2.1. Frenkel Excitation and Charge Transfer States of Pigment–Protein Complex
We represent the reaction center of PSII as a pigment–protein complex (PPC), a molecular aggregate of
M pigments being held in a protein scaffolding. Molecular excitations of the pigments in the PPC are described in terms of electrons and holes residing in the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) within each pigment [
19]. The ground state of the PPC,
, is defined as the state in which all of the HOMOs are occupied and all of the LUMOs unoccupied (see
Figure 2a). The excited state of the PPC,
is defined to have one hole in the HOMO at pigment
m and one electron in the LUMO at pigment
n (see
Figure 2b). In what follows, we refer to the excited state for
as the charge transfer (CT) state and that for
as the Frenkel exciton (FE) state which is denoted by
(see
Figure 2c). The total charge is conserved under these excitations so the PPC as a whole remains neutral.
The pigments in the PPC are affected by vibrational fluctuations originating in the protein scaffolding and surrounding solvent. To incorporate this phenomenon, the Hamiltonian of the PPC is divided into three parts as
, where
is the system Hamiltonian describing the excitation dynamics of the pigment sites,
is the bath Hamiltonian describing the environmental vibrations, and
is the system–bath Hamiltonian that determines the way that the pigments are influenced by the vibrations. For the system Hamiltonian, we employ the tight-binding model of PPC [
19], whose matrix elements are specified by
where
are the tunneling matrix elements [
23],
is the (renormalized) Coulomb interaction potential between the hole in the HOMO at pigment
m and the electron in the LUMO at pigment
n, and
is the Förster resonance coupling between the FEs at pigments
m and
n, satisfying
. For later convenience, we define the site energy
by the diagonal elements (
and
) of
,
This represents the amount of energy it takes to excite the ground state
to
. The matrix element between two FE states is obtained from Equation (
1) as
, indicating that the coupling between the FE states are determined only by
. In contrast, coupling between CT states and between FE and CT states are determined only by the off-diagonal elements of the tunneling matrix,
of
. These are associated with the overlap integrals of the electronic wavefunctions related to the molecular orbitals which are well approximated by the exponential function of distance [
19,
23]. We therefore model the tunneling matrix by
where
is the coefficient representing the overall strength of coupling satisfying
,
is the representative position of pigment
m, and
is a length scale that generally depends on the pigment pair [
19].
The bath of vibrations described by
originates in the protein structure and surrounding solvent [
23]. The influence of these vibrations on the system described by
gives rise to a renormalization of the interaction potentials
in Equation (
1). Such renormalization takes the form of
where
is the (bare) Coulomb potential of the hole–electron interactions and
is the reorganization energy of the excited state
(including
for
). The Coulomb interaction potential we use here is
where
is the dielectric constant, and
is a cutoff parameter yielding the exciton binding energy for
that we denote by
.
With the detailed form of
, the physical content of the diagonal elements of the tunneling matrix,
, is considered. Let us denote the energy level of the HOMO by
and that of the LUMO by
for pigment
m. These energy levels are measured from the vacuum level (infinitely far from the PPC) which defines the zero-energy level. The HOMO-LUMO gap,
, is by definition the amount of energy it takes to excite an electron from the HOMO to the LUMO state [
1]. This value is identical to the amount of energy required to create the FE at pigment
m at the ground state. Thus, the binding energy of FE,
, is included in the HOMO-LUMO gap. Based on this, we assign
the amount of energy required to create a hole in the HOMO of pigment
m by removing an electron from the ground state to the vacuum level, that is
, as illustrated in
Figure 3a. Likewise, as shown in
Figure 3b, we assign
the amount of energy it takes to bring an electron from the vacuum level to the LUMO at pigment
m while its HOMO is occupied, so that
. Here, the biding energy is subtracted from the energy level of the LUMO level because an electron, instead of a hole, resides in the HOMO.
Incorporating our argument above, we introduce an alternative expression for the site energy. Substituting
obtained above for Equation (
2), the site energies are written as
. Hereafter, we set the HOMO level of pigment 1 (
) as the reference level and define
as illustrated in
Figure 3c. We refer to this quantity as the level shift of pigment
m. Rearranging the expression of site energy of the FE for the energy level of the LUMO,
, we express Equation (
2) in terms of the level shift as
where
which we refer to as the zero-shift site energy of
. Since
holds, the site energies of Equation (
6) depend on
variables which are
, provided the values of
are all determined.
