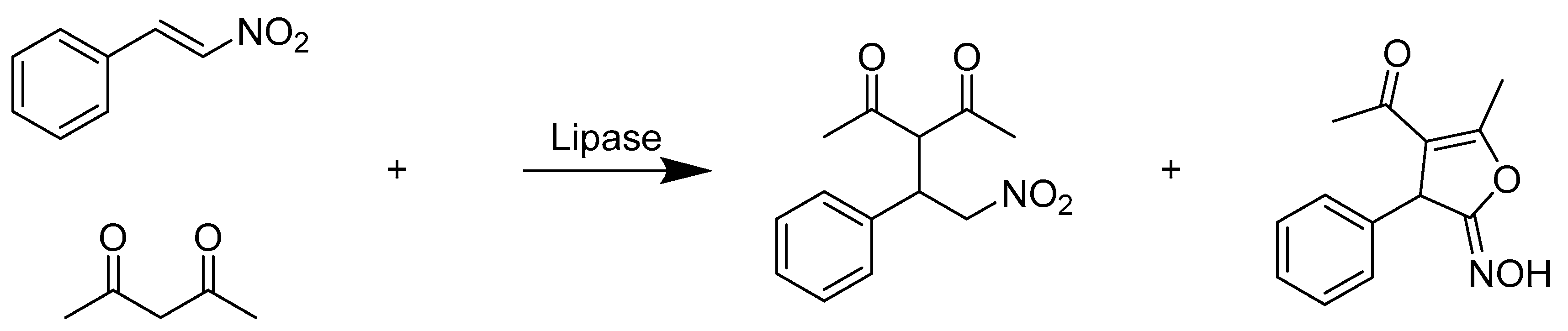{kind=link}
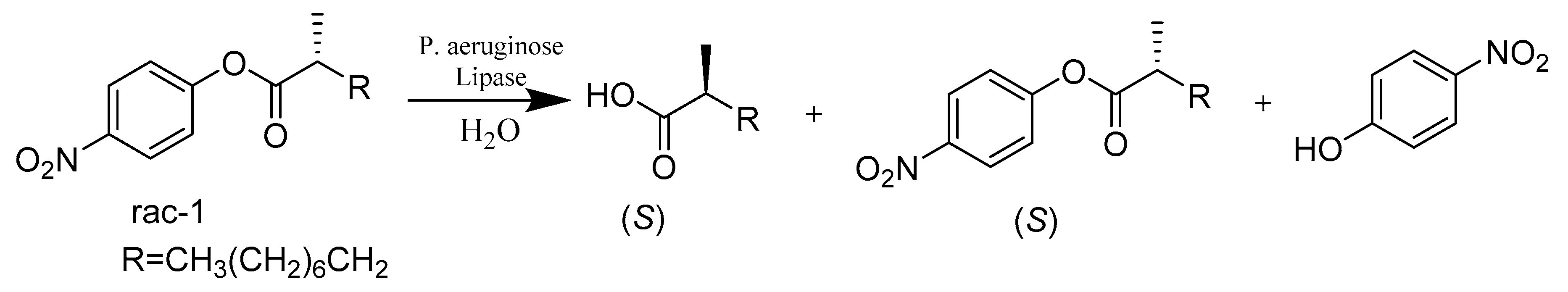{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. A Brief History of Enzymes and Their Application
1.2. A Brief Introduction to Regioselective and Stereoselective Reactions
2. Examples of Enzyme Application in Regioselective and Stereoselective Reactions
2.1. Oxidoreductases E.C.1
2.1.1. Enzymes Applied for Reduction
2.1.2. Enzymes Applied for Oxidation
2.2. Transferases E.C.2
2.2.1. Enzymes Applied for Alkylation
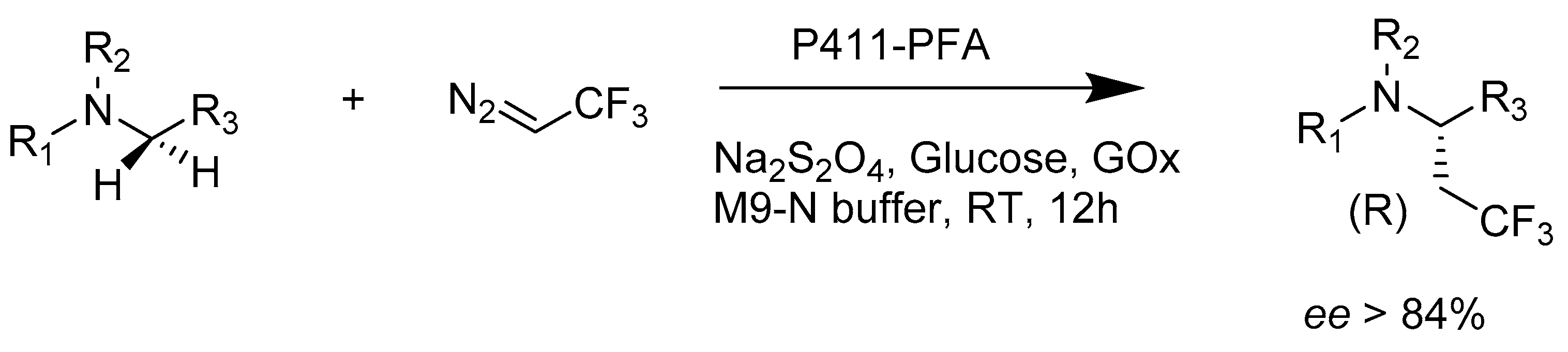
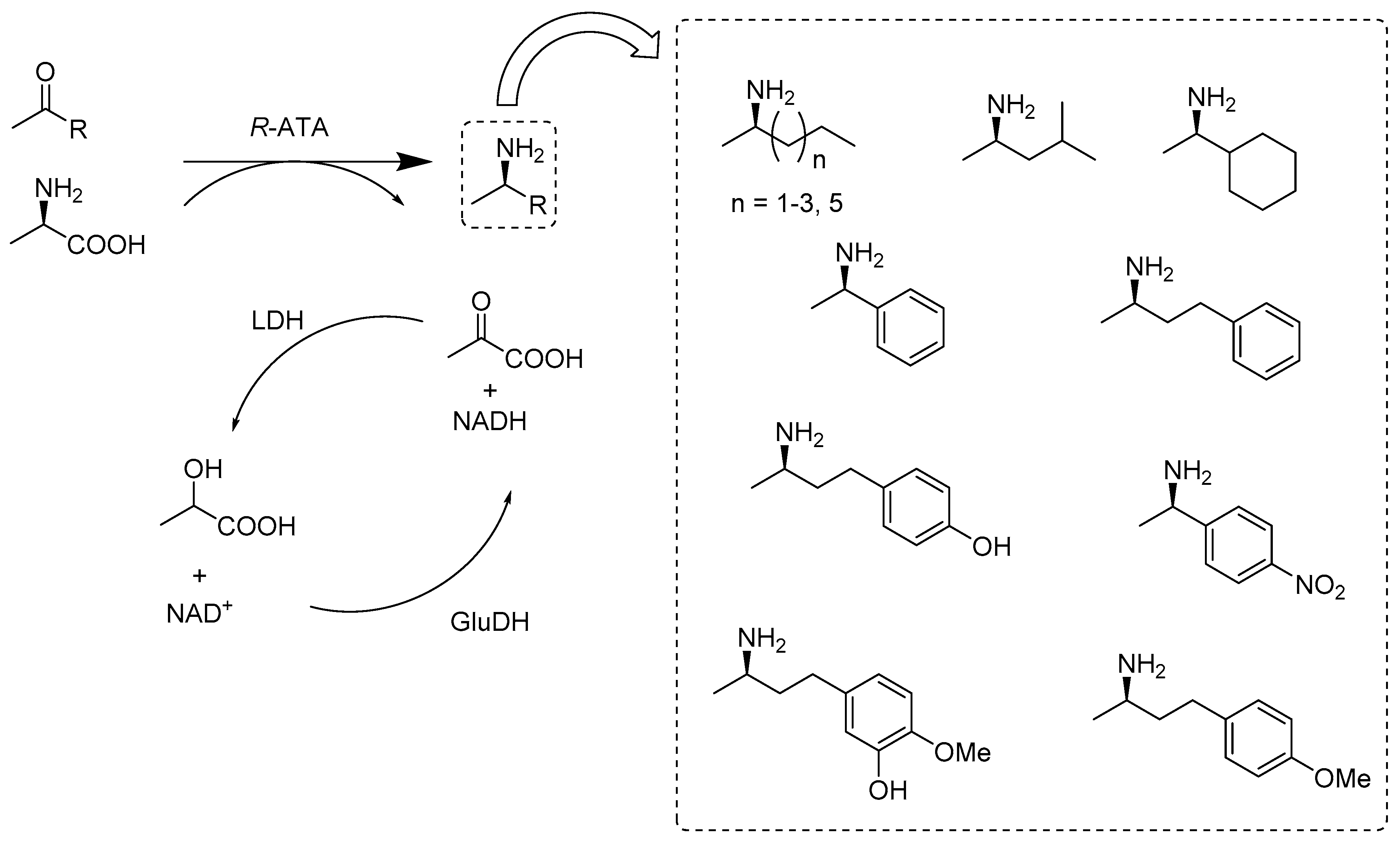
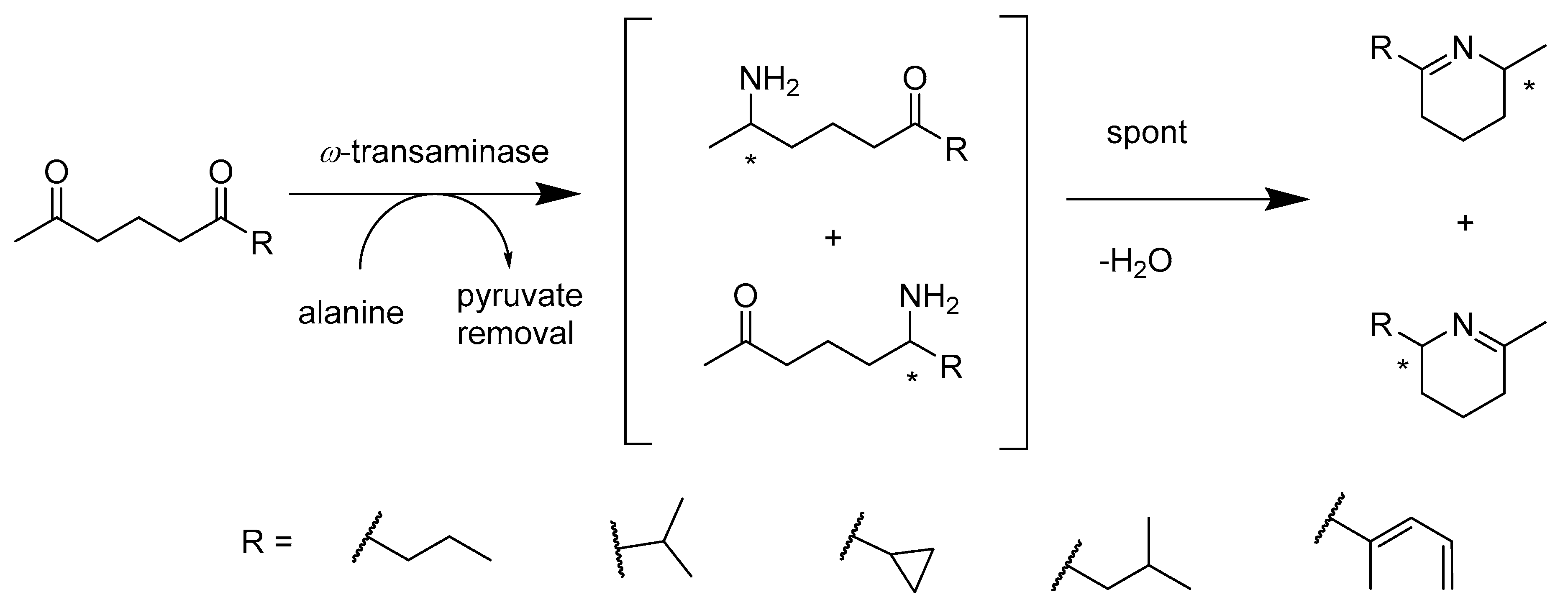
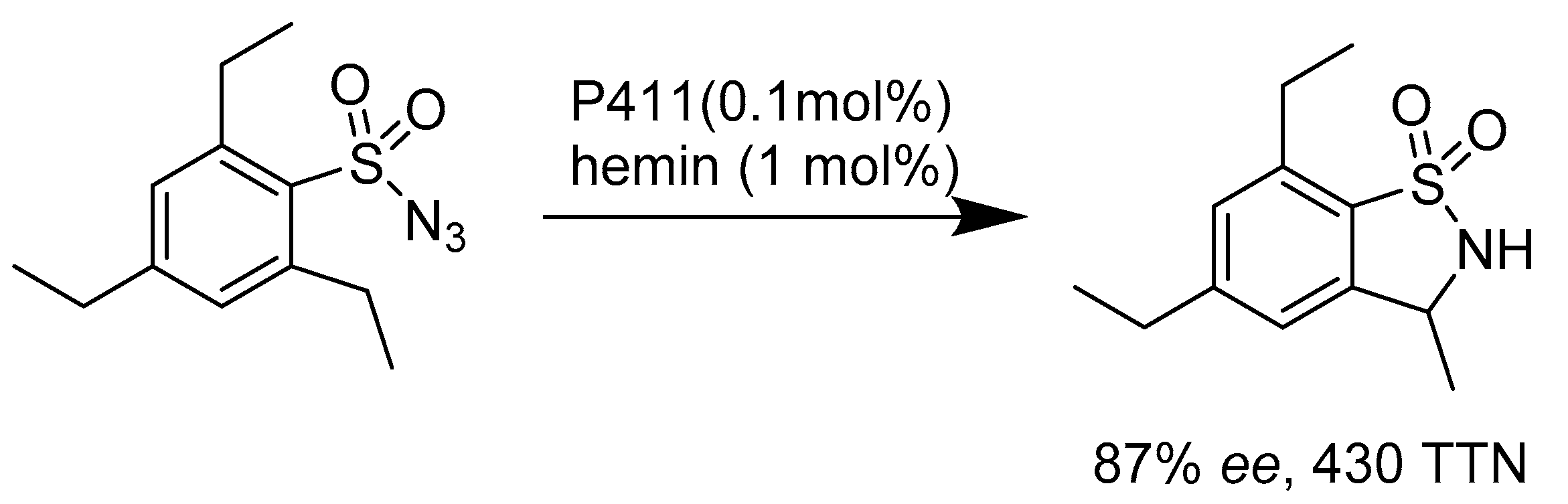
2.2.2. Enzymes Applied for Amination
2.2.3. Enzymes Applied for Glycosylation
2.2.4. Enzymes Applied for Halogenation
2.3. Hydrolases E.C.3
2.4. Lyases E.C.4
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Punekar, N.S. Enzymes: Catalysis, Kinetics and Mechanisms; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Rao, D.B.; Sane, P.G.; Georgiev, E.L. Collagenase in the treatment of dermal and decubitus ulcers. J. Am. Geriatr. Soc. 1975, 23, 22–30. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Wei, Y. Application of Fluorescence in Studying Therapeutic Enzymes. Adv. Exp. Med. Biol. 2019, 1148, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Hollmann, F.; Park, J.B.; Bühler, B. The use of enzymes in the chemical industry in Europe. Curr. Opin. Biotechnol. 2002, 13, 359–366. [Google Scholar] [CrossRef]
- Sunar, K.; Kumar, U.; Deshmukh, S.K. Recent Applications of Enzymes in Personal Care Products. In Agro-Industrial Wastes as Feedstock for Enzyme Production; Elsevier: Philadelphia, PA, USA, 2016; pp. 279–298. [Google Scholar] [CrossRef]
- Singh, R.; Kumar, M.; Mittal, A.; Mehta, P.K. Microbial enzymes: Industrial progress in 21st century. 3 Biotech 2016, 6, 174. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Wombacher, R.; Keiper, S.; Suhm, S.; Serganov, A.; Patel, D.J.; Jaschke, A. Control of stereoselectivity in an enzymatic reaction by backdoor access. Angew. Chem. Int. Ed. Engl. 2006, 45, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; McCallum, N.C.; Ni, Q.Z.; Li, W.; Boyce, H.; Mao, H.; Zhou, X.; Sun, H.; Thompson, M.P.; Battistella, C.; et al. Selenomelanin: An Abiotic Selenium Analogue of Pheomelanin. J. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef]
- Taofiq, O.; González-Paramás, A.M.; Martins, A.; Barreiro, M.F.; Ferreira, I.C.F.R. Mushrooms extracts and compounds in cosmetics, cosmeceuticals and nutricosmetics—A review. Ind. Crop. Prod. 2016, 90, 38–48. [Google Scholar] [CrossRef]
- Weinshilboum, R.; Axelrod, J. Serum dopamine-beta-hydroxylase activity. Circ. Res. 1971, 28, 307–315. [Google Scholar] [CrossRef]
