Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants

 , and
, and
Abstract
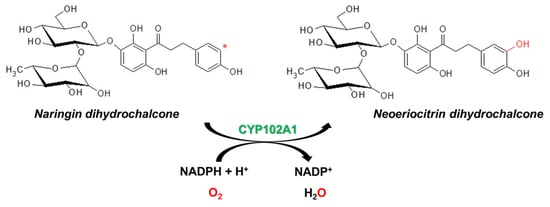
1. Introduction
2. Results and Discussion
2.1. Hydroxylation of Naringin DC by CYP102A1 Mutants
2.2. Optimal Expression of CYP102A1 M221 Mutant
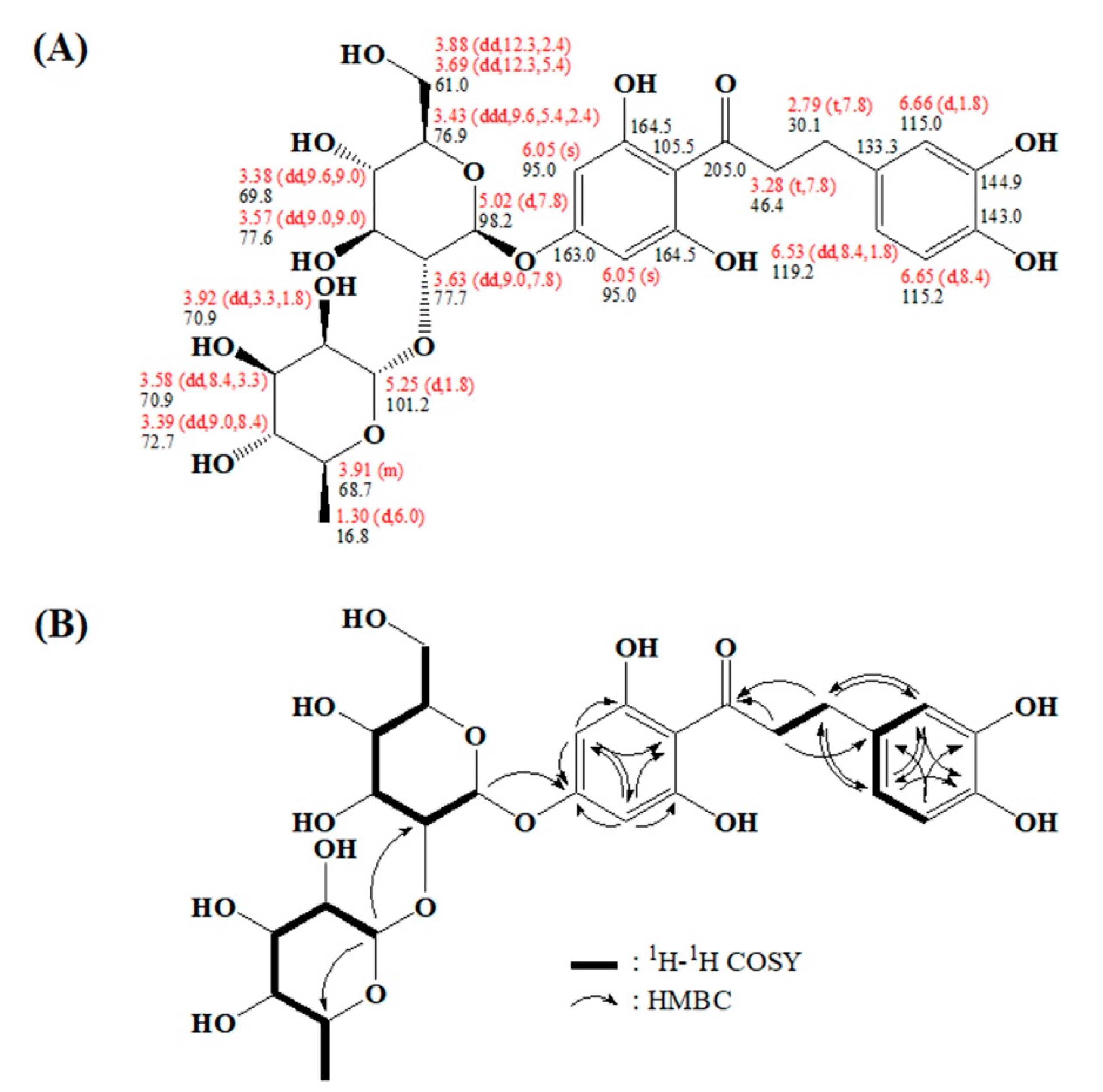
2.3. Characterizing a Major Product of Naringin DC by CYP102A1 Mutants
2.4. Kinetic Parameters and TTNs of Naringin DC Hydroxylation by CYP102A1 Mutants
2.5. Spectral Titration of Naringin DC toward CYP102A1 Mutants
3. Materials and Methods
3.1. Materials
3.2. Optimal Expression of CYP102A1 M221 Mutant
3.3. CYP102A1 Mutants Used to Screen Highly Active Naringin DC Hydroxylases
3.4. Hydroxylation of Naringin Dihydrochalcone by CYP102A1 Mutants
3.5. LC-MS Analysis
3.6. NMR Spectroscopy
3.7. Spectral Binding Titration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Szliszka, E.; Czuba, Z.P.; Mazur, B.; Paradysz, A.; Krol, W. Chalcones and Dihydrochalcones Augment TRAIL-Mediated Apoptosis in Prostate Cancer Cells. Molecules 2010, 15, 5336–5353. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhang, Y.; Chen, X.; Wang, Y.; Chen, W.; Xu, Q.; Li, P.; Ma, F. Extraction, identification, and antioxidant and anticancer tests of seven dihydrochalcones from Malus “Red Splendor” fruit. Food Chem. 2017, 231, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, M.; Dugé de Bernonville, T.; Lohou, D.; Guyot, S.; Guillemette, T.; Brisset, M.-N.; Dat, J.F. Histolocalization and physico-chemical characterization of dihydrochalcones: Insight into the role of apple major flavonoids. Phytochemistry 2013, 90, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- Ehrenkranz, J.R.L.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes/Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Dugé de Bernonville, T.; Guyot, S.; Paulin, J.-P.; Gaucher, M.; Loufrani, L.; Henrion, D.; Derbré, S.; Guilet, D.; Richomme, P.; Dat, J.F.; et al. Dihydrochalcones: Implication in resistance to oxidative stress and bioactivities against advanced glycation end-products and vasoconstriction. Phytochemistry 2010, 71, 443–452. [Google Scholar] [CrossRef]
- Tang, N.; Yan, W. Solubilities of Naringin Dihydrochalcone in Pure Solvents and Mixed Solvents at Different Temperatures. J. Chem. Eng. Data 2016, 61, 4085–4089. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, K.; Zhou, Y.; An, Y.; Hu, T.; Lu, J.; Huang, S.; Pei, G. Naringin Dihydrochalcone Ameliorates Cognitive Deficits and Neuropathology in APP/PS1 Transgenic Mice. Front. Aging Neurosci. 2018, 10, 169. [Google Scholar] [CrossRef]
- Río, J.A.D.; Fuster, M.D.; Sabater, F.; Porras, I.; García-Lidón, A.; Ortuño, A. Selection of citrus varieties highly productive for the neohesperidin dihydrochalcone precursor. Food Chem. 1997, 59, 433–437. [Google Scholar] [CrossRef]
