Exploiting Direct Current Plasma Electrolytic Oxidation to Boost Photoelectrocatalysis


 ,
,
Abstract
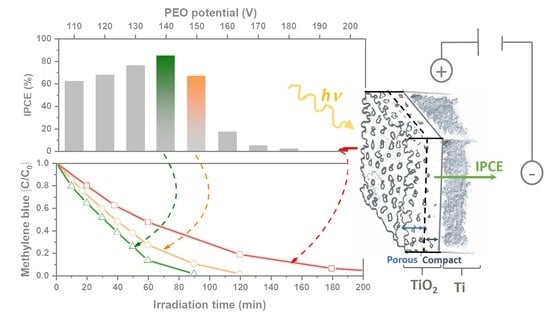
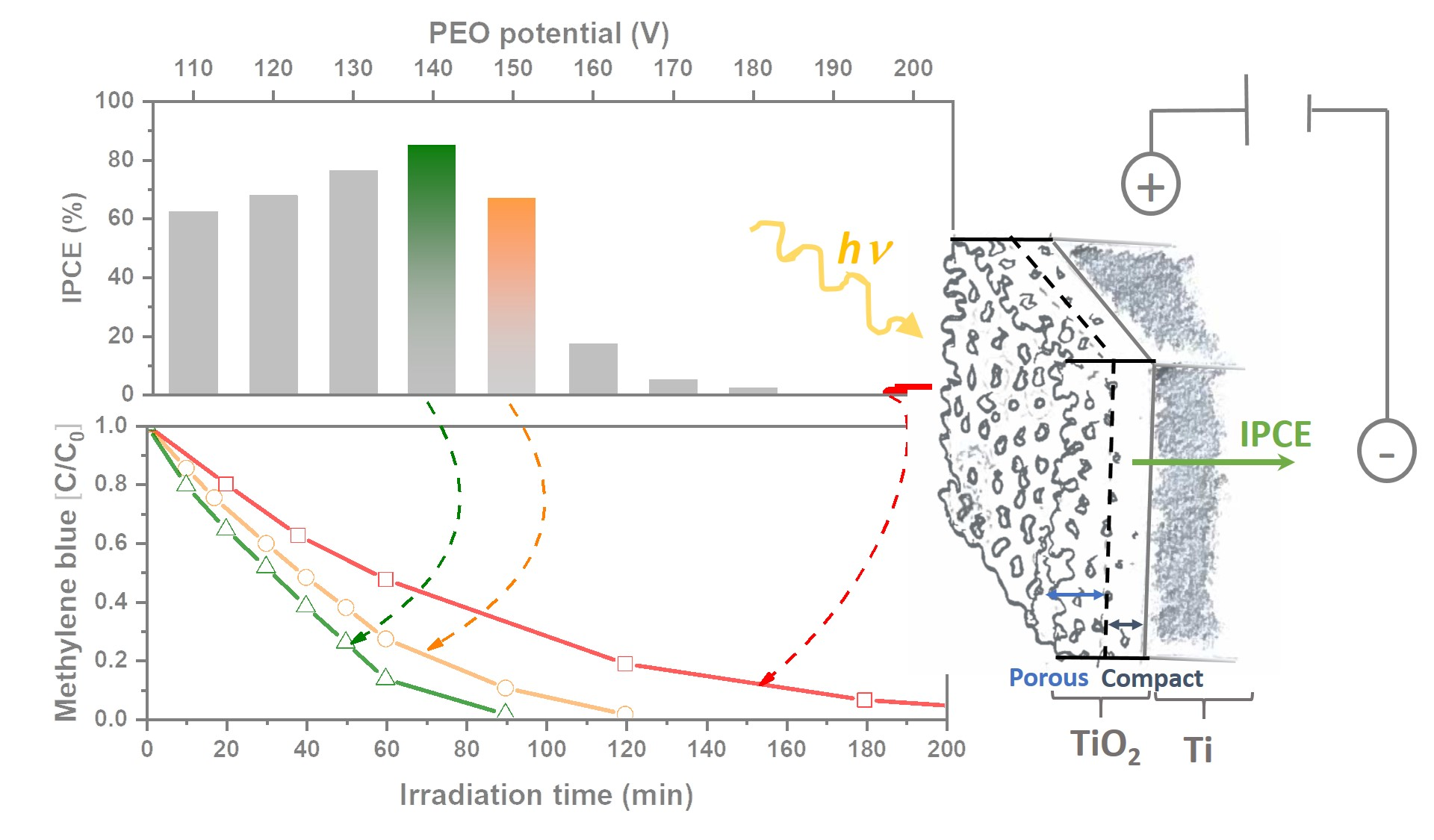{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
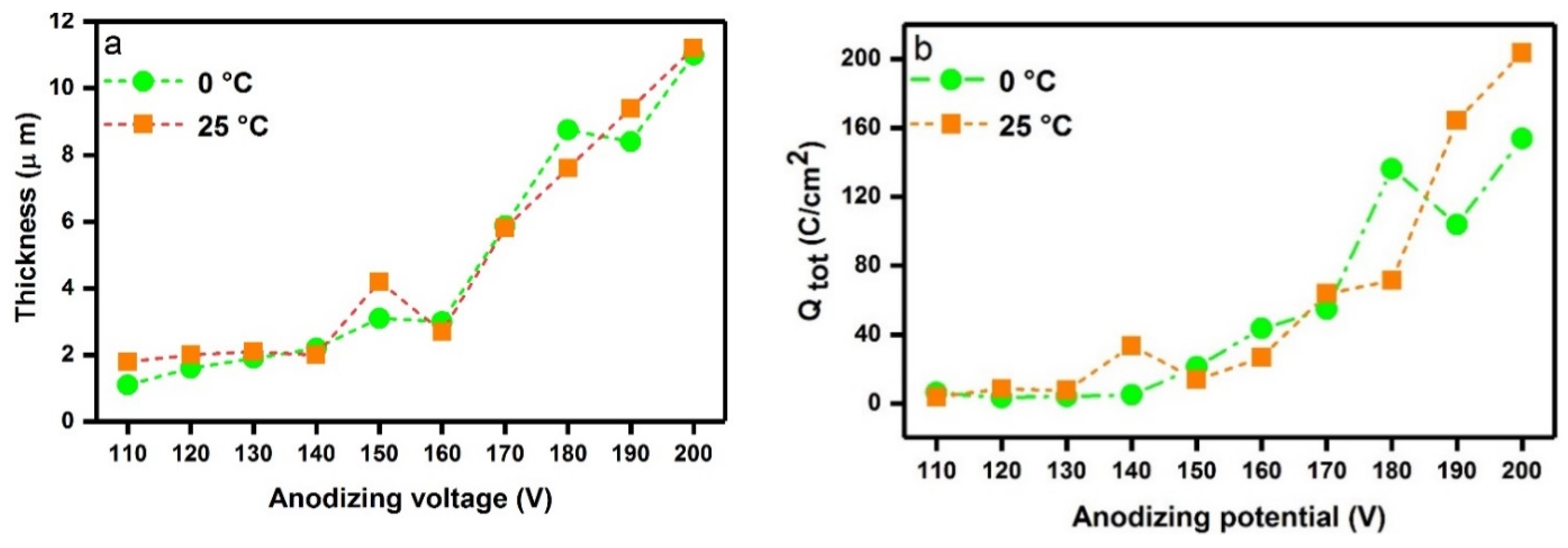
2.1. Oxide Thickness and Surface Morphology
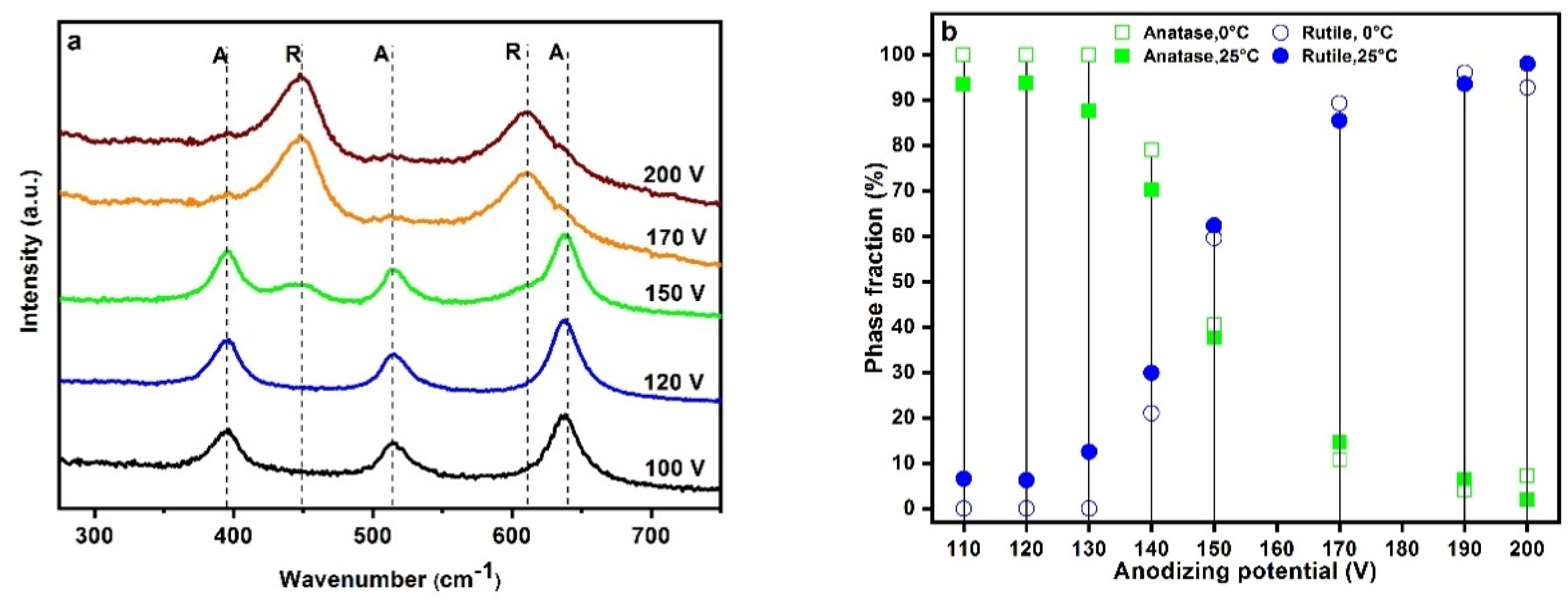
2.2. Crystal Structure
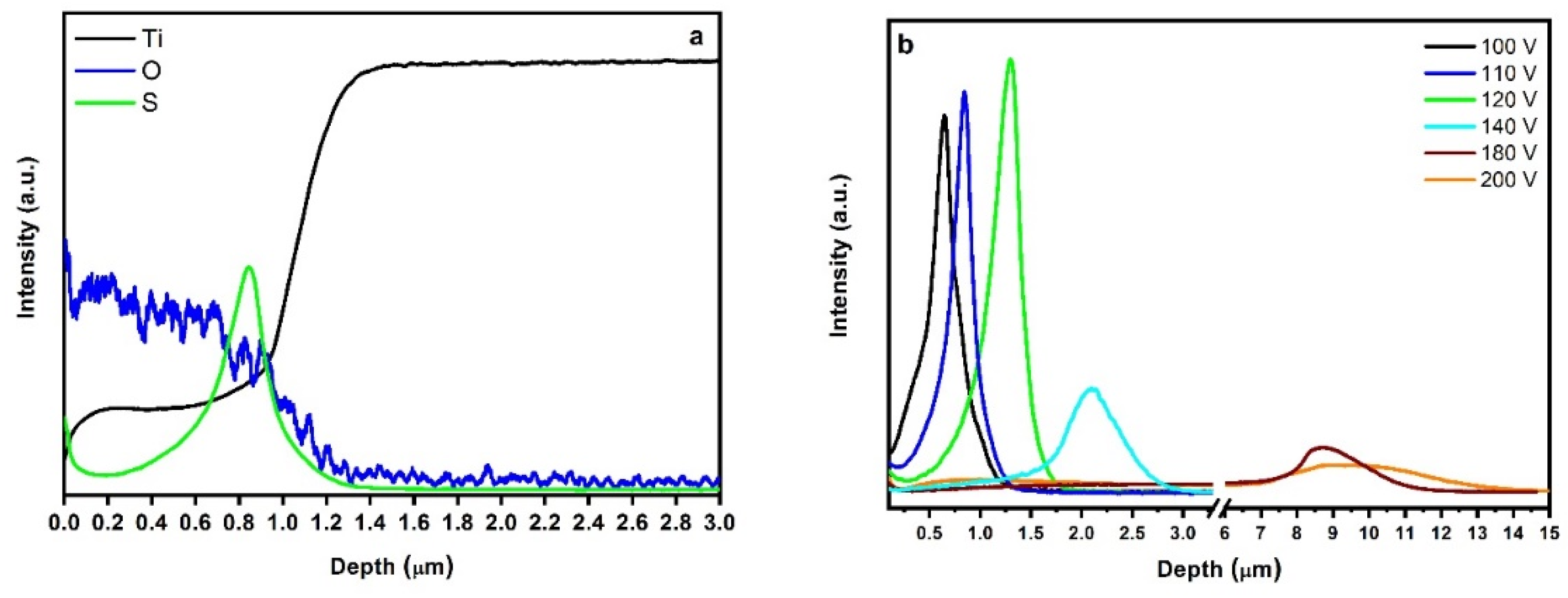
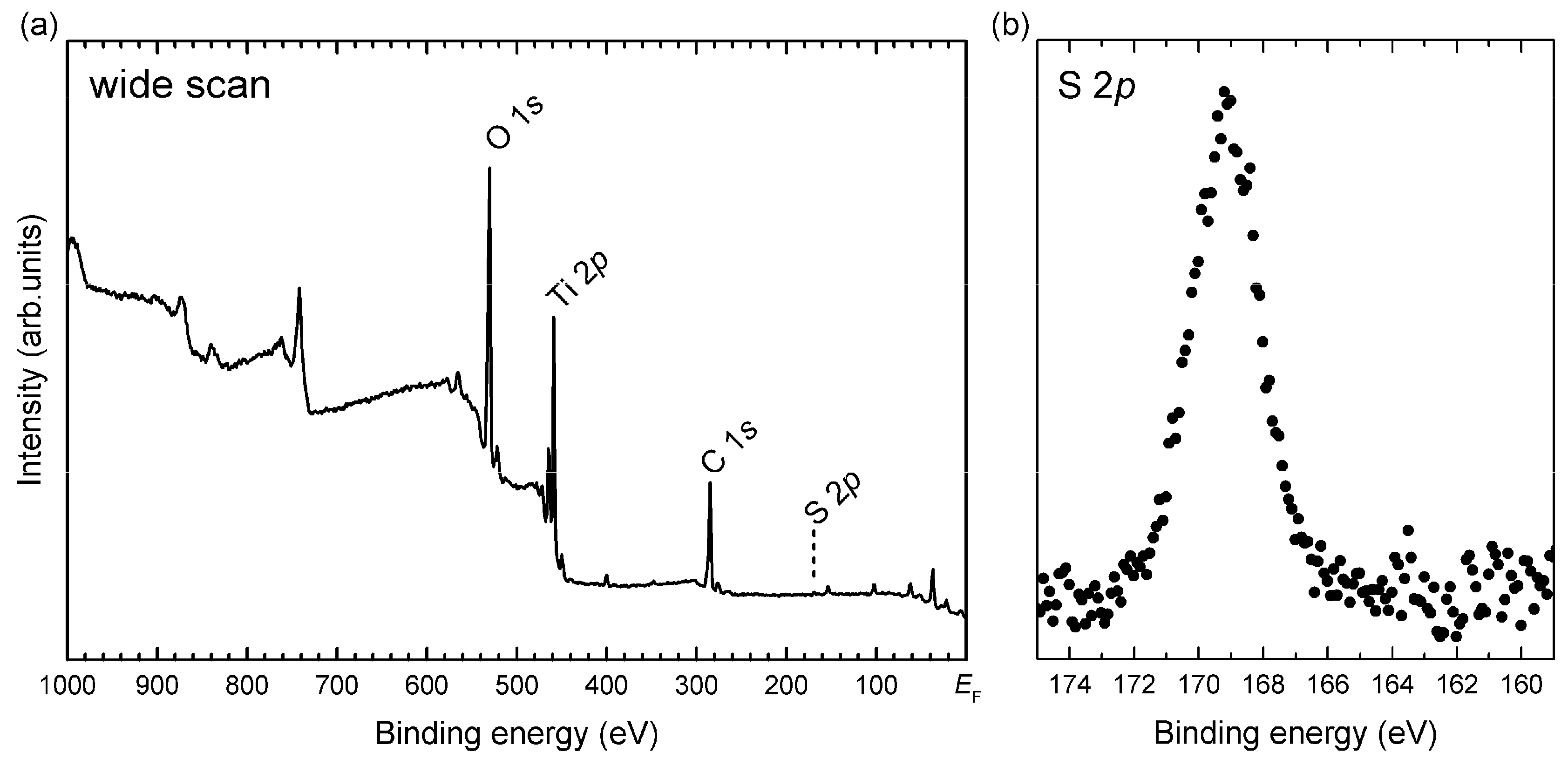
2.3. In-Depth Composition Profile
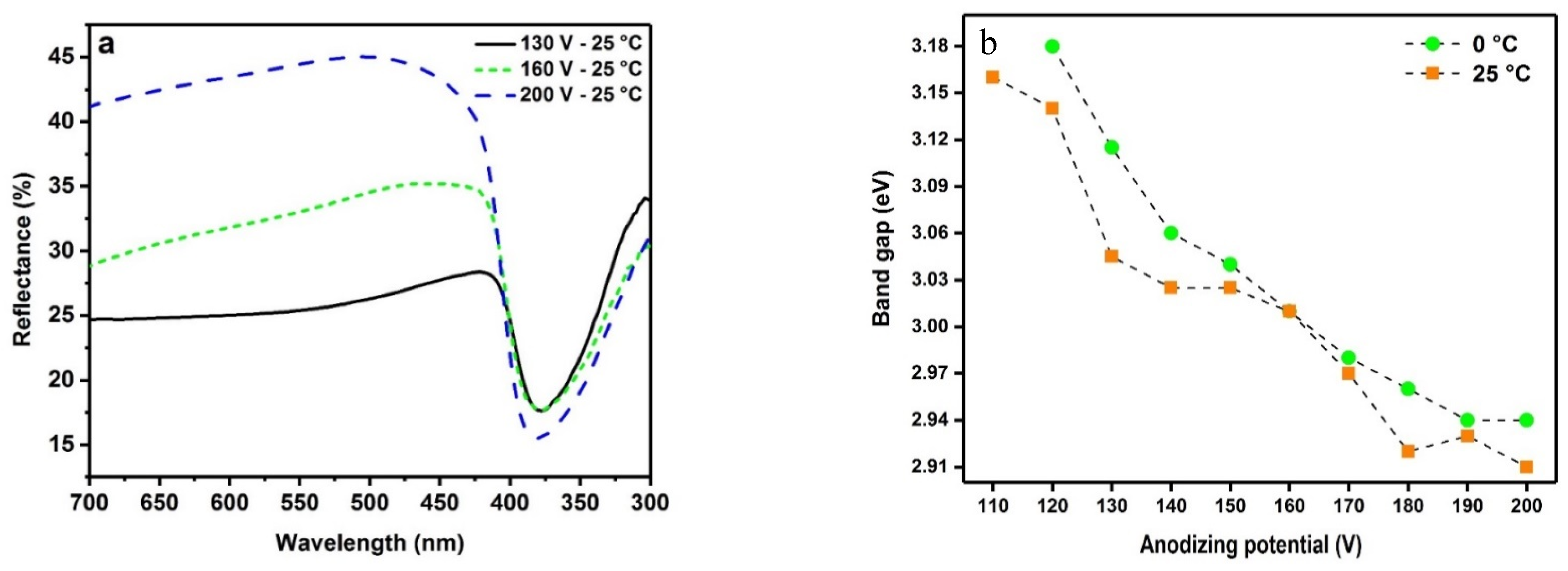
2.4. Optical Properties
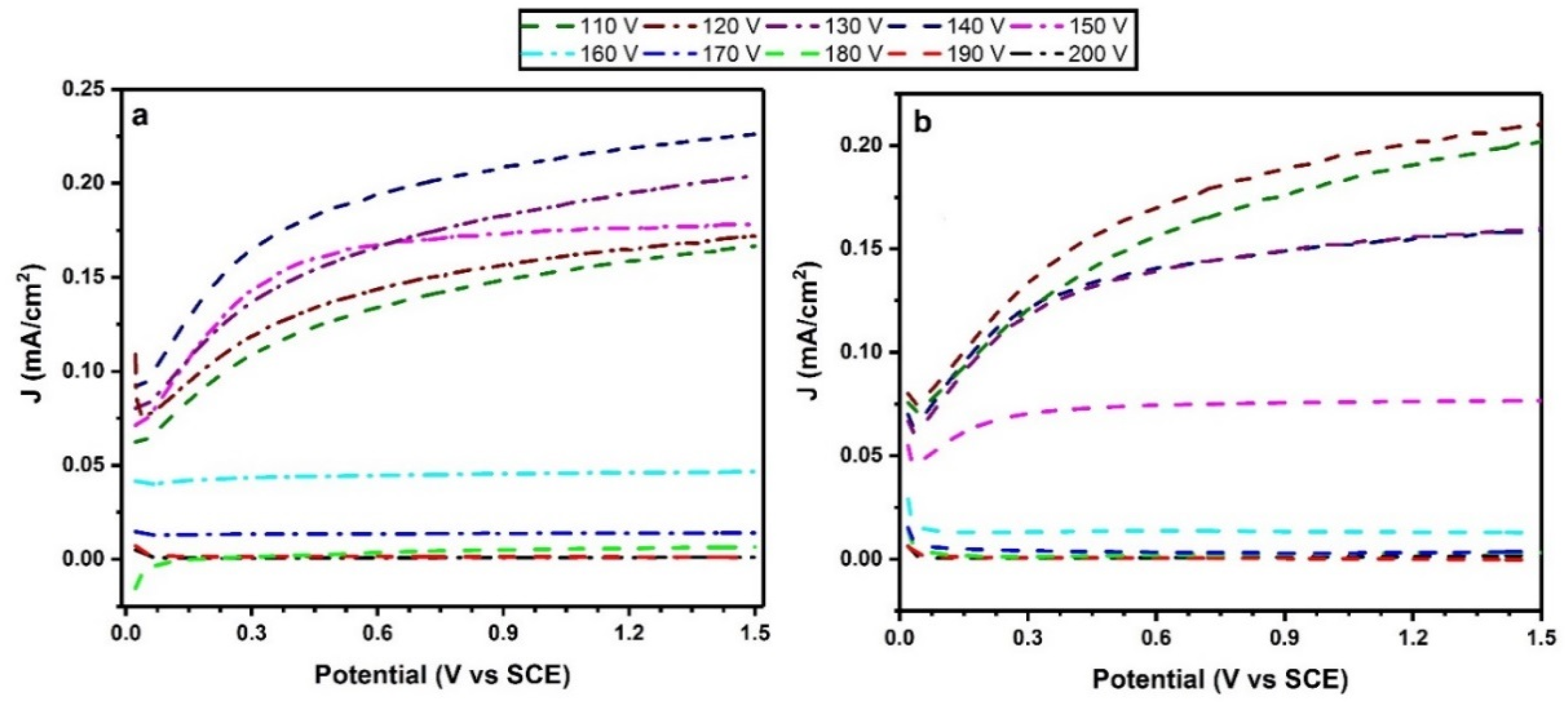
2.5. Photoelectrochemical Activity
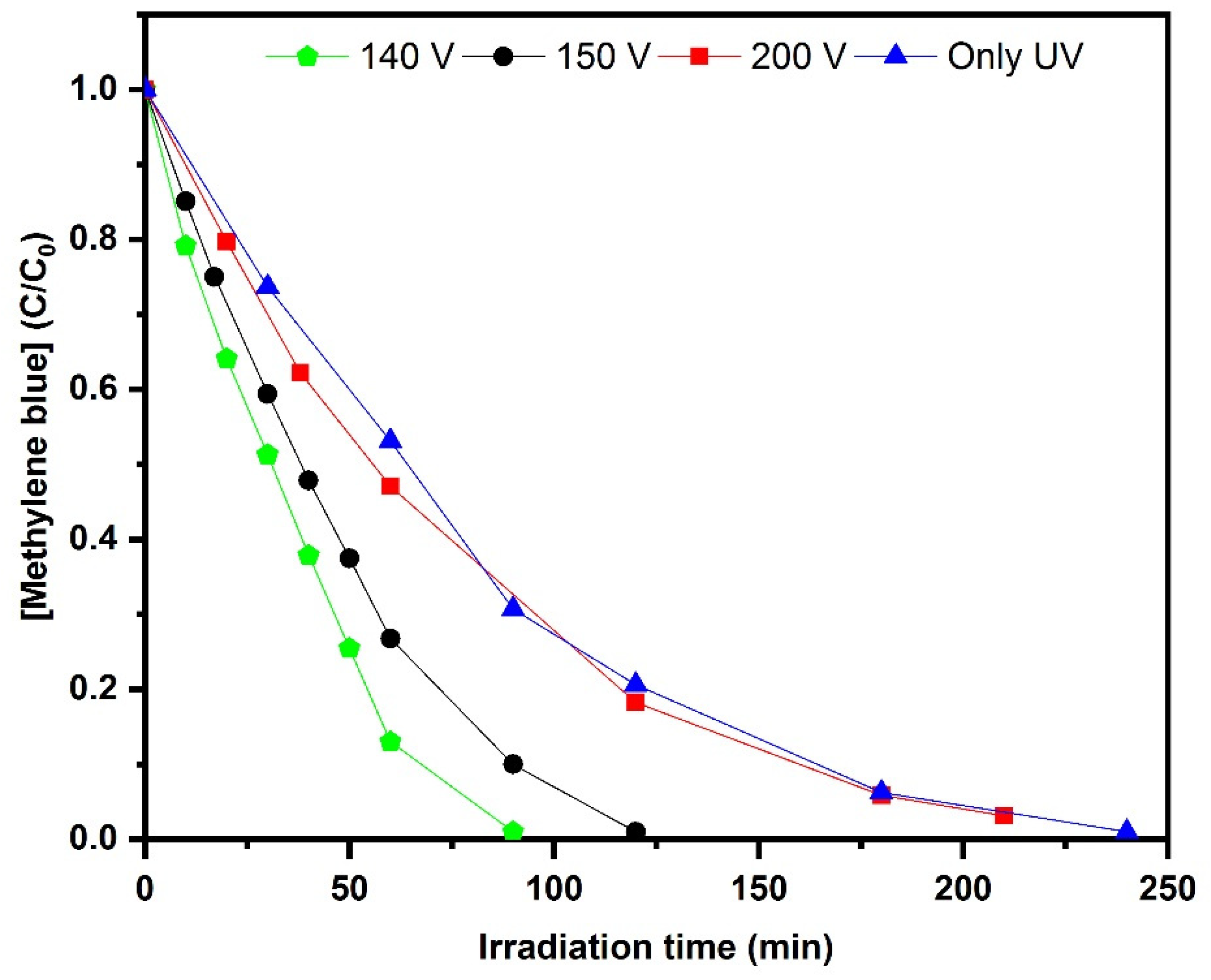
2.6. Photoelectrocatalytic Decolorization of Methylene blue
3. Materials and Methods
3.1. Synthesis of TiO2 Films