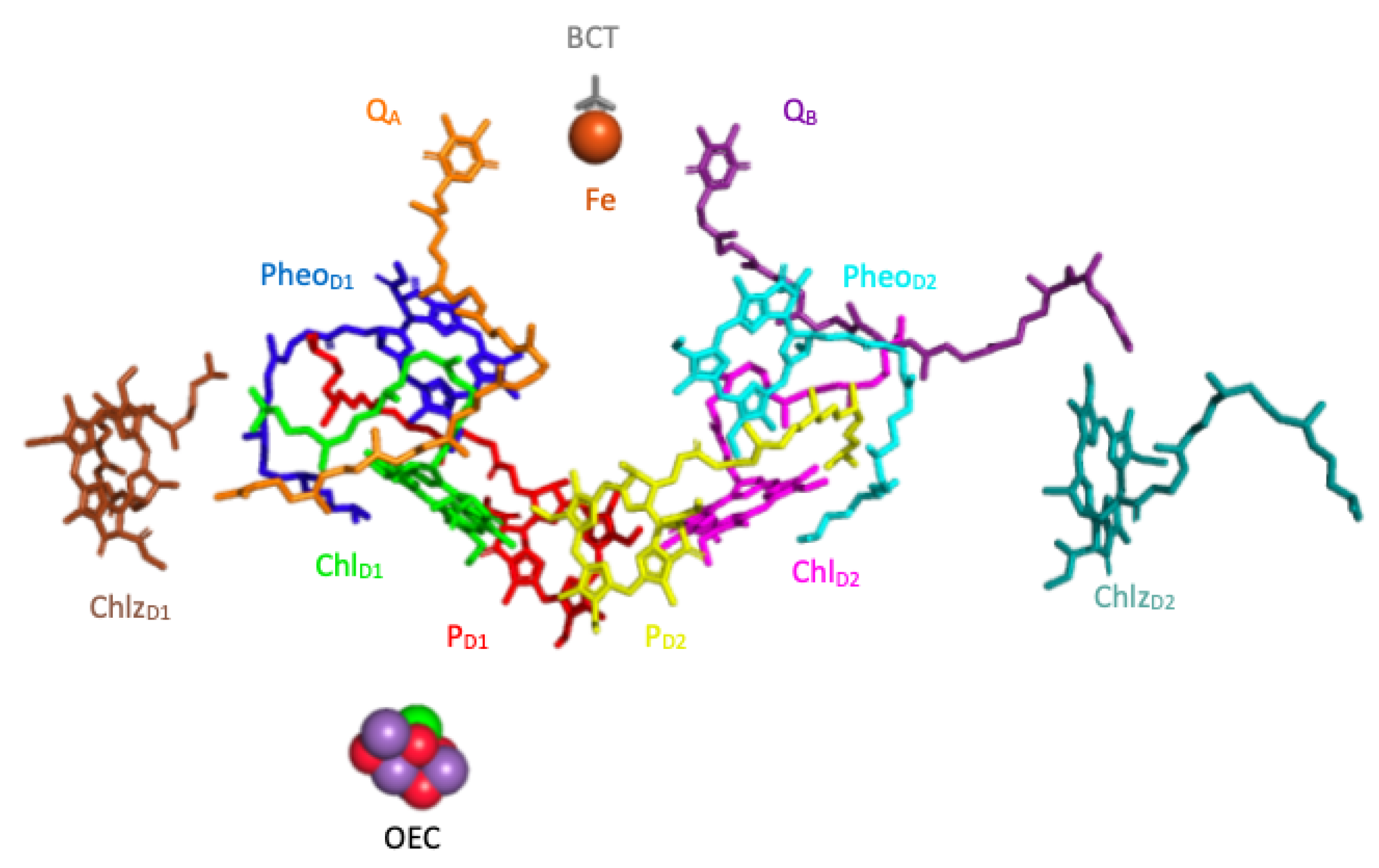
2.2. The Model of the PSII Reaction Center
The reaction center of the PSII contains four chlorophylls (
,
,
and
), two pheophytins (
and
), and two plastoquinones (
and
). The timescale of the kinetics of the charge transfer between the pigments is on the order of a few pico-seconds (ps) whereas the timescale of
reduction is on the order of 100 ps [
24]. For this reason we exclude the plastoquinones from our model, focusing on the first few pico-seconds (ps) after the photo-excitation of the complex. In what follows, for the sake of notational simplicity, we also refer to each of the six pigments by numbers (1 through 6) assigned as shown in
Figure 4.