- Nakamura, Y.; Watanabe, S.; Miyake, N.; Kohno, H.; Osawa, T. Dihydrochalcones: Evaluation as Novel Radical Scavenging Antioxidants. J. Agric. Food Chem. 2003, 51, 3309–3312. [Google Scholar] [CrossRef]
- Liu, B.; Zhu, X.; Zeng, J.; Zhao, J. Preparation and physicochemical characterization of the supramolecular inclusion complex of naringin dihydrochalcone and hydroxypropyl-β-cyclodextrin. Food Res. Int. 2013, 54, 691–696. [Google Scholar] [CrossRef]
- Park, S.-H.; Kim, D.-H.; Kim, D.; Kim, D.-H.; Jung, H.-C.; Pan, J.-G.; Ahn, T.; Kim, D.; Yun, C.-H. Engineering Bacterial Cytochrome P450 (P450) BM3 into a Prototype with Human P450 Enzyme Activity Using Indigo Formation. Drug Metab. Dispos. 2010, 38, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 2002, 1, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Kim, K.-H.; Kim, D.-H.; Liu, K.-H.; Jung, H.-C.; Pan, J.-G.; Yun, C.-H. Generation of human metabolites of 7-ethoxycoumarin by bacterial cytochrome P450 BM3. Drug Metab. Dispos. 2008, 36, 2166–2170. [Google Scholar] [CrossRef]
- Narhi, L.O.; Fulco, A.J. Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 1986, 261, 7160–7169. [Google Scholar]
- Girvan, H.M.; Waltham, T.N.; Neeli, R.; Collins, H.F.; McLean, K.J.; Scrutton, N.S.; Leys, D.; Munro, A.W. Flavocytochrome P450 BM3 and the origin of CYP102 fusion species. Biochem. Soc. Trans. 2006, 34, 1173–1177. [Google Scholar] [CrossRef]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends Biotechnol. 2012, 30, 26–36. [Google Scholar] [CrossRef]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. P450(BM3) (CYP102A1): Connecting the dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef]
- Le, T.-K.; Jang, H.-H.; Nguyen, H.; Doan, T.; Lee, G.-Y.; Park, K.; Ahn, T.; Joung, Y.; Kang, H.-S.; Yun, C.-H. Highly regioselective hydroxylation of polydatin, a resveratrol glucoside, for one-step synthesis of astringin, a piceatannol glucoside, by P450 BM3. Enzym. Microb. Technol. 2016, 97, 34–42. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef]
- Kang, J.-Y.; Kim, S.-Y.; Kim, D.; Kim, D.; Sun-Mi, S.; Park, S.-H.; Kim, K.-H.; Jung, H.; Pan, J.-G.; Joung, Y.; et al. Characterization of diverse natural variants of CYP102A1 found within a species of Bacillus megaterium. AMB Express 2011, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Ahn, T.; Jung, H.-C.; Pan, J.-G.; Yun, C.-H. Generation of the human metabolite piceatannol from the anticancer-preventive agent resveratrol by bacterial cytochrome P450 BM3. Drug Metab. Dispos. 2009, 37, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Kang, J.-Y.; Kim, D.; Park, S.-H.; Park, S.; Kim, D.; Park, K.; Lee, Y.J.; Jung, H.; Pan, J.-G.; et al. Generation of Human Chiral Metabolites of Simvastatin and Lovastatin by Bacterial CYP102A1 Mutants. Drug Metab. Dispos. Biol. Fate Chem. 2010, 39, 140–150. [Google Scholar] [CrossRef]