3.2. Morphologies and Crystal Structures of TiO2 Films
3.3. Photoelectrochemical Properties of TiO2 Films
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garcia-Segura, S.; Brillas, E. Applied photoelectrocatalysis on the degradation of organic pollutants in wastewaters. J. Photochem. Photobiol. C Photochem. Rev. 2017, 31, 1–35. [Google Scholar] [CrossRef]
- Turolla, A.; Fumagalli, M.; Bestetti, M.; Antonelli, M. Electrophotocatalytic decolorization of an azo dye on TiO2 self-organized nanotubes in a laboratory scale reactor. Desalination 2012, 285, 377–382. [Google Scholar] [CrossRef]
- Franz, S.; Perego, D.; Marchese, O.; Bestetti, M. Photoelectrochemical advanced oxidation processes on nanostructured TiO2 catalysts: Decolorization of a textile azo-dye. J. Water Chem. Technol. 2015, 37, 108–115. [Google Scholar] [CrossRef]
- Murgolo, S.; Franz, S.; Arab, H.; Bestetti, M.; Falletta, E.; Mascolo, G. Degradation of emerging organic pollutants in wastewater effluents by electrochemical photocatalysis on nanostructured TiO2 meshes. Water Res. 2019, 164, 114920. [Google Scholar] [CrossRef]
- Franz, S.; Falletta, E.; Arab, H.; Murgolo, S.; Bestetti, M.; Mascolo, G. Degradation of carbamazepine by photo(electro)catalysis on nanostructured TiO2 meshes: Transformation products and reaction pathways. Catalysts 2020, 10, 169. [Google Scholar] [CrossRef]
- Collivignarelli, M.C.; Abbà, A.; Carnevale Miino, M.; Arab, H.; Bestetti, M.; Franz, S. Decolorization and biodegradability of a real pharmaceutical wastewater treated by H2O2-assisted photoelectrocatalysis on TiO2 meshes. J. Hazard. Mater. 2020, 387, 121668. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef]
