Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds


Abstract
1. Introduction
2. Results and Discussion
2.1. Asymmetric Michael Addition of α, β-Unsaturated Nitroolefins and Ketones
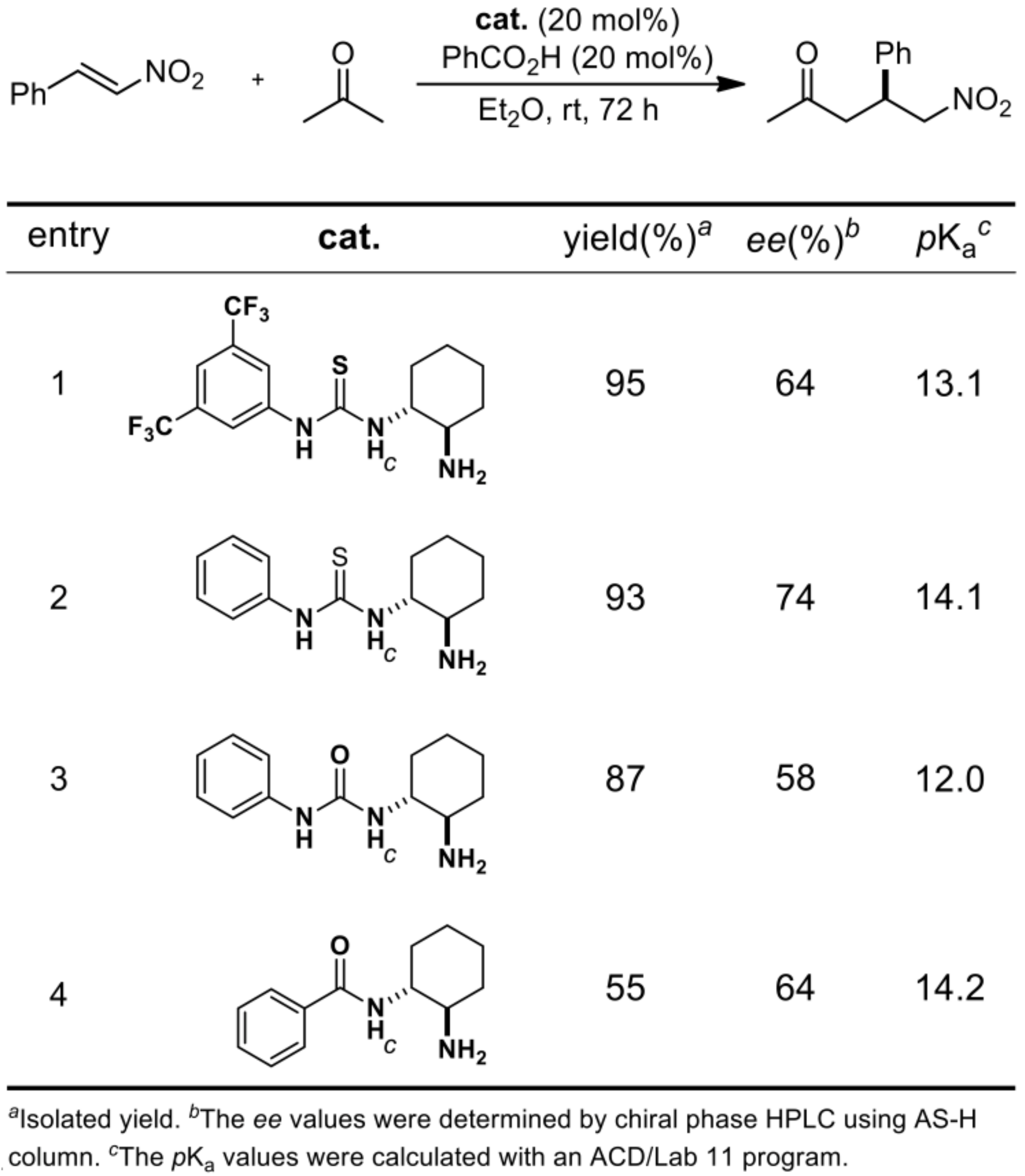
2.1.1. Catalyst Screening
2.1.2. Solvent Effects
2.1.3. Additive Effect
2.1.4. Equivalence Effect
2.1.5. Temperature and Time Effects
2.1.6. Reaction Conditions According to the Type of Aromatic Ketones
2.1.7. Reaction by Unsaturated β-Nitroalkenes
2.1.8. Reaction of Acetone with α, β-Unsaturated Nitroalkenes
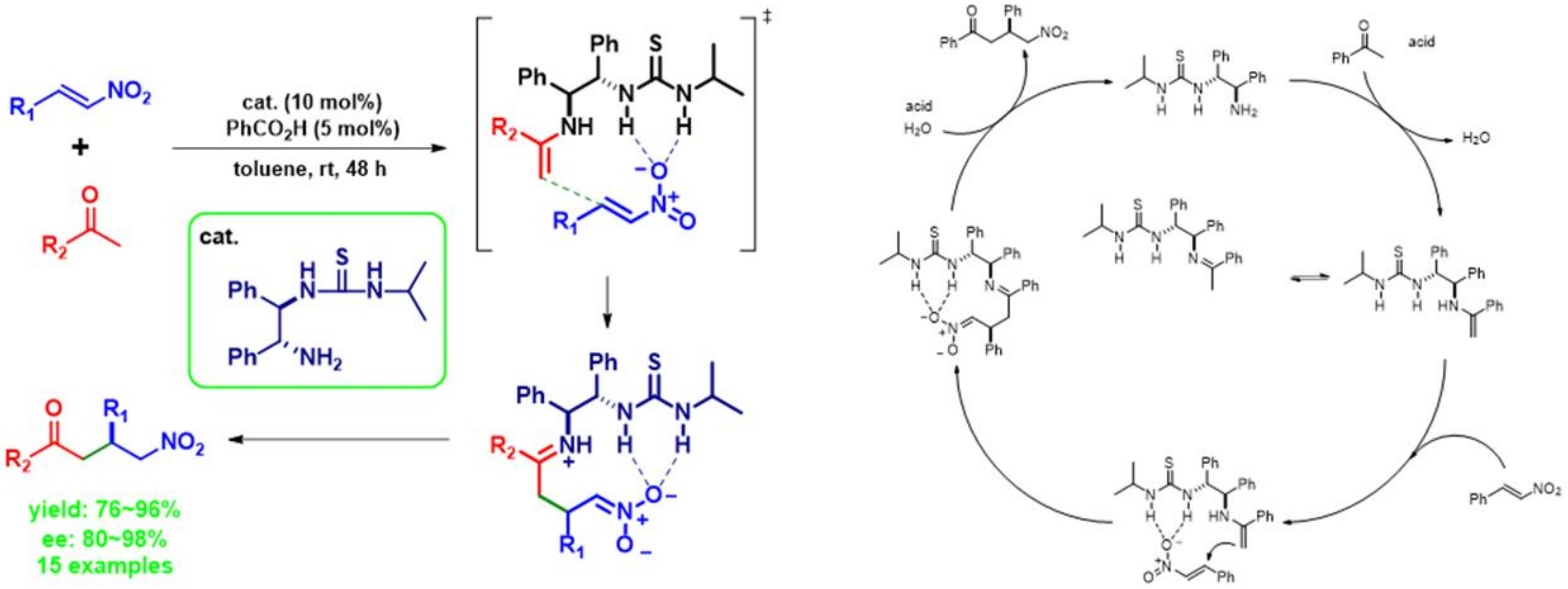
2.1.9. Reaction Mechanism to Predicted Transition State
2.1.10. Synthesis of (S)-GABA Derivatives and Pyrrolidinone Derivatives
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Catalysts (1a-n); General Procedure
3.2.1. Synthesis of Monoalkylated Thiourea Catalysts
3.2.2. Synthesis of Arylated Thiourea Catalysts
3.3. Synthesis of Product Compounds
3.3.1. General Procedure of the Asymmetric Michael Addition
3.3.2. Synthesis of (S)-Phenibut® and Baclofen®
3.4. Results of DFT Calculations and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I.; Moisan, L. In the golden age of organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Seayad, J.; List, B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org. Chem. 2002, 12, 1877–1894. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 1701–1716. [Google Scholar] [CrossRef]
- Guo, Z.-L.; Xue, J.-H.; Fu, L.-N.; Zhang, S.-E.; Guo, Q.-X. The direct asymmetric alkylation of α-amino aldehydes with 3-indolylmethanols by enamine catalysis. Org. Lett. 2014, 16, 6472–6475. [Google Scholar] [CrossRef]
- Tan, B.; Hernández-Torres, G.; Barbas, C.F. Rationally designed amide donors for organocatalytic asymmetric Michael reactions. Angew. Chem. Int. Ed. 2012, 51, 5381–5385. [Google Scholar] [CrossRef]
- Curti, C.; Rassu, G.; Zambrano, V.; Pinna, L.; Pelosi, G.; Sartori, A.; Battistini, L.; Zanardi, F.; Casiraghi, G. Bifunctional Cinchona Alkaloid/Thiourea Catalyzes Direct and Enantioselective Vinylogous Michael Addition of 3-Alkylidene Oxindoles to Nitroolefins. Angew. Chem. Int. Ed. 2012, 51, 6200–6204. [Google Scholar] [CrossRef]