- Kang, J.-Y.; Ryu, S.H.; Park, S.-H.; Cha, G.S.; Kim, D.-H.; Kim, K.-H.; Hong, A.W.; Ahn, T.; Pan, J.-G.; Joung, Y.H.; et al. Chimeric cytochromes P450 engineered by domain swapping and random mutagenesis for producing human metabolites of drugs. Biotechnol. Bioeng. 2014, 111, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-H.; Ryu, S.-H.; Le, T.-K.; Doan, T.T.M.; Nguyen, T.H.H.; Park, K.D.; Yim, D.-E.; Kim, D.-H.; Kang, C.-K.; Ahn, T.; et al. Regioselective C-H hydroxylation of omeprazole sulfide by Bacillus megaterium CYP102A1 to produce a human metabolite. Biotechnol. Lett. 2017, 39, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and Other Phenolic Compounds from Medicinal Plants for Pharmaceutical and Medical Aspects: An Overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Ovesná, Z.; Kozics, K.; Bader, Y.; Saiko, P.; Handler, N.; Erker, T.; Szekeres, T. Antioxidant activity of resveratrol, piceatannol and 3,3′,4,4′,5,5′-hexahydroxy-trans-stilbene in three leukemia cell lines. Oncol. Rep. 2006, 16, 617–624. [Google Scholar] [CrossRef]
- Lai, T.N.H.; Herent, M.-F.; Quetin-Leclercq, J.; Nguyen, T.B.T.; Rogez, H.; Larondelle, Y.; André, C.M. Piceatannol, a potent bioactive stilbene, as major phenolic component in Rhodomyrtus tomentosa. Food Chem. 2013, 138, 1421–1430. [Google Scholar] [CrossRef]
- Arai, D.; Kataoka, R.; Otsuka, S.; Kawamura, M.; Maruki-Uchida, H.; Sai, M.; Ito, T.; Nakao, Y. Piceatannol is superior to resveratrol in promoting neural stem cell differentiation into astrocytes. Food Funct. 2016, 7, 4432–4441. [Google Scholar] [CrossRef]
- Kershaw, J.; Kim, K.-H. The Therapeutic Potential of Piceatannol, a Natural Stilbene, in Metabolic Diseases: A Review. J. Med. Food 2017, 20, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.-H.; Peng, C.; Zhang, H. Polydatin: A review of pharmacology and pharmacokinetics. Pharm. Biol. 2013, 51, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Mérillon, J.-M.; Fauconneau, B.; Teguo, P.W.; Barrier, L.; Vercauteren, J.; Huguet, F. Antioxidant Activity of the Stilbene Astringin, Newly Extracted from Vitis vinifera Cell Cultures. Clin. Chem. 1997, 43, 1092–1093. [Google Scholar] [CrossRef]
- Waffo-Teguo, P.; Lee, D.; Cuendet, M.; Mérillon, J.; Pezzuto, J.M.; Kinghorn, A.D. Two new stilbene dimer glucosides from grape (Vitis vinifera) cell cultures. J. Nat. Prod. 2001, 64, 136–138. [Google Scholar] [CrossRef]