- Shen, Z.; Lv, C.; Zeng, S. Significance and challenges of stereoselectivity assessing methods in drug metabolism. J. Pharm. Anal. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. C Embryo Today 2015, 105, 140–156. [Google Scholar] [CrossRef]
- Miller, M.T. Thalidomide embryopathy: A model for the study of congenital incomitant horizontal strabismus. Trans. Am. Ophthalmol. Soc. 1991, 89, 623–674. [Google Scholar] [PubMed]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Hua, L. Enantioselective enzymatic reductions of sterically bulky aryl alkyl ketones catalyzed by a NADPH-dependent carbonyl reductase. J. Org. Chem. 2006, 71, 9484–9486. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, P.; Sinisterra, J.V.; Molinari, F.; Alcantara, A.R.; de Maria, P.D. Biocatalytic strategies for the asymmetric synthesis of alpha-hydroxy ketones. Acc. Chem. Res. 2010, 43, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Kaluzna, I.A.; Feske, B.D.; Wittayanan, W.; Ghiviriga, I.; Stewart, J.D. Stereoselective, biocatalytic reductions of alpha-chloro-beta-keto esters. J. Org. Chem. 2005, 70, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Yang, Y.; Hua, L. Stereoselective enzymatic synthesis of chiral alcohols with the use of a carbonyl reductase from Candida magnoliae with anti-Prelog enantioselectivity. J. Org. Chem. 2006, 71, 4202–4205. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lee, T.S.; Khosla, C. Engineered biosynthesis of regioselectively modified aromatic polyketides using bimodular polyketide synthases. PLoS Biol. 2004, 2, e31. [Google Scholar] [CrossRef]
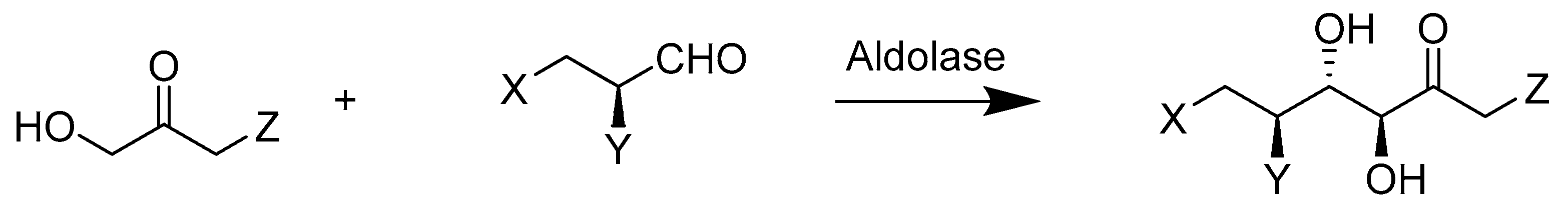
- Classen, T.; Korpak, M.; Schölzel, M.; Pietruszka, J. Stereoselective Enzyme Cascades: An Efficient Synthesis of Chiral γ-Butyrolactones. ACS Catal. 2014, 4, 1321–1331. [Google Scholar] [CrossRef]
- Oroz-Guinea, I.; Garcia-Junceda, E. Enzyme catalysed tandem reactions. Curr. Opin. Chem. Biol. 2013, 17, 236–249. [Google Scholar] [CrossRef]
- Groger, H.; Hummel, W.; Buchholz, S.; Drauz, K.; Nguyen, T.V.; Rollmann, C.; Husken, H.; Abokitse, K. Practical asymmetric enzymatic reduction through discovery of a dehydrogenase-compatible biphasic reaction media. Org. Lett. 2003, 5, 173–176. [Google Scholar] [CrossRef] [PubMed]
- De Gonzalo, G.; Lavandera, I.; Faber, K.; Kroutil, W. Enzymatic reduction of ketones in "micro-aqueous" media catalyzed by ADH-A from Rhodococcus ruber. Org. Lett. 2007, 9, 2163–2166. [Google Scholar] [CrossRef]
- Boratynski, F.; Smuga, M.; Wawrzenczyk, C. Lactones 42. Stereoselective enzymatic/microbial synthesis of optically active isomers of whisky lactone. Food Chem. 2013, 141, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Kulig, J.; Simon, R.C.; Rose, C.A.; Husain, S.M.; Häckh, M.; Lüdeke, S.; Zeitler, K.; Kroutil, W.; Pohl, M.; Rother, D. Stereoselective synthesis of bulky 1,2-diols with alcohol dehydrogenases. Catal. Sci. Technol. 2012, 2, 1580–1589. [Google Scholar] [CrossRef]