- Yan, X.Y.; Huang, X.; Gou, S.; Huang, J.; Wen, Y.; Feng, X. Enantioselective Cyanosilylation of Ketones Catalyzed by a Nitrogen-Containing Bifunctional Catalyst. Adv. Synth. Catal. 2006, 348, 538–544. [Google Scholar]
- Kwiatkowski, P.; Dudzinski, K.; Łyzwa, D. Effect of high pressure on the organocatalytic asymmetric Michael reaction: Highly enantioselective synthesis of γ-nitroketones with quaternary stereogenic centers. Org. Lett. 2011, 13, 3624–3627. [Google Scholar] [CrossRef]
- Gu, Q.; You, S.L. Desymmetrization of cyclohexadienones via cinchonine derived thiourea-catalyzed enantioselective aza-Michael reaction and total synthesis of (-)-Mesembrine. Chem. Sci. 2011, 2, 1519–1522. [Google Scholar] [CrossRef]
- Corey, E.J.; Zhang, F.Y. Enantioselective Michael Addition of Nitromethane to α, β-Enones Catalyzed by Chiral Quaternary Ammonium Salts. A Simple Synthesis of (R)-Baclofen. Org. Lett. 2000, 2, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Sigman, M.S.; Jacobsen, E.N. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Hiemstra, H.; Wynberg, H. Addition of aromatic thiols to conjugated cycloalkenones, catalyzed by chiral. beta.-hydroxy amines. A mechanistic study of homogeneous catalytic asymmetric synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Dolling, U.H.; Davis, P.; Grabowski, E.J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. [Google Scholar] [CrossRef]
- Oku, J.-I.; Inoue, S. Asymmetric cyanohydrin synthesis catalysed by a synthetic cyclic dipeptide. J. Chem. Soc. Chem. Commun. 1981, 229–230. [Google Scholar] [CrossRef]
- McGilvra, J.D.; Unni, A.K.; Modi, K.; Rawal, V.H. Highly Diastereo-and Enantioselective Mukaiyama Aldol Reactions Catalyzed by Hydrogen Bonding. Angew. Chem. Int. Ed. 2006, 45, 6130–6133. [Google Scholar] [CrossRef]
- Huang, Y.; Unni, A.K.; Thadani, A.N.; Rawal, V.H. Single enantiomers from a chiral-alcohol catalyst. Nature 2003, 424, 146. [Google Scholar] [CrossRef]
- Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed. 2004, 43, 1566–1568. [Google Scholar] [CrossRef]
- Uraguchi, D.; Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jacobsen, E.N. Highly enantioselective direct conjugate addition of ketones to nitroalkenes promoted by a chiral primary amine− thiourea catalyst. J. Am. Chem. Soc. 2006, 128, 7170–7171. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cui, H.F.; Nie, J.; Dong, K.Y.; Li, X.J.; Ma, J.A. Highly enantioselective Michael addition of aromatic ketones to nitroolefins promoted by chiral bifunctional primary amine-thiourea catalysts based on saccharides. Org. Lett. 2007, 9, 923–925. [Google Scholar] [CrossRef]
- Lao, J.H.; Zhang, X.J.; Wang, J.J.; Li, X.M.; Yan, M.; Luo, H.B. The effect of hydrogen bond donors in asymmetric organocatalytic conjugate additions. Tetrahedron Asymmetry 2009, 20, 2818–2822. [Google Scholar] [CrossRef]
- Chen, J.R.; Lu, H.H.; Li, X.Y.; Cheng, L.; Wan, J.; Xiao, W. Readily Tunable and Bifunctional l-Prolinamide Derivatives: Design and Application in the Direct Enantioselective Aldol Reactions. Org. Lett. 2005, 7, 4543–4545. [Google Scholar] [CrossRef] [PubMed]
- Pansare, S.V.; Kirby, R.L. Secondary–secondary diamine catalysts for the enantioselective Michael addition of cyclic ketones to nitroalkenes. Tetrahedron 2009, 65, 4557–4561. [Google Scholar] [CrossRef]
- Sun, Z.W.; Peng, F.Z.; Li, Z.Q.; Zou, L.W.; Zhang, S.X.; Li, X.; Shao, Z.H. Enantioselective conjugate addition of both aromatic ketones and acetone to nitroolefins catalyzed by chiral primary amines bearing multiple hydrogen-bonding donors. J. Org. Chem. 2012, 77, 4103–4110. [Google Scholar] [CrossRef]
- Tsakos, M.; Kokotos, C.G.; Kokotos, G. Primary amine-thioureas with improved catalytic properties for “difficult” Michael reactions: Efficient organocatalytic syntheses of (S)-Baclofen,(R)-Baclofen and (S)-Phenibut. Adv. Synth. Catal. 2012, 354, 740–746. [Google Scholar] [CrossRef]
- Yalalov, D.A.; Tsogoeva, S.B.; Schmatz, S. Chiral thiourea-based bifunctional organocatalysts in the asymmetric nitro-Michael addition: A joint experimental-theoretical study. Adv. Synth. Catal. 2006, 348, 826–832. [Google Scholar] [CrossRef]