- Lee, D.E.; Lee, K.W.; Byun, S.; Jung, S.K.; Song, N.; Lim, S.H.; Heo, Y.-S.; Kim, J.E.; Kang, N.J.; Kim, B.Y.; et al. 7,3′,4′-Trihydroxyisoflavone, a Metabolite of the Soy Isoflavone Daidzein, Suppresses Ultraviolet B-induced Skin Cancer by Targeting Cot and MKK4. J. Biol. Chem. 2011, 286, 14246–14256. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Jang, J.; Le, T.-K.; Nguyen, T.H.H.; Woo, S.-M.; Yoo, S.-K.; Lee, Y.J.; Park, K.D.; Yeom, S.-J.; Kim, G.-J.; et al. Biocatalytic Production of a Potent Inhibitor of Adipocyte Differentiation from Phloretin Using Engineered CYP102A1. J. Agric. Food Chem. 2020, 68, 6683–6691. [Google Scholar] [CrossRef]
- Pandey, B.P.; Roh, C.; Choi, K.-Y.; Lee, N.; Kim, E.J.; Ko, S.; Kim, T.; Yun, H.; Kim, B.-G. Regioselective hydroxylation of daidzein using P450 (CYP105D7) from Streptomyces avermitilis MA4680. Biotechnol. Bioeng. 2010, 105, 697–704. [Google Scholar] [PubMed]
- Thibodeaux, C.J.; Melançon, C.E.; Liu, H. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. Engl. 2008, 47, 9814–9859. [Google Scholar] [CrossRef]
- Gutmann, A.; Bungaruang, L.; Weber, H.; Leypold, M.; Breinbauer, R.; Nidetzky, B. Towards the synthesis of glycosylated dihydrochalcone natural products using glycosyltransferase-catalysed cascade reactions. Green Chem. 2014, 16, 4417–4425. [Google Scholar] [CrossRef]
- Bowles, D.; Isayenkova, J.; Lim, E.-K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef]
- Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 2009, 583, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Plaza, M.; Pozzo, T.; Liu, J.; Gulshan Ara, K.Z.; Turner, C.; Nordberg Karlsson, E. Substituent effects on in vitro antioxidizing properties, stability, and solubility in flavonoids. J. Agric. Food Chem. 2014, 62, 3321–3333. [Google Scholar] [CrossRef] [PubMed]
- Slámová, K.; Kapešová, J.; Valentová, K. “Sweet Flavonoids”: Glycosidase-Catalyzed Modifications. Int. J. Mol. Sci. 2018, 19, 2126. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qi, T.; Xu, L.; Lu, L.; Xiao, M. Recent progress in the enzymatic glycosylation of phenolic compounds. J. Carbohydr. Chem. 2016, 35, 1–23. [Google Scholar] [CrossRef]
- Devi, M.A.; Das, N.P. In vitro effects of natural plant polyphenols on the proliferation of normal and abnormal human lymphocytes and their secretions of interleukin-2. Cancer Lett. 1993, 69, 191–196. [Google Scholar] [CrossRef]
- Nelson, J.A.; Falk, R.E. The efficacy of phloridzin and phloretin on tumor cell growth. Anticancer. Res. 1993, 13, 2287–2292. [Google Scholar]
- Alsanea, S.; Gao, M.; Liu, D. Phloretin Prevents High-Fat Diet-Induced Obesity and Improves Metabolic Homeostasis. AAPS J. 2017, 19, 797–805. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, X.; Xu, X.-G.; Li, Y.-H.; Wang, B.; Gao, X.-H.; Chen, H.-D.; Yatskayer, M.; Oresajo, C. Protective effects of a topical antioxidant complex containing vitamins C and E and ferulic acid against ultraviolet irradiation-induced photodamage in Chinese women. J. Drugs Dermatol. 2013, 12, 464–468. [Google Scholar]