- Bestetti, M.; Franz, S.; Cuzzolin, M.; Arosio, P.; Cavallotti, P.L. Structure of nanotubular titanium oxide templates prepared by electrochemical anodization in H2SO4/HF solutions. Thin Solid Films 2007, 515, 5253–5258. [Google Scholar] [CrossRef]
- Eskandarloo, H.; Hashempour, M.; Vicenzo, A.; Franz, S.; Badiei, A.; Behnajady, M.A.; Bestetti, M. High-temperature stable anatase-type TiO2 nanotube arrays: A study of the structure-activity relationship. Appl. Catal. B Environ. 2016, 185, 119–132. [Google Scholar] [CrossRef]
- Yerokhin, A.L.; Nie, X.; Leyland, A.; Matthews, A.; Dowey, S.J. Plasma electrolysis for surface engineering. Surf. Coat. Technol. 1999, 122, 73–93. [Google Scholar] [CrossRef]
- Sundararajan, G.; Rama Krishna, L. Mechanisms underlying the formation of thick alumina coatings through the MAO coating technology. Surf. Coat. Technol. 2003, 167, 269–277. [Google Scholar] [CrossRef]
- Bayati, M.R.; Moshfegh, A.Z.; Golestani-Fard, F. In situ growth of vanadia-titania nano/micro-porous layers with enhanced photocatalytic performance by micro-arc oxidation. Electrochim. Acta 2010, 55, 3093–3102. [Google Scholar] [CrossRef]
- He, J.; Cai, Q.Z.; Ji, Y.G.; Luo, H.H.; Li, D.J.; Yu, B. Influence of fluorine on the structure and photocatalytic activity of TiO2 film prepared in tungstate-electrolyte via micro-arc oxidation. J. Alloys Compd. 2009, 482, 476–481. [Google Scholar] [CrossRef]
- Li, J.F.; Wan, L.; Feng, J.Y. Study on the preparation of titania films for photocatalytic application by micro-arc oxidation. Sol. Energy Mater. Sol. Cells 2006, 90, 2449–2455. [Google Scholar] [CrossRef]