- Li, B.L.; Wang, Y.F.; Luo, S.P.; Zhong, A.G.; Li, Z.B.; Du, X.H.; Xu, D.Q. Enantioselective Michael addition of aromatic ketones to nitroolefins catalyzed by bifunctional thioureas and mechanistic insight. Eur. J. Org. Chem. 2010, 656–662. [Google Scholar] [CrossRef]
- Felluga, F.; Gombac, V.; Pitacco, G.; Valentin, E. A short and convenient chemoenzymatic synthesis of both enantiomers of 3-phenylGABA and 3-(4-chlorophenyl) GABA (Baclofen). Tetrahedron Asymmetry 2005, 16, 1341–1345. [Google Scholar] [CrossRef]
- Dambrova, M.; Zvejniece, L.; Liepinsh, E.; Cirule, H.; Zharkova, O.; Veinberg, G.; Kalvinsh, I. Comparative pharmacological activity of optical isomers of phenibut. Eur. J. Pharmacol. 2008, 583, 128–134. [Google Scholar] [CrossRef]
- Perfumi, M.; Santoni, M.; Ciccocioppo, R.; Massi, M. Blockade of γ-aminobutyric acid receptors does not modify the inhibition of ethanol intake induced by Hypericum perforatum in rats. Alcohol. Alcohol. 2002, 37, 540–546. [Google Scholar] [CrossRef]
- Bowery, N.G. GABAB receptor pharmacology. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 109–147. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cat. | Yield [a] | ee [b] |
|---|---|---|---|
| 1 | 1a | N.R. [c] | - |
| 2 | 1b | N.R. [c] | - |
| 3 | 1c | 73 | 98 |
| 4 | 1d | 85 | 98 |
| 5 | 1e | 76 | 90 |
| 6 | 1f | 58 | 92 |
| 7 | 1g | 46 | 90 |
| 8 | 1h | 78 | 94 |
| 9 | 1i | 60 | 94 |
| 10 | 1j | 51 | 92 |
| 11 | 1k | 53 | 90 |

| Entry | Solvent | Yield [a] | ee [b] |
|---|---|---|---|
| 1 | H2O | 63 | 88 |
| 2 | neat | 91 | 92 |
| 3 | toluene | 85 | 98 |
| 4 | ether | 80 | 98 |
| 5 | CH2Cl2 | 79 | 98 |
| 6 | Tetrahydrofuran | 74 | 94 |
| 7 | methanol | 60 | 94 |
| 8 | CH3CN | 54 | 94 |

| Entry | Additive | Yield [a] | ee [b] |
|---|---|---|---|
| 1 | - | N.R. [c] | - |
| 2 | Trifluoroacetic acid | N.R. [c] | - |
| 3 | Oxalic acid | N.R. [c] | - |
| 4 | Salicylic acid | 80 | 90 |
| 5 | Terephthalic acid | 73 | 86 |
| 6 | Benzoic acid | 85 | 98 |