- O’Reilly, E.; Iglesias, C.; Ghislieri, D.; Hopwood, J.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. A regio- and stereoselective omega-transaminase/monoamine oxidase cascade for the synthesis of chiral 2,5-disubstituted pyrrolidines. Angew. Chem. Int. Ed. Engl. 2014, 53, 2447–2450. [Google Scholar] [CrossRef]
- Willistein, M.; Haas, J.; Fuchs, J.; Estelmann, S.; Ferlaino, S.; Muller, M.; Ludeke, S.; Boll, M. Enantioselective Enzymatic Naphthoyl Ring Reduction. Chemistry 2018, 24, 12505–12508. [Google Scholar] [CrossRef] [PubMed]
- Oura, T.; Kajiwara, S. Substrate specificity and regioselectivity of delta12 and omega3 fatty acid desaturases from Saccharomyces kluyveri. Biosci. Biotechnol. Biochem. 2008, 72, 3174–3179. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.W.; Meinhold, P.; Glieder, A.; Arnold, F.H. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc. 2003, 125, 13442–13450. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Nguyen, D.; Gonzalez, M.; Cortez, A.; Mullen, S.E.; Cheruzel, L.E. Regio- and stereoselective hydroxylation of 10-undecenoic acid with a light-driven P450 BM3 biocatalyst yielding a valuable synthon for natural product synthesis. Bioorg. Med. Chem. 2014, 22, 5687–5691. [Google Scholar] [CrossRef]
- Hu, S.; Huang, J.; Mei, L.; Yu, Q.; Yao, S.; Jin, Z. Altering the regioselectivity of cytochrome P450 BM-3 by saturation mutagenesis for the biosynthesis of indirubin. J. Mol. Catal. B Enzym. 2010, 67, 29–35. [Google Scholar] [CrossRef]
- Landwehr, M.; Hochrein, L.; Otey, C.R.; Kasrayan, A.; Backvall, J.E.; Arnold, F.H. Enantioselective alpha-hydroxylation of 2-arylacetic acid derivatives and buspirone catalyzed by engineered cytochrome P450 BM-3. J. Am. Chem. Soc. 2006, 128, 6058–6059. [Google Scholar] [CrossRef] [PubMed]
- Negretti, S.; Narayan, A.R.; Chiou, K.C.; Kells, P.M.; Stachowski, J.L.; Hansen, D.A.; Podust, L.M.; Montgomery, J.; Sherman, D.H. Directing group-controlled regioselectivity in an enzymatic C-H bond oxygenation. J. Am. Chem. Soc. 2014, 136, 4901–4904. [Google Scholar] [CrossRef]
- Kubo, T.; Peters, M.W.; Meinhold, P.; Arnold, F.H. Enantioselective epoxidation of terminal alkenes to (R)- and (S)-epoxides by engineered cytochromes P450 BM-3. Chemistry 2006, 12, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Bartlett, M.J.; Wang, Y.; Lu, L.; Hartwig, J.F.; Zhao, H. Development of a One-Pot Tandem Reaction Combining Ruthenium-Catalyzed Alkene Metathesis and Enantioselective Enzymatic Oxidation To Produce Aryl Epoxides. ACS Catal. 2015, 5, 3817–3822. [Google Scholar] [CrossRef]