- Wu, X.; Ding, X.; Qin, W.; He, W.; Jiang, Z. Enhanced photo-catalytic activity of TiO2 films with doped La prepared by micro-plasma oxidation method. J. Hazard. Mater. 2006, 137, 192–197. [Google Scholar] [CrossRef]
- Wu, X.; Wei, Q.; Zhaohua, J. Influence of Fe3+ ions on the photocatalytic activity of TiO2 films prepared by micro-plasma oxidation method. Thin Solid Films 2006, 496, 288–292. [Google Scholar] [CrossRef]
- Franz, S.; Perego, D.; Marchese, O.; Lucotti, A.; Bestetti, M. Photoactive TiO2 coatings obtained by Plasma Electrolytic Oxidation in refrigerated electrolytes. Appl. Surf. Sci. 2016, 385, 498–505. [Google Scholar] [CrossRef]
- Mirelman, L.K.; Curran, J.A.; Clyne, T.W. The production of anatase-rich photoactive coatings by plasma electrolytic oxidation. Surf. Coat. Technol. 2012, 207, 66–71. [Google Scholar] [CrossRef]
- Yerokhin, A.L.; Nie, X.; Leyland, A.; Matthews, A. Characterisation of oxide films produced by plasma electrolytic oxidation of a Ti-6Al-4V alloy. Surf. Coat. Technol. 2000, 130, 195–206. [Google Scholar] [CrossRef]
- Bayati, M.R.; Golestani-Fard, F.; Moshfegh, A.Z. Visible photodecomposition of methylene blue over micro arc oxidized WO3-loaded TiO2 nano-porous layers. Appl. Catal. A Gen. 2010, 382, 322–331. [Google Scholar] [CrossRef]
- Bayati, M.R.; Moshfegh, A.Z.; Golestani-Fard, F. On the photocatalytic activity of the sulfur doped titania nano-porous films derived via micro-arc oxidation. Appl. Catal. A Gen. 2010, 389, 60–67. [Google Scholar] [CrossRef]
- He, J.; Luo, Q.; Cai, Q.Z.; Li, X.W.; Zhang, D.Q. Microstructure and photocatalytic properties of WO3/TiO2 composite films by plasma electrolytic oxidation. Mater. Chem. Phys. 2011, 129, 242–248. [Google Scholar] [CrossRef]
- Lukiyanchuk, I.V.; Rudnev, V.S.; Tyrina, L.M. Plasma electrolytic oxide layers as promising systems for catalysis. Surf. Coat. Technol. 2016, 307, 1183–1193. [Google Scholar] [CrossRef]
- Vasilyeva, M.S.; Rudnev, V.S.; Sergeev, A.A.; Sergeeva, K.A.; Nepomnyaschiy, A.V.; Ustinov, A.Y.; Zvereva, A.A.; Kilin, K.N.; Voznesenskiy, S.S. Composition, structure, and photocatalytic properties of Fe-containing oxide layers on titanium. Prot. Met. Phys. Chem. Surfaces 2017, 53, 879–888. [Google Scholar] [CrossRef]
- Vasilić, R.; Stojadinović, S.; Radić, N.; Stefanov, P.; Dohčević-Mitrović, Z.; Grbić, B. One-step preparation and photocatalytic performance of vanadium doped TiO2 coatings. Mater. Chem. Phys. 2015, 151, 337–