For the FE–FE sector of Equation (
1), we adopt the site energies,
for
, from Gelzinis et al. [
21] and the FE couplings
from Shibata et al. [
25]. The relevant values are illustrated in
Figure 4b and are also summarized in
Table A1. For the CT–CT and FE–CT sectors of Equation (
1), we must obtain the zero-shift site energies
and the off-diagonal elements of tunneling matrix,
. The parameters specifying
are
,
,
, and
, among which
are the previously determinded site energies of the FEs. For the protein environment of the PSII RC, we use
as suggested by Müh and Renger [
11]. For
, we examined the zero-phonon lines given by Novoderezhkin et al. [
15] and found that
is an optimum value. This
yields the binding energy of FE,
, for all of the pigment sites. The positions
have been adopted from Gelzinis et al. [
21]. These values are summarized in
Table A2. The reorganization energy for the FE states are obtained once the spectral density of the pigment–protein interaction is determined. As discussed in more depth later on in the Methods section, we set
for all of the FE states and (
)
for all of the CT states. The zero-shift site energies are then evaluated for each of the 36 states (6 FE states and 30 CT states), the results from which are shown in
Figure 5a. Note that the site energies of the FEs are identical to their zero-shift site energies,
, due to Equation (
6). To specify the CT–CT and FE–CT couplings, we further require the inclusion of
and
in the tunneling matrix elements of Equation (
3). For these parameters, we aim to adjust the coupling between
and
to be
as given by Novoderezhkin et al. [
26]. This can be achieved by taking
assuming
Å [
19]. These values are used for all
m and
n of
for simplicity. The results of the coupling strength,
for
, are shown in
Figure 5b.
We now investigate how the population dynamics of the excited states depend on the level shifts of the pigments in the PSII RC. The populations of excited states are represented as the diagonal elements of the reduced density matrix,
. In order to see correlations among the FE and CT populations, we quantify the transfer efficiency by the time average of population for each state,
where
is the time over which the average is computed. It has been observed in past experimental studies that the PSII RC strongly prefers the reduction to occur at
, the pheophytin in the D1 branch. To characterize this asymmetry, we define the efficiency of electron transfer (ET) to
by
, and likewise the efficiency to
by
. Their difference,
is a measure of the asymmetry.
stipulates that ET is more likely to occur at the D1 branch, as observed experimentally, while
specifies that ET is expected to occur at the D2 branch.
2.3. Level Shifts in the D1 Branch
As the starting point of the analysis of charge transfer phenomena in the PSII RC, we first set all of the level shifts to zero so that
. Then, a simulation of the FE and CT states after the photo-excitation of the PSII RC by a
pulse was run to visualize the population dynamics within the system. The result for the first ps of the simulation is shown in
Figure 6. Although each of the 36 states (see
Figure 5) are included in the computation, the populations are dominated by FE states while the contribution from CT states is limited. Within the FE states, a major transition is observed to occur during the first 0.4 ps. After this event,
and
steadily increase while the others decrease. This is mainly because
and
have the lowest site energies (see
Figure 4b) within the D1 and D2 branches respectively and therefore act as sink sites of the exciton in the PSII RC. Similarly, the two CT populations of
and
seen in
Figure 6 have the first and the second lowest zero-shift site energies respectively amongst all of the CT states (see
Figure 5a). Considering this similarity, we therefore expect the CT states to become more excited as their site energies are lowered by manipulating the level shifts.