- Loskot, S.A.; Romney, D.K.; Arnold, F.H.; Stoltz, B.M. Enantioselective Total Synthesis of Nigelladine A via Late-Stage C-H Oxidation Enabled by an Engineered P450 Enzyme. J. Am. Chem. Soc. 2017, 139, 10196–10199. [Google Scholar] [CrossRef]
- Farwell, C.C.; McIntosh, J.A.; Hyster, T.K.; Wang, Z.J.; Arnold, F.H. Enantioselective imidation of sulfides via enzyme-catalyzed intermolecular nitrogen-atom transfer. J. Am. Chem. Soc. 2014, 136, 8766–8771. [Google Scholar] [CrossRef]
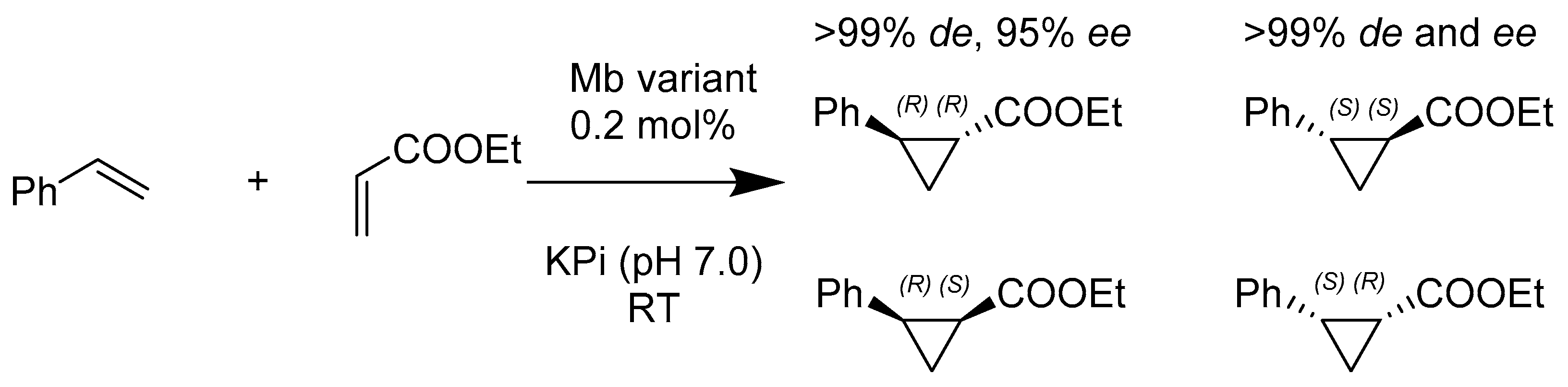
- Brandenberg, O.F.; Prier, C.K.; Chen, K.; Knight, A.M.; Wu, Z.; Arnold, F.H. Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes. ACS Catal. 2018, 8, 2629–2634. [Google Scholar] [CrossRef]
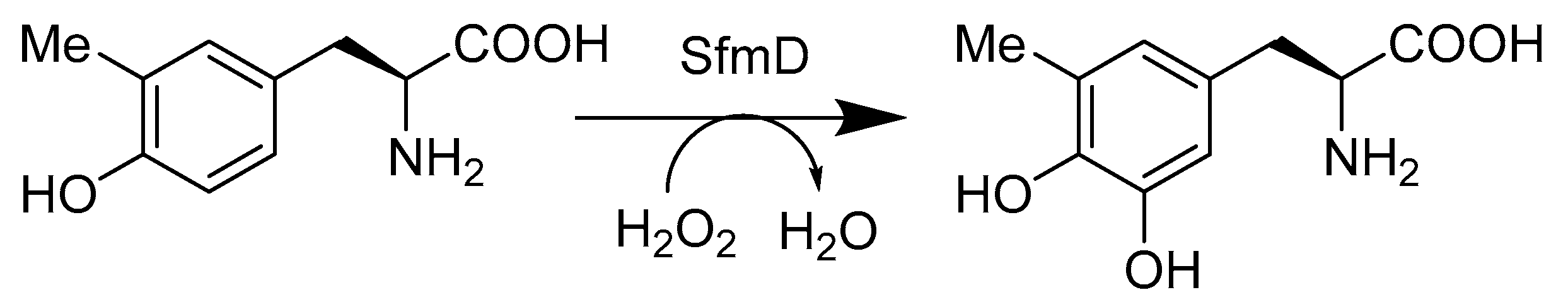
- Tang, M.C.; Fu, C.Y.; Tang, G.L. Characterization of SfmD as a Heme peroxidase that catalyzes the regioselective hydroxylation of 3-methyltyrosine to 3-hydroxy-5-methyltyrosine in saframycin A biosynthesis. J. Biol. Chem. 2012, 287, 5112–5121. [Google Scholar] [CrossRef]
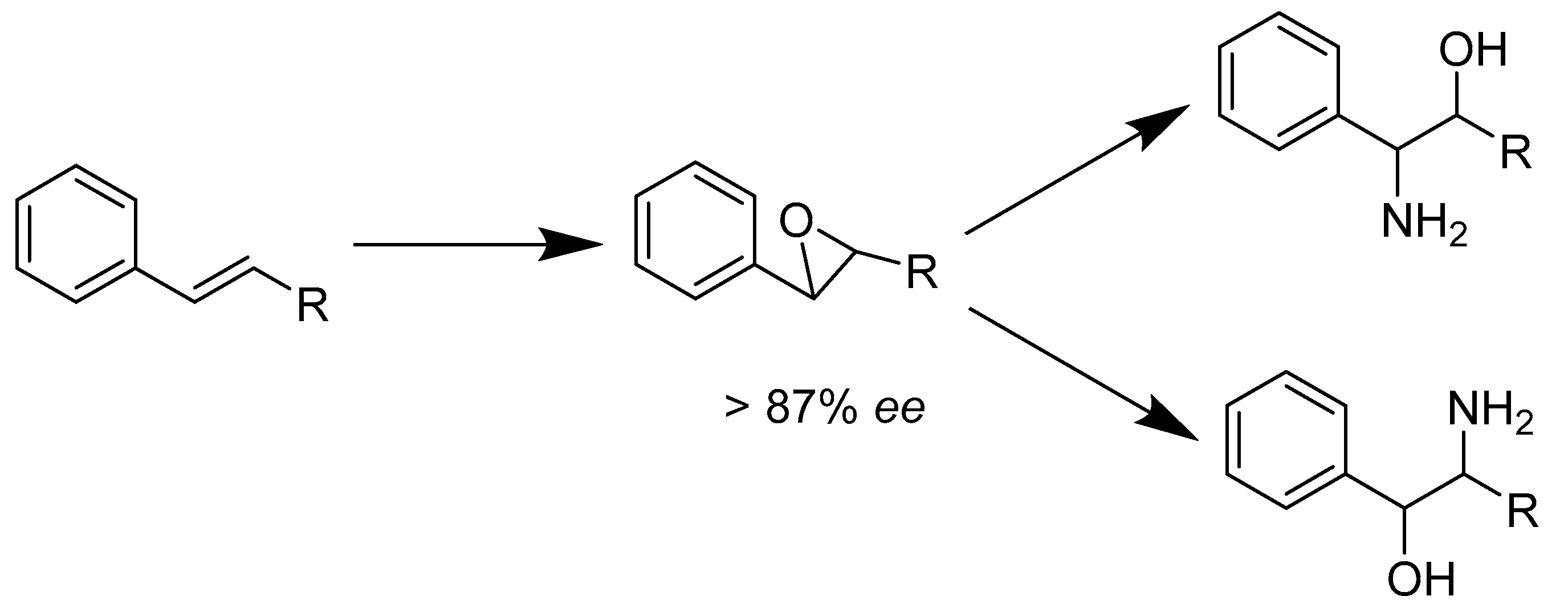
- Cho, I.; Prier, C.K.; Jia, Z.J.; Zhang, R.K.; Gorbe, T.; Arnold, F.H. Enantioselective Aminohydroxylation of Styrenyl Olefins Catalyzed by an Engineered Hemoprotein. Angew. Chem. Int. Ed. Engl. 2019, 58, 3138–3142. [Google Scholar] [CrossRef]
- Sello, G.; Orsini, F.; Bernasconi, S.; Gennaro, P.D. Synthesis of enantiopure 2-amino-1-phenyl and 2-amino-2-phenyl ethanols using enantioselective enzymatic epoxidation and regio- and diastereoselective chemical aminolysis. Tetrahedron Asymmetry 2006, 17, 372–376. [Google Scholar] [CrossRef]
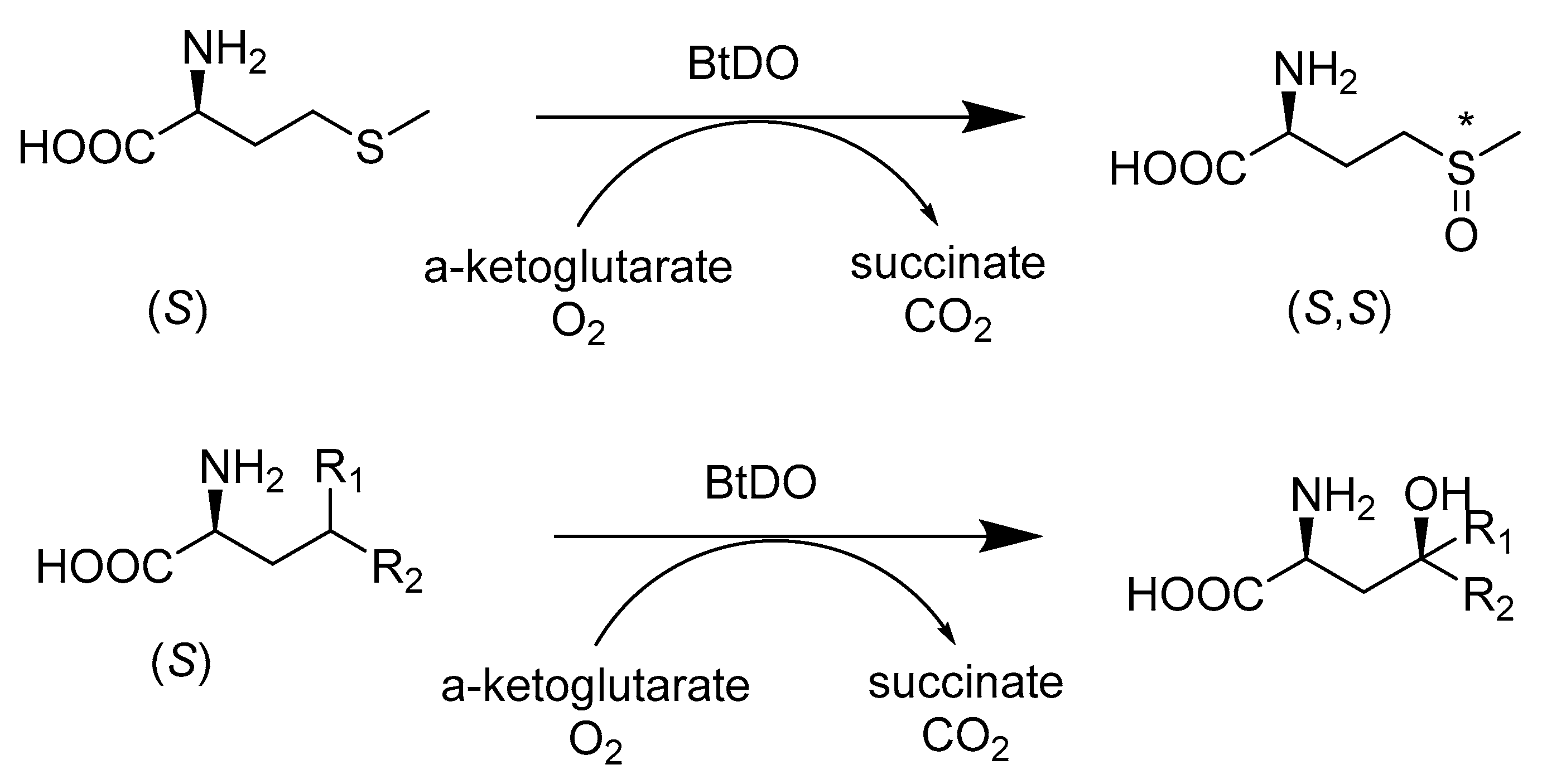
- Enoki, J.; Meisborn, J.; Muller, A.C.; Kourist, R. A Multi-Enzymatic Cascade Reaction for the Stereoselective Production of gamma-Oxyfunctionalyzed Amino Acids. Front. Microbiol. 2016, 7, 425. [Google Scholar] [CrossRef] [PubMed]
- Sib, A.; Gulder, T.A.M. Stereoselective Total Synthesis of Bisorbicillinoid Natural Products by Enzymatic Oxidative Dearomatization/Dimerization. Angew. Chem. Int. Ed. Engl. 2017, 56, 12888–12891. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, P.; Sreenilayam, G.; Tyagi, V.; Fasan, R. Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed. Engl. 2016, 55, 16110–16114. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Iwata, C.; Toda, H. Molecular cloning and characterization of a flavonoid-O-methyltransferase with broad substrate specificity and regioselectivity from Citrus depressa. BMC Plant Biol. 2016, 16, 180. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, X.; Zhang, R.K.; Arnold, F.H. Enantiodivergent alpha-Amino C-H Fluoroalkylation Catalyzed by Engineered Cytochrome P450s. J. Am. Chem. Soc. 2019, 141, 9798–9802. [Google Scholar] [CrossRef]