- Julsing, M.K.; Cornelissen, S.; Bühler, B.; Schmid, A. Heme-iron oxygenases: Powerful industrial biocatalysts? Curr. Opin. Chem. Biol. 2008, 12, 177–186. [Google Scholar] [CrossRef]
- Yim, S.-K.; Kim, D.-H.; Jung, H.-C.; Pan, J.-G.; Kang, H.-S.; Ahn, T.; Yun, C.-H. Surface display of heme- and diflavin-containing cytochrome P450 BM3 in Escherichia coli: A whole cell biocatalyst for oxidation. J. Microbiol. Biotechnol. 2010, 20, 712–717. [Google Scholar] [CrossRef]
- Kaderbhai, M.A.; Ugochukwu, C.C.; Kelly, S.L.; Lamb, D.C. Export of Cytochrome P450 105D1 to the Periplasmic Space of Escherichia coli. Appl. Environ. Microbiol. 2001, 67, 2136–2138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Remsberg, C.M.; Yáñez, J.A.; Vega-Villa, K.R.; Miranda, N.D.; Andrews, P.K.; Davies, N.M. HPLC-UV Analysis of Phloretin in Biological Fluids and Application to Pre-Clinical Pharmacokinetic Studies. J. Chromatogr. Sep. Tech. 2010, 1, 101. [Google Scholar] [CrossRef]
- Hosea, N.A.; Miller, G.P.; Guengerich, F.P. Elucidation of Distinct Ligand Binding Sites for Cytochrome P450 3A4. Biochemistry 2000, 39, 5929–5939. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | kcat (min−1) | Km (μM) | kcat/Km (min−1μM−1) |
|---|---|---|---|
| M16V2 | 7.8 ± 0.5 | 160 ± 25 | 0.049 ± 0.008 |
| G1 | 11.0 ± 0.3 | 73 ± 5 | 0.151 ± 0.011 |
| M179 | 5.3 ± 0.2 | 76 ± 9 | 0.070 ± 0.008 |
| M221 | 10.8 ± 0.2 | 79 ± 6 | 0.137 ± 0.011 |
| M620 | 5.7 ± 0.7 | 441 ± 101 | 0.013 ± 0.003 |
| M850 | 4.5 ± 0.4 | 243 ± 43 | 0.019 ± 0.004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.H.H.; Woo, S.-M.; Nguyen, N.A.; Cha, G.-S.; Yeom, S.-J.; Kang, H.-S.; Yun, C.-H. Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants. Catalysts 2020, 10, 823. https://doi.org/10.3390/catal10080823
Nguyen THH, Woo S-M, Nguyen NA, Cha G-S, Yeom S-J, Kang H-S, Yun C-H. Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants. Catalysts. 2020; 10(8):823. https://doi.org/10.3390/catal10080823
Chicago/Turabian StyleNguyen, Thi Huong Ha, Su-Min Woo, Ngoc Anh Nguyen, Gun-Su Cha, Soo-Jin Yeom, Hyung-Sik Kang, and Chul-Ho Yun. 2020. "Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants" Catalysts 10, no. 8: 823. https://doi.org/10.3390/catal10080823
APA StyleNguyen, T. H. H., Woo, S.-M., Nguyen, N. A., Cha, G.-S., Yeom, S.-J., Kang, H.-S., & Yun, C.-H. (2020). Regioselective Hydroxylation of Naringin Dihydrochalcone to Produce Neoeriocitrin Dihydrochalcone by CYP102A1 (BM3) Mutants. Catalysts, 10(8), 823. https://doi.org/10.3390/catal10080823