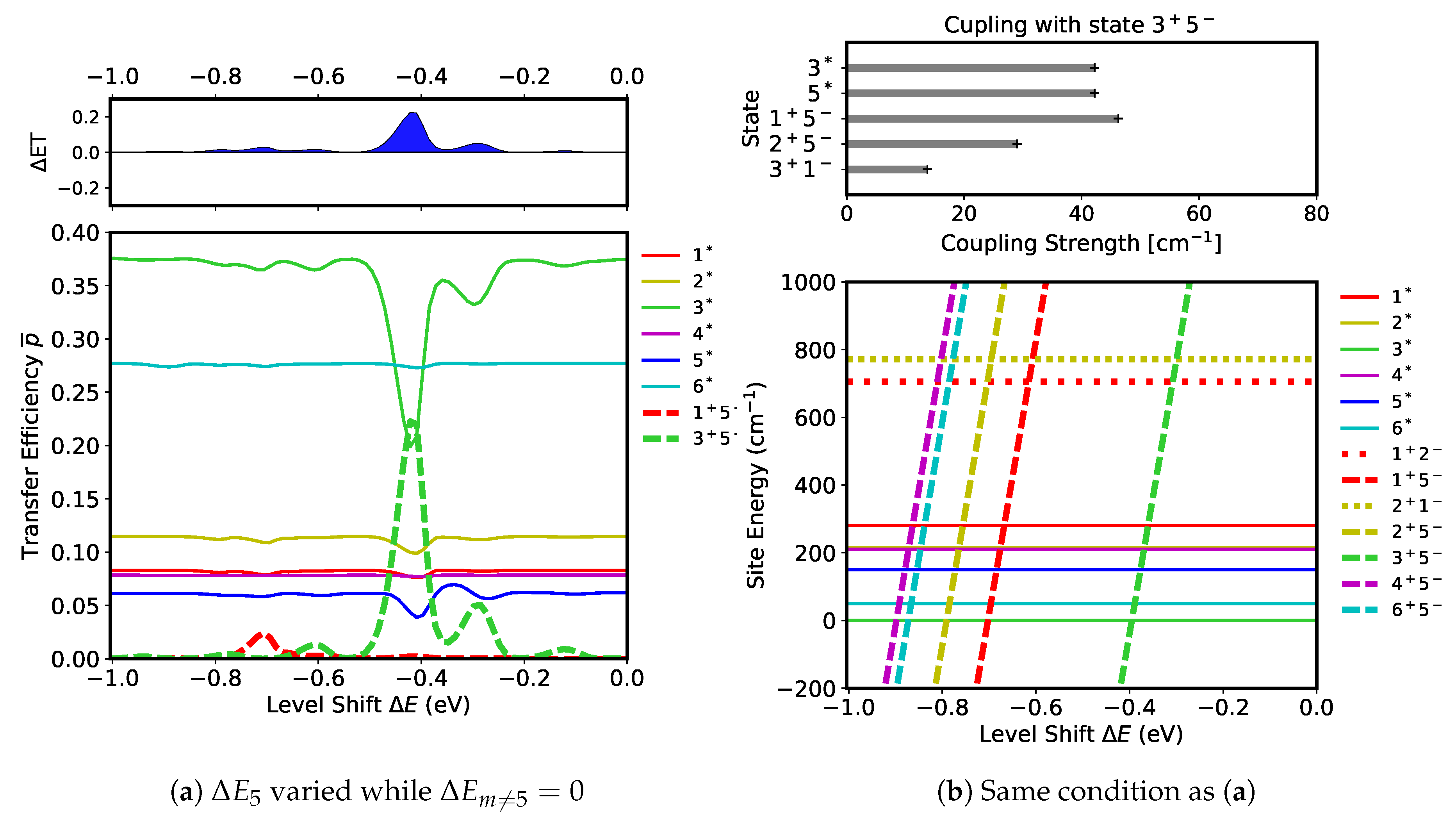
First, we evaluate which CT states can be excited if the level shifts are changed in the D1 branch. We begin by observing how the level shift at
, that is
, affects the transfer efficiency by maintaining all other level shifts at zero,
. Simulations were run to obtain a range of
values to secure transfer efficiencies for
(in Equation (
7)). The results of the simulations are shown in
Figure 7a. A remarkable feature can be seen in the data collected of the transfer efficiencies in the form of a large peak of
(dashed green) at
(
) accompanied by large fractional decreases in
(solid green) and in
(solid blue). This peak indicates that the CT state
is being activated by the two FE states,
and
. This is interpreted as a formation of the radical pair
by the photo-induced charge transfer process occurring between the excited molecules
and
. In the upper panel of
Figure 7a, there is a prominent positive peak in
associated with the formation of
, representing an efficient electron transfer to
. In addition to the large peak in
, there is a very small peak of
(dashed red) at
(
) corresponding to a slight ET increase in the D1 branch.