| 7 | Acetic acid | 68 | 94 |
| 8 | H2O | 74 | 98 |
| 9 | H2O (1 eq.) | 70 | 98 |

| Entry | X (equiv) | Y (mol%) | Z (mol%) | Yield (%) [a] | ee (%) [b] |
|---|---|---|---|---|---|
| 1 | 2 | 10 | 15 | 85 | 98 |
| 2 | 5 | 10 | 15 | 86 | 96 |
| 3 | 2 | 5 | 15 | 51 | 98 |
| 4 | 2 | 20 | 15 | 88 | 96 |
| 5 | 2 | 10 | 5 | 87 | 98 |
| 6 | 2 | 10 | 10 | 85 | 98 |

| Entry | Temp (℃) | Time (h) | Yield (%) [a] | ee (%) [b] |
|---|---|---|---|---|
| 1 | r.t. | 24 | 55 | 98 |
| 2 | r.t. | 48 | 87 | 98 |
| 3 | r.t. | 72 | 88 | 98 |
| 4 | 40 | 48 | 92 | 86 |
| 5 | 50 | 48 | 91 | 80 |

| Entry | Ar | Product | Yield (%) [a] | ee (%) [b] |
|---|---|---|---|---|
| 1 | 4-FC6H4 | 3a | 89 | 92 |
| 2 | 2-FC6H4 | 3b | 81 | 96 |
| 3 | 4-NO2C6H4 | 3c | 85 | 94 |
| 4 | 4-MeOC6H4 | 3d | 91 | 86 |
| 5 | 2-naphthyl | 3e | 84 | 80 |

| Entry | Ar | Product | Yield (%) [a] | ee (%) [b] |
|---|---|---|---|---|
| 1 | 4-MeC6H4 | 4a | 95 | 88 |
| 2 | 4-BrC6H4 | 4b | 89 | 92 |
| 3 | 4-ClC6H4 | 4c | 83 | 92 |
| 4 | 4-MeOC6H4 | 4d | 85 | 94 |
| 5 | 2-MeOC6H4 | 4e | 76 | 82 |

| Entry | Ar | Product | Yield (%) [a] | ee (%) [b] |
|---|---|---|---|---|
| 1 | C6H5 | 5a | 88 | 94 |
| 2 | 4-MeC6H4 | 5b | 81 | 86 |
| 3 | 4-MeOC6H4 | 5c | 85 | 82 |
| 4 | 4-ClC6H4 | 5d | 76 | 86 |
| 5 | 2-MeOC6H4 | 5e | 83 | 94 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.H.; Nam, S.H.; Kim, B.-S.; Ha, D.-C. Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds. Catalysts 2020, 10, 618. https://doi.org/10.3390/catal10060618
Shim JH, Nam SH, Kim B-S, Ha D-C. Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds. Catalysts. 2020; 10(6):618. https://doi.org/10.3390/catal10060618
Chicago/Turabian StyleShim, Jae Ho, Si Hun Nam, Byeong-Seon Kim, and Deok-Chan Ha. 2020. "Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds" Catalysts 10, no. 6: 618. https://doi.org/10.3390/catal10060618
APA StyleShim, J. H., Nam, S. H., Kim, B.-S., & Ha, D.-C. (2020). Organocatalytic Asymmetric Michael Addition of Ketones to α, β-Unsaturated Nitro Compounds. Catalysts, 10(6), 618. https://doi.org/10.3390/catal10060618