- Wollinsky, B.; Ludwig, L.; Xie, X.; Li, S.M. Breaking the regioselectivity of indole prenyltransferases: Identification of regular C3-prenylated hexahydropyrrolo[2,3-b]indoles as side products of the regular C2-prenyltransferase FtmPT1. Org. Biomol. Chem. 2012, 10, 9262–9270. [Google Scholar] [CrossRef]
- Schätzle, S.; Steffen-Munsberg, F.; Thontowi, A.; Höhne, M.; Robins, K.; Bornscheuer, U.T. Enzymatic Asymmetric Synthesis of Enantiomerically Pure Aliphatic, Aromatic and Arylaliphatic Amines with (R)-Selective Amine Transaminases. Adv. Synth. Catal. 2011, 353, 2439–2445. [Google Scholar] [CrossRef]
- Simon, R.C.; Grischek, B.; Zepeck, F.; Steinreiber, A.; Belaj, F.; Kroutil, W. Regio- and stereoselective monoamination of diketones without protecting groups. Angew. Chem. Int. Ed. Engl. 2012, 51, 6713–6716. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Coelho, P.S.; Farwell, C.C.; Wang, Z.J.; Lewis, J.C.; Brown, T.R.; Arnold, F.H. Enantioselective intramolecular C-H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2013, 52, 9309–9312. [Google Scholar] [CrossRef]
- Li, J.; Pan, J.; Zhang, J.; Xu, J.-H. Stereoselective synthesis of l-tert-leucine by a newly cloned leucine dehydrogenase from Exiguobacterium sibiricum. J. Mol. Catal. B Enzym. 2014, 105, 11–17. [Google Scholar] [CrossRef]
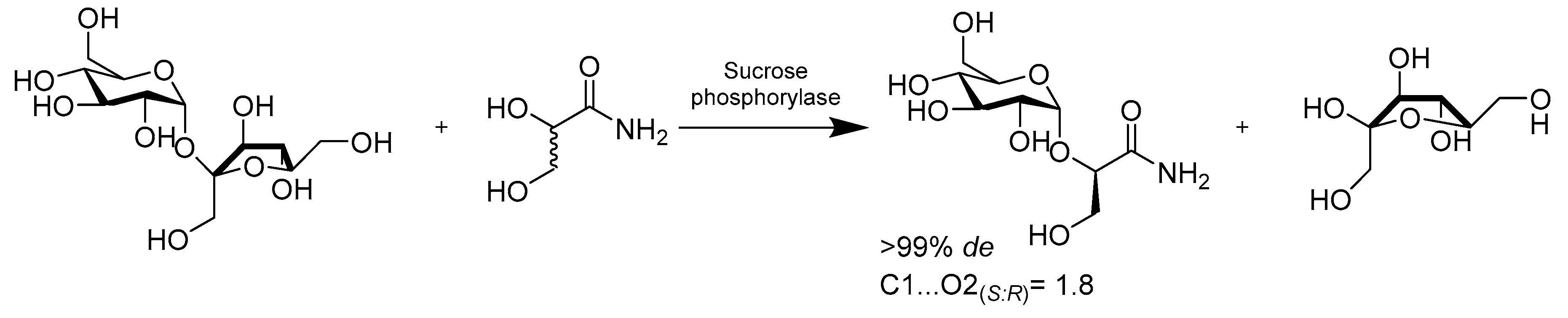
- Wildberger, P.; Brecker, L.; Nidetzky, B. Chiral resolution through stereoselective transglycosylation by sucrose phosphorylase: Application to the synthesis of a new biomimetic compatible solute, (R)-2-O-alpha-D-glucopyranosyl glyceric acid amide. Chem. Commun. 2014, 50, 436–438. [Google Scholar] [CrossRef] [PubMed][Green Version]
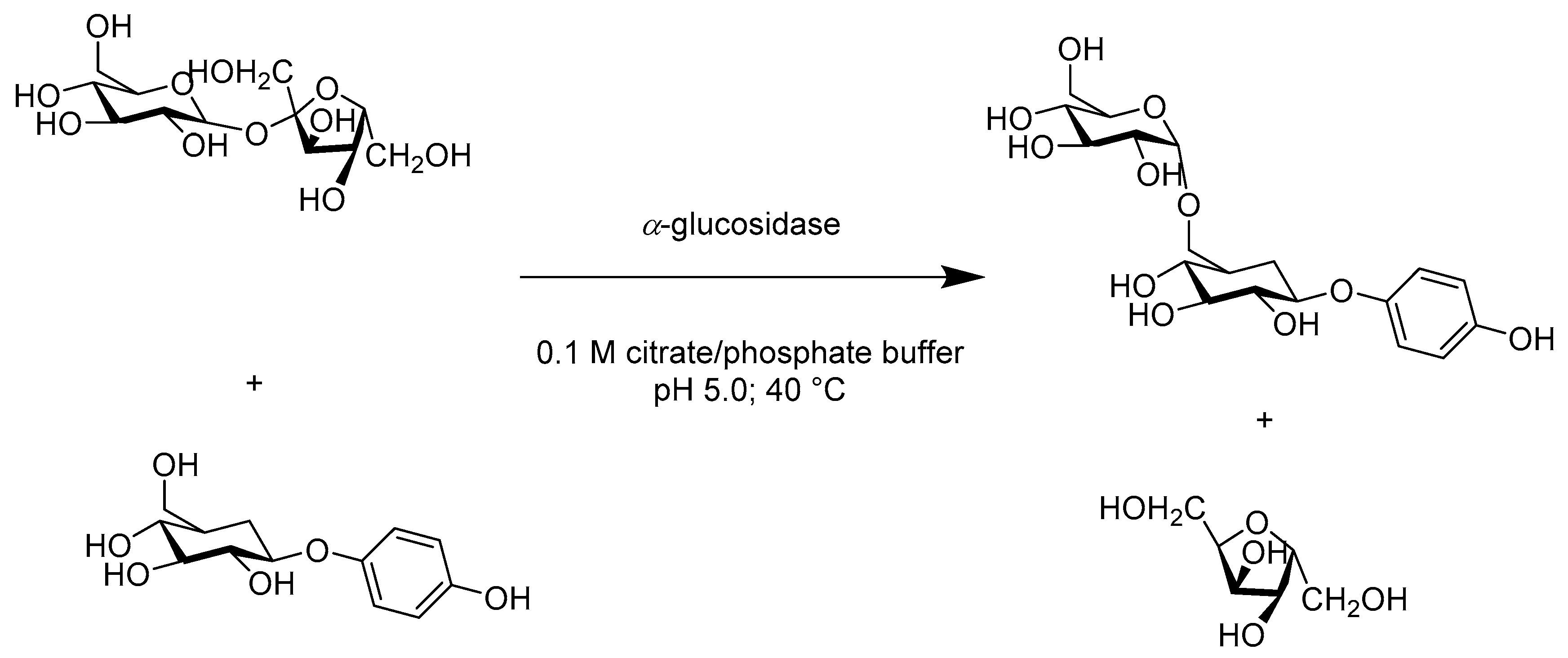
- Milosavić, N.B.; Prodanović, R.M.; Jankov, R.M. A simple and efficient one-step, regioselective, enzymatic glucosylation of arbutin by α-glucosidase. Tetrahedron Lett. 2007, 48, 7222–7224. [Google Scholar] [CrossRef]
- Chen, X.; Xu, L.; Jin, L.; Sun, B.; Gu, G.; Lu, L.; Xiao, M. Efficient and Regioselective Synthesis of beta-GalNAc/GlcNAc-Lactose by a Bifunctional Transglycosylating beta-N-Acetylhexosaminidase from Bifidobacterium bifidum. Appl. Environ. Microbiol. 2016, 82, 5642–5652. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, S.; Chokhawala, H.; Sun, M.; Zheng, H.; Chen, X. Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural alpha-2,6-linked sialosides: A P. damsela alpha-2,6-sialyltransferase with extremely flexible donor-substrate specificity. Angew. Chem. Int. Ed. Engl. 2006, 45, 3938–3944. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yao, W.; Cheng, J.; Zhang, X.; Jin, L.; Yu, H.; Chen, X.; Wang, F.; Cao, H. Regioselective chemoenzymatic synthesis of ganglioside disialyl tetrasaccharide epitopes. J. Am. Chem. Soc. 2014, 136, 5205–5208. [Google Scholar] [CrossRef] [PubMed]