The peaks in the transfer efficiencies can be interpreted in terms of resonance and coupling strength among the excited states. The site energies of the states for the range of
values are shown in
Figure 7b. Resonance between the states can be induced when their site energies are close together, illustrated by the crossing points of the site energies in
Figure 7b. The five CT states
for
whose site energies depend on
as
, have site energies which are seen to cross with all of the FE states within the parameter window. Inspecting
Figure 7a, however, only two of them, (
and
), are found to be active in the actual dynamics. Such selection of CT states can be understood in terms of coupling strength between the excited states. The CT states strongly coupled with
are shown in the upper panel of
Figure 7b. The FE states,
and
, are both strongly coupled with
, thus active transfers between these states can occur around the crossing point,
. This is exactly what is observed in
Figure 7a where
is lower than this value by the amount of ∼
. This shift of the resonance point is on the order of
, indicting that it can be induced by pigment-pigment and pigment–protein interactions. The CT state
is also strongly coupled with
. However, because their site energies do not cross each other, the charge transfer will not occur between them. Likewise, a transfer between
and
does not occur because their site energies also do not cross each other. The small peak of
in
Figure 7a is another result of resonance between
and the FE states,
and
, around
. However, the charge transfer here is not as active as that of
because the populations of
and
are small (see
Figure 7a). Additionally, the coupling strength between these FE states and
is weak at
(see
Figure 5b), further restricting the CT activity. The rest of the linearly changing states in
Figure 7b, which are
,
, and
, remain inactive because none are coupled strongly enough with the FE states, as can be noted in
Figure 5b. There is an additional small peak of
at
in
Figure 7a whose origin is yet to be identified.
Next, the effects of the level shift at
on the transfer efficiencies are examined. Simulations were run for a range of
keeping all other level shifts unchanged at zero,
. The results from these simulations are shown in
Figure S1a of the Supplementary Materials. Remarkably, three large peaks of the CT states are seen in the transfer efficiency,
(dash-dotted red) at
), of
(dash-dotted blue) at
), and of
(dash-dotted yellow) at
). As shown in
Figure S1b of the Supplementary Materials, these peaks correspond to the resonance of the three CT states,
,
, and
, with FE states at their crossing points. Thus three radical pairs
,
, and
are formed by this resonance. The site energies of
and
are also crossing with those of FE states (shown in
Figure S1b), but they remain inactive because they are coupled only weakly with FE states, as can be seen in
Figure 5b.
Now it must be considered how the population of the radical pair
(
) can dominate the terminal sate of the PSII RC. We do this by seeking charge transfer pathways that maximize the transfer efficiency
. As can be seen in
Figure 5b, the FE states do not couple strongly with
, so
needs to be excited by other CT states. We refer to such states as the primary CT states. Inspecting
Figure 5b,
is strongly coupled with
,
,
, and
. Meanwhile,
and
are not as active because they are only slightly coupled with the FE states. Since no coupling exists between
and
, they independently couple with
. Thus, we have narrowed down the possibilities to the following two pathways:
and
. The first pathway depends on the activity of
, which can be excited efficiently by the FE states around the resonance peak at
as seen in
Figure S1a of Supplementary Material. To activate the pathway, the
value is kept constant at
(
) while
is adjusted to bring
in resonance with
. From
, we expect the resonance to happen around
. To see how
reacts to this adjustment, we ran simulations for a range of
values around the expected resonance value. The results are shown in
Figure 8a. The peak of
(dashed red) is seen at
which is around the expected point though shifted by
). It is also evident that the increase in
(dashed red) is correlated to the decrease in
(dash-dotted red), demonstrating the resonant charge transfer between
and
.
The second possible pathway to excite
is
. This pathway is expected to be active when these CT states are mutually in resonance by
. From
Figure 7a, we have gathered that the transfer efficiency of
is maximized when
. Using Equation (
6), this implies that the condition
will induce the highest activity of
. To examine the activity of
in the pathway, simulations were run for a range of
values under this condition. The results from these simulations are shown in
Figure 8b. The peak of
(dashed red) is observed at
(
) accompanied by the largest decrease in
, indicating that the pathway is most active at this point. Interestingly, we have obtained the same
value as that of the first pathway. This leads to
for the activation of the second pathway, which only deviates
from that of the first pathway.
The radical pair,
(
), can thus be excited efficiently from the FE states mediated by the two primary CT states,
(
) and
(
). To visualize how these efficient pathways impact the population dynamics, simulations were run for an extended time up to 10 ps at
. The result for the first pathway,
, is shown in
Figure 9a. A very quick excitation of the radical pair is observed as its population (dashed red) reaches the greatest value within the first 2 ps. Paying close attention on the first 0.4 ps, the population dynamics of the FE states shown in the inset of
Figure 9a are observed to be similar to those in
Figure 6, except that the population of
in
Figure 9a (solid green) does not increase monotonically but begins to decrease around 0.1 ps. This difference indicates that
is the primary donor of the excitation energy for the three CT states,
,
, and
. From
to
, the population of
continues to grow until it reaches its maximum value of 0.38 at 1.9 ps, whereas the populations of
and
continue to decrease exponentially. Hence the radical pair
causes the dominant population. After the first 2 ps, the excited CT states remain active by keeping their populations mostly steady but with a slow decrease in
over time. Note that the population of
(solid cyan) appears to behave differently from other states because it belongs to the D2 branch. During the course of population change,
is always positive and increasing until
, indicating that the election continues to be transferred to the D1 branch through the interplay between
and other states. As a result of this,
and
reaches 0.664 and 0.003, respectively, at 10 ps. This produces a
value of 0.661 in
Figure 9a. Viewing the asymmetry by the ratio of these ET populations, that is
, it is suggested that ET in the PSII RC is extremely biased toward the D1 branch by the charge transfer pathway.