- Nishihachijo, M.; Hirai, Y.; Kawano, S.; Nishiyama, A.; Minami, H.; Katayama, T.; Yasohara, Y.; Sato, F.; Kumagai, H. Asymmetric synthesis of tetrahydroisoquinolines by enzymatic Pictet-Spengler reaction. Biosci. Biotechnol. Biochem. 2014, 78, 701–707. [Google Scholar] [CrossRef]
- Bonamore, A.; Rovardi, I.; Gasparrini, F.; Baiocco, P.; Barba, M.; Molinaro, C.; Botta, B.; Boffi, A.; Macone, A. An enzymatic, stereoselective synthesis of (S)-norcoclaurine. Green Chem. 2010, 12, 1623–1627. [Google Scholar] [CrossRef]
- Ding, W.; Williams, D.R.; Northcote, P.; Siegel, M.M.; Tsao, R.; Ashcroft, J.; Morton, G.O.; Alluri, M.; Abbanat, D.; Maiese, W.M.; et al. Pyrroindomycins, novel antibiotics produced by Streptomyces rugosporus sp. LL-42D005. I. Isolation and structure determination. J. Antibiot. 1994, 47, 1250–1257. [Google Scholar] [CrossRef]
- Zehner, S.; Kotzsch, A.; Bister, B.; Sussmuth, R.D.; Mendez, C.; Salas, J.A.; van Pee, K.H. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. [Google Scholar] [CrossRef]
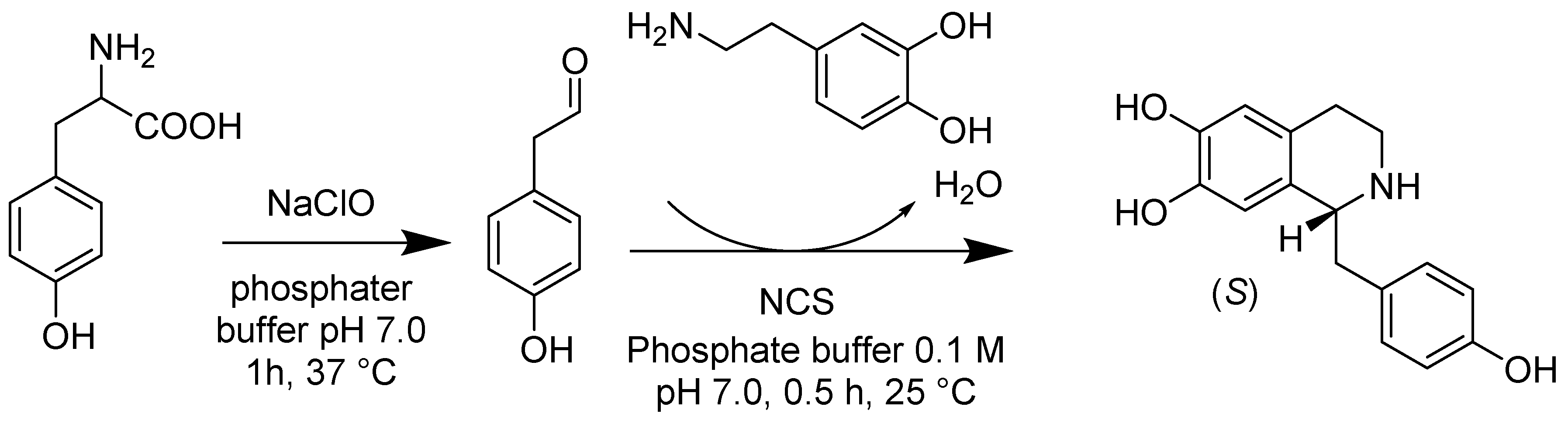
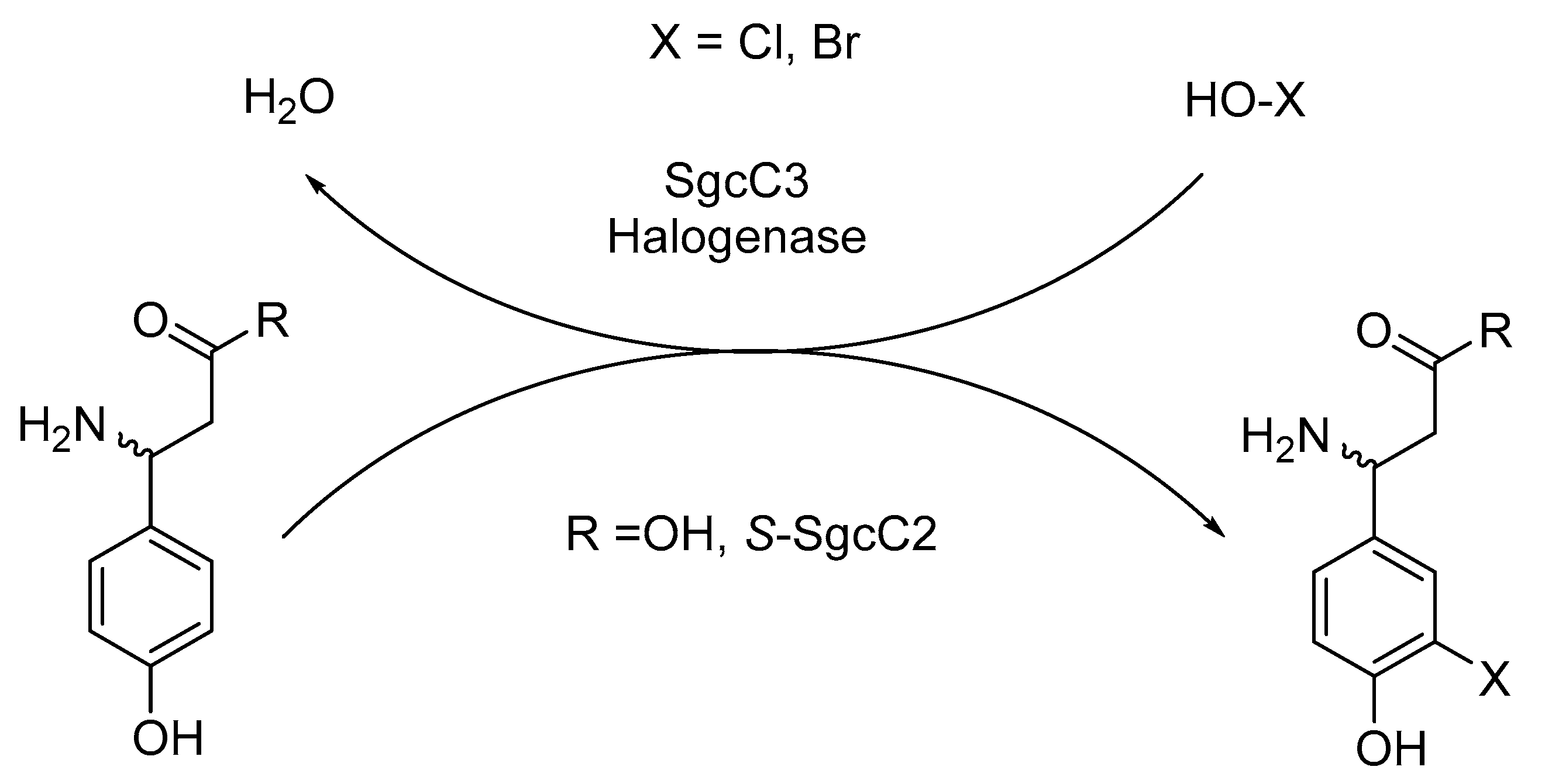
- Lin, S.; Van Lanen, S.G.; Shen, B. Regiospecific chlorination of (S)-beta-tyrosyl-S-carrier protein catalyzed by SgcC3 in the biosynthesis of the enediyne antitumor antibiotic C-1027. J. Am. Chem. Soc. 2007, 129, 12432–12438. [Google Scholar] [CrossRef]
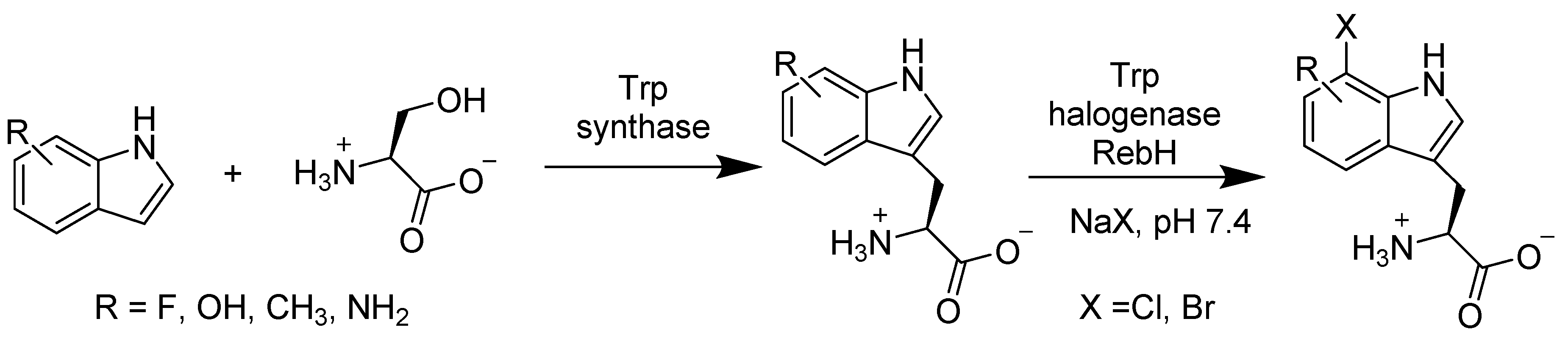
- Frese, M.; Guzowska, P.H.; Voss, H.; Sewald, N. Regioselective Enzymatic Halogenation of Substituted Tryptophan Derivatives using the FAD-Dependent Halogenase RebH. ChemCatChem 2014, 6, 1270–1276. [Google Scholar] [CrossRef]
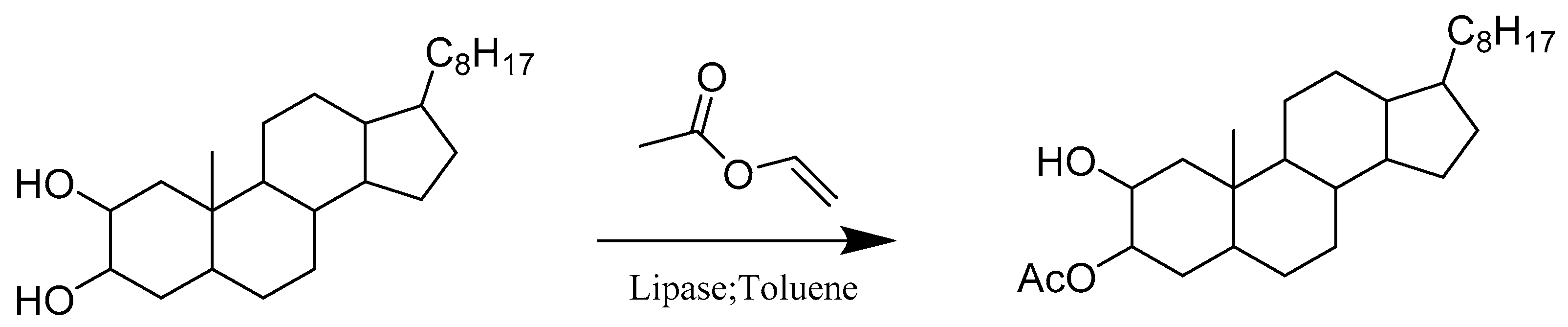
- Silva, M.M.C.; Riva, S.; e Melo, M.L.S. Regioselective enzymatic acylation of vicinal diols of steroids. Tetrahedron 2005, 61, 3065–3073. [Google Scholar] [CrossRef]
- Simeó, Y.; Sinisterra, J.V.; Alcántara, A.R. Regioselective enzymatic acylation of pharmacologically interesting nucleosides in 2-methyltetrahydrofuran, a greener substitute for THF. Green Chem. 2009, 11, 855–862. [Google Scholar] [CrossRef]
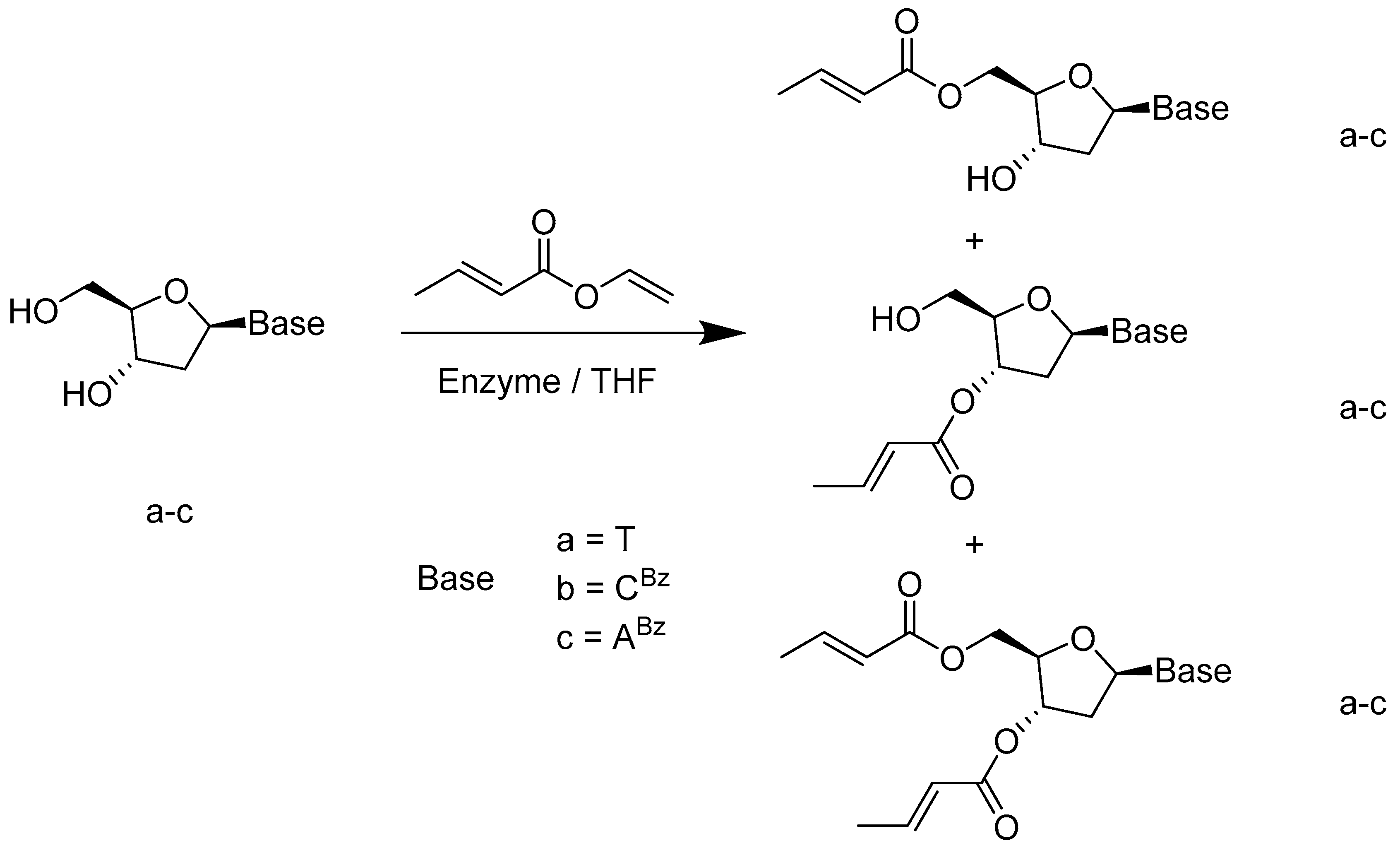
- Díaz-Rodríguez, A.; Fernández, S.; Lavandera, I.; Ferrero, M.; Gotor, V. Novel and efficient regioselective enzymatic approach to 3′-, 5′- and 3′,5′-di-O-crotonyl 2′-deoxynucleoside derivatives. Tetrahedron Lett. 2005, 46, 5835–5838. [Google Scholar] [CrossRef]
- Kaar, J.L.; Jesionowski, A.M.; Berberich, J.A.; Moulton, R.; Russell, A.J. Impact of ionic liquid physical properties on lipase activity and stability. J. Am. Chem. Soc. 2003, 125, 4125–4131. [Google Scholar] [CrossRef]