At the higher temperature of 300 K, the populations shown in
Figure 9b appear to behave similarly to those at 77 K. The major transition at both temperatures happens within the first 3 ps and the population of
is the largest. However, the population of
is much lower with a maximum value of only 0.16 at 2.7 ps while other CT states appear to be more active at the higher temperature. In particular, the populations of
(dotted red) and
(dotted yellow) are more pronounced compared to those at 77 K, possibly due to thermal excitations induced in the FE and CT states via the system–bath coupling. In regard to the ET dynamics, the overall value of
is also lower but is still positive and increasing over time. At 10 ps,
and
are 0.411 and 0.012 respectively, making the asymmetry ratio of
. This large value indicates ET in the PSII RC is still substantially biased toward the D1 branch even at 300 K by the active pathway.
The result of our simulation for the second pathway,
, is shown in
Figure S2 of Supplementary Material. The dynamics of
are similar to those of the first pathway, but its initial ascent progresses slightly slower and overall population is about 3/4 as much of that of the first pathway. The population reaches its maximum value of 0.29 at
, and then decays slowly over time. Despite the lower population in the radical pair, the ET and its asymmetry are comparable to that of the first pathway. At
,
and
reaches 0.611 and 0.003 at 77 K, respectively, which amounts to the asymmetry ratio of
. At 300 K,
and
reach 0.476 and 0.011 respectively which produces a asymmetry ratio of
. Thus, the ET is highly biased toward the D1 branch at both 77 K and 300 K.
2.4. Level Shifts in the D2 Branch
We now turn our attention to the other branch of the PSII RC, the D2 branch, which has been experimentally observed to be mostly inactive in ET dynamics. Here, we examine to what extent the D2 branch could be active in the PSII RC. In particular, we seek a pathway to activate the radical pair
, the counter part of
in the D2 branch. As we have done for the D1 branch, we change the level shifts of the pigments in the D2 branch to maximize the transfer efficiency
. For simplicity, we keep
for
, and adjust
and
to form a resonance that activates
. The results of the simulations for a range of
values are shown in
Figure S3a of the Supplementary Materials. By analogy with the
pathway of the D1 branch, we consider the
pathway here. The resonance peak of
(dash-dotted yellow) is found at
, and thus we use this to search for the
value that maximizes
. The results of the simulations are shown in
Figure S3b of Supplementary Material. We locate the peak of
(dashed yellow) at
.
With these values of level shifts, we see how the populations of the radical pair
are formed through dynamics. The first 10 ps of the simulation run at 77 K is shown in
Figure 10. Comparing this with
Figure 9a, the behavior of active CT states are observed to be similar to their counter parts in the D1 branch, which are
,
, and
. In particular, the major transitions that occur during the first 3 ps of the simulation and the population is dominated by the radical pair,
. However, the maximum population of
is recorded at 0.29 in
Figure 10, so it is lower than its D1 counterpart,
in
Figure 9a, by a factor of 0.76. This ratio roughly holds for their subsequent populations after 3 ps. This difference in the populations of the radical pairs can be caused by the difference in molecular species of their primary donor sites. The donor of
is the FE of a pheophytin
, whereas the donor of
is the FE of a chlorophyll
. As can be seen in
Figure 6, the initial populations of
and
in our simulations are 0.115 and 0.193 respectively which implies that the population of
is lower by a factor of 0.6. This is partly the consequence of the transition dipole moment of pheophytin and that of chlorophyll, which are
and
, respectively, making the ratio of oscillator strengths roughly
. Thus, the activity of the radical pairs is largely affected by the intrinsic photo-activity of their donor FE states. Although the population of
is low in
Figure 10, the asymmetry in ET is strongly biased toward the D2 branch. This is quantified by the ET populations in the D1 and D2 branches,
and
which are 0.03 and 0.49 at 10 ps, respectively, yielding the asymmetry ratio of
, indicating the electron is almost certainly found in the D2 branch. This is the complete opposite to what is observed in experiments.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