- Azuma, H.; Miyasaka, K.; Yokotani, T.; Tachibana, T.; Kojima-Yuasa, A.; Matsui-Yuasa, I.; Ogino, K. Lipase-catalyzed preparation of optically active 1′-acetoxychavicol acetates and their structure-activity relationships in apoptotic activity against human leukemia HL-60 cells. Bioorg. Med. Chem. 2006, 14, 1811–1818. [Google Scholar] [CrossRef]
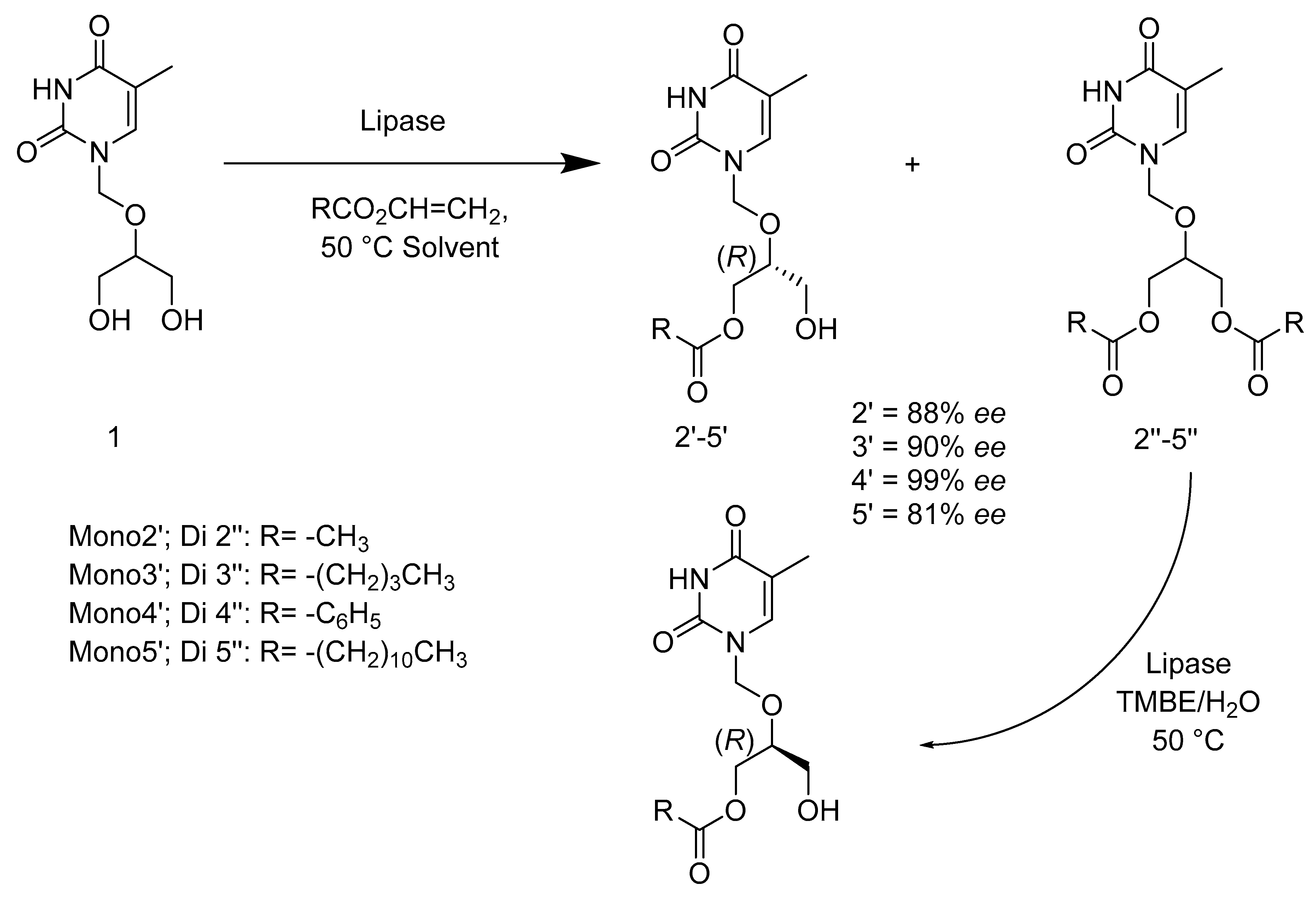
- Kołodziejska, R.; Górecki, M.; Frelek, J.; Dramiński, M. Enantioselective enzymatic desymmetrization of the prochiral pyrimidine acyclonucleoside. Tetrahedron Asymmetry 2012, 23, 683–689. [Google Scholar] [CrossRef]
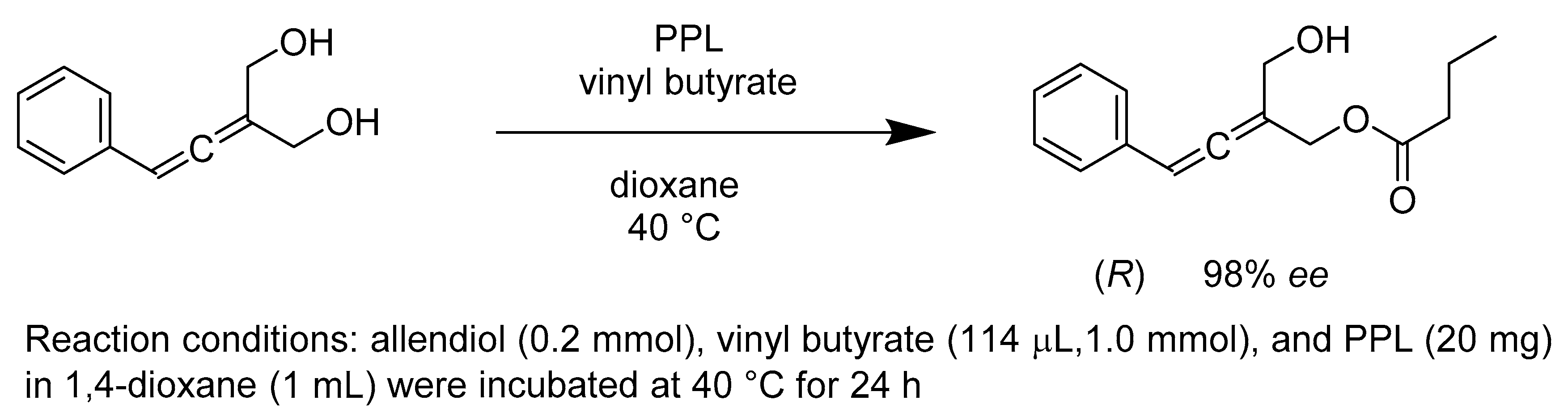
- Manzuna Sapu, C.; Backvall, J.E.; Deska, J. Enantioselective enzymatic desymmetrization of prochiral allenic diols. Angew. Chem. Int. Ed. Engl. 2011, 50, 9731–9734. [Google Scholar] [CrossRef]
- Lim, R.; Mitsunobu, K. Effect of dibutyryl cyclic AMP on nucleic acid and protein synthesis in neuronal and glial tumor cells. Life Sci. II 1972, 11, 1063–1070. [Google Scholar] [CrossRef]
- Nishino, S.F. Direct acridine orange counting of bacteria preserved with acidified lugol iodine. Appl. Environ. Microbiol. 1986, 52, 602–604. [Google Scholar] [CrossRef]
- Liguori, A.; Sindona, G.; Uccella, N. Sequence effect on the slow degradations of dinucleotides by fast atom bombardment tandem mass spectrometry. Biomed. Environ. Mass Spectrom. 1988, 16, 451–454. [Google Scholar] [CrossRef] [PubMed]
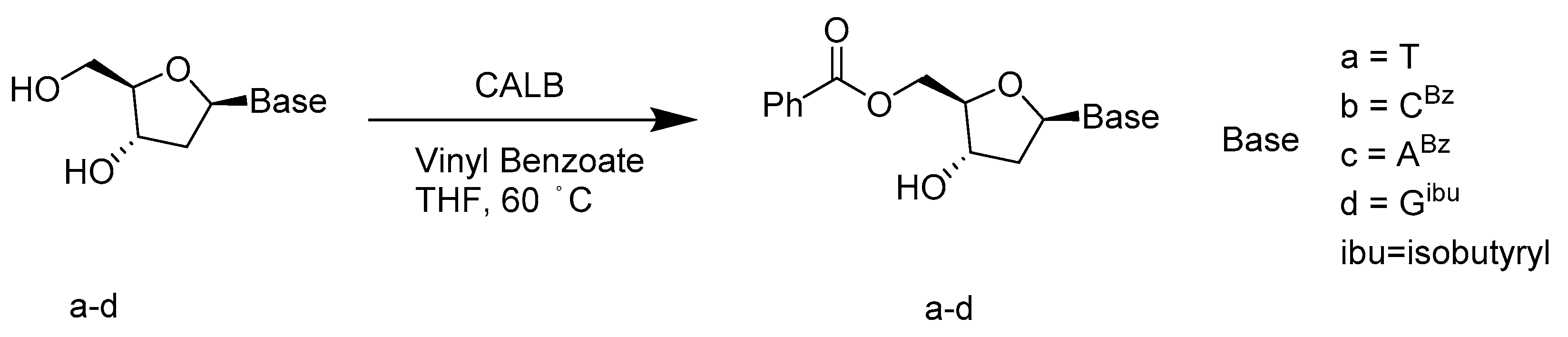
- García, J.; Fernández, S.; Ferrero, M.; Sanghvi, Y.S.; Gotor, V. A mild, efficient and regioselective enzymatic procedure for 5′-O-benzoylation of 2′-deoxynucleosides. Tetrahedron Lett. 2004, 45, 1709–1712. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, S.; Ma, H.; Zhang, A.; Lv, X.; Zheng, L. Stereoselective synthesis of caffeic acid amides via enzyme-catalyzed asymmetric aminolysis reaction. J. Biotechnol. 2013, 168, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Farwell, C.C.; Zhang, R.K.; McIntosh, J.A.; Hyster, T.K.; Arnold, F.H. Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent. Sci. 2015, 1, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Li, K.; He, T.; Feng, X.-W.; Wang, N.; Wang, X.-Y.; Yu, X.-Q. A novel enzymatic tandem process: Utilization of biocatalytic promiscuity for high stereoselective synthesis of 5-hydroxyimino-4,5-dihydrofurans. Tetrahedron 2011, 67, 2681–2688. [Google Scholar] [CrossRef]
- Reetz, M.T.; Prasad, S.; Carballeira, J.D.; Gumulya, Y.; Bocola, M. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: Rigorous comparison with traditional methods. J. Am. Chem. Soc. 2010, 132, 9144–9152. [Google Scholar] [CrossRef] [PubMed]
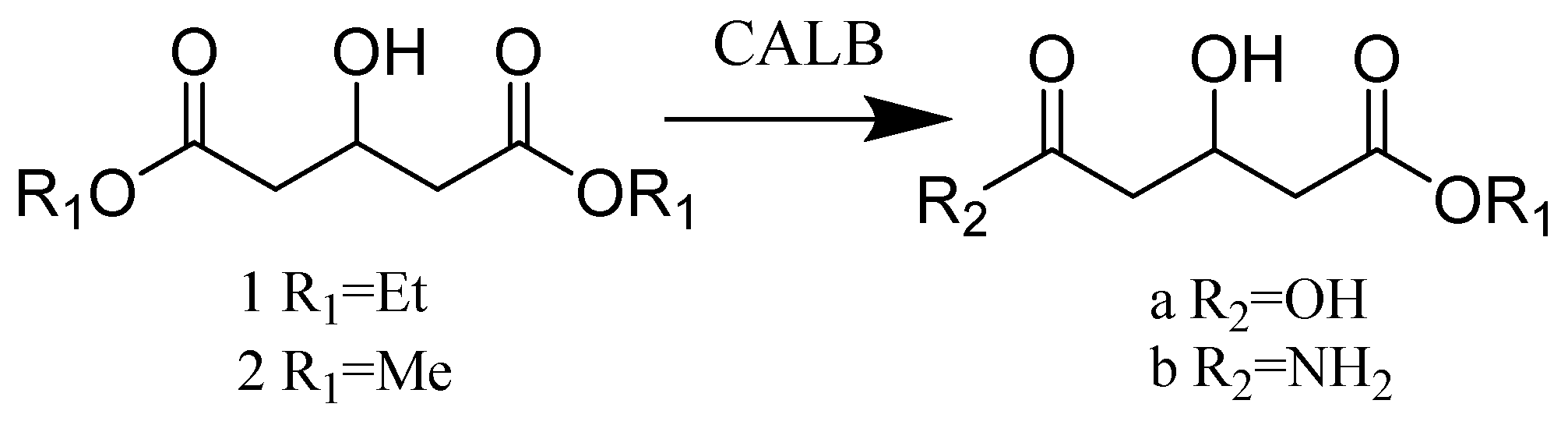
- Jacobsen, E.E.; Hoff, B.H.; Moen, A.R.; Anthonsen, T. Enatioselective enzymatic preparation of chiral glutaric monocarboxylic acids and amides. J. Mol. Catal. B Enzym. 2003, 21, 55–58. [Google Scholar] [CrossRef]
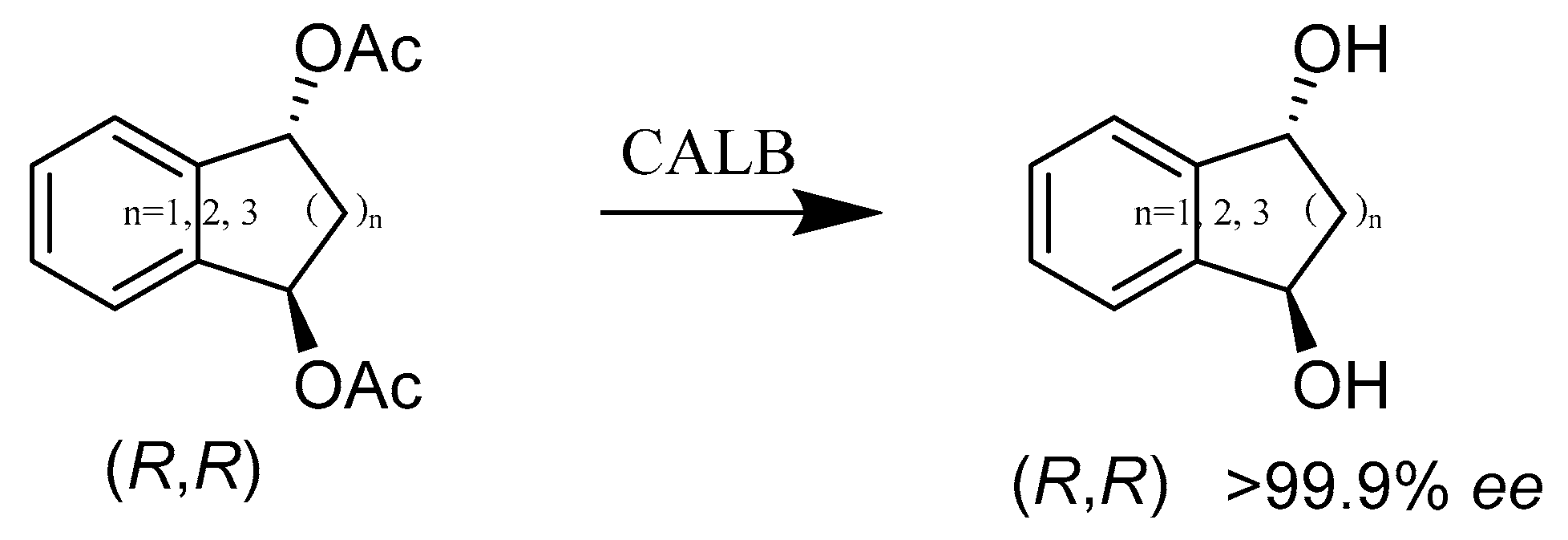
- Krumlinde, P.; Bogar, K.; Backvall, J.E. Asymmetric synthesis of bicyclic diol derivatives through metal and enzyme catalysis: Application to the formal synthesis of sertraline. Chemistry 2010, 16, 4031–4036. [Google Scholar] [CrossRef]
- Wuensch, C.; Glueck, S.M.; Gross, J.; Koszelewski, D.; Schober, M.; Faber, K. Regioselective enzymatic carboxylation of phenols and hydroxystyrene derivatives. Org. Lett. 2012, 14, 1974–1977. [Google Scholar] [CrossRef]
- Williams, G.J.; Domann, S.; Nelson, A.; Berry, A. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 3143–3148. [Google Scholar] [CrossRef]
- Husain, S.M.; Stillger, T.; Dünkelmann, P.; Lödige, M.; Walter, L.; Breitling, E.; Pohl, M.; Bürchner, M.; Krossing, I.; Müller, M.; et al. Stereoselective Reduction of 2-Hydroxy Ketones towards syn- and anti-1,2-Diols. Adv. Synth. Catal. 2011, 353, 2359–2362. [Google Scholar] [CrossRef]
- Wu, X.; Fei, M.; Chen, Y.; Wang, Z.; Chen, Y. Enzymatic synthesis of L-norephedrine by coupling recombinant pyruvate decarboxylase and omega-transaminase. Appl. Microbiol. Biotechnol. 2014, 98, 7399–7408. [Google Scholar] [CrossRef] [PubMed]
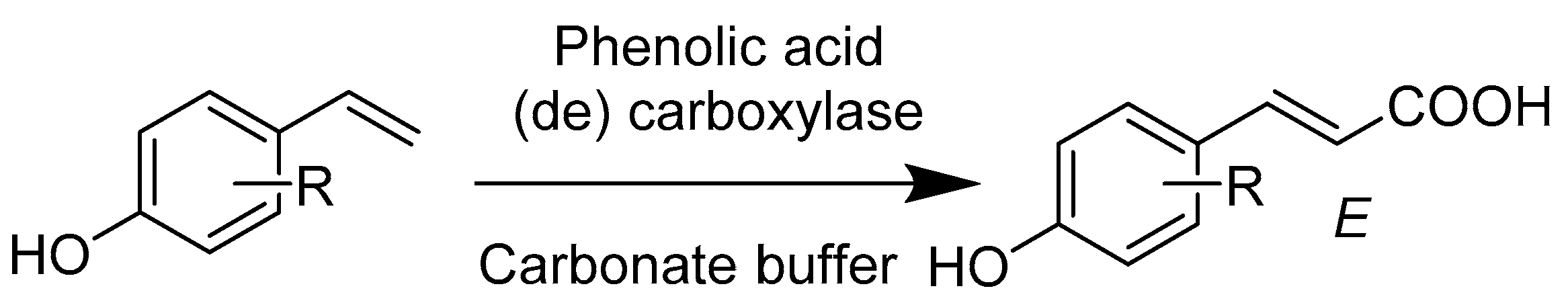
- Wuensch, C.; Pavkov-Keller, T.; Steinkellner, G.; Gross, J.; Fuchs, M.; Hromic, A.; Lyskowski, A.; Fauland, K.; Gruber, K.; Glueck, S.M.; et al. Regioselective Enzymatic beta-Carboxylation of para-Hydroxy-styrene Derivatives Catalyzed by Phenolic Acid Decarboxylases. Adv. Synth. Catal. 2015, 357, 1909–1918. [Google Scholar] [CrossRef] [PubMed]



































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, R.; Wang, Z.; Wamsley, M.C.; Duke, C.N.; Lii, P.H.; Epley, S.E.; Todd, L.C.; Roberts, P.J. Application of Enzymes in Regioselective and Stereoselective Organic Reactions. Catalysts 2020, 10, 832. https://doi.org/10.3390/catal10080832
Mu R, Wang Z, Wamsley MC, Duke CN, Lii PH, Epley SE, Todd LC, Roberts PJ. Application of Enzymes in Regioselective and Stereoselective Organic Reactions. Catalysts. 2020; 10(8):832. https://doi.org/10.3390/catal10080832
Chicago/Turabian StyleMu, Ruipu, Zhaoshuai Wang, Max C. Wamsley, Colbee N. Duke, Payton H. Lii, Sarah E. Epley, London C. Todd, and Patty J. Roberts. 2020. "Application of Enzymes in Regioselective and Stereoselective Organic Reactions" Catalysts 10, no. 8: 832. https://doi.org/10.3390/catal10080832
APA StyleMu, R., Wang, Z., Wamsley, M. C., Duke, C. N., Lii, P. H., Epley, S. E., Todd, L. C., & Roberts, P. J. (2020). Application of Enzymes in Regioselective and Stereoselective Organic Reactions. Catalysts, 10(8), 832. https://doi.org/10.3390/catal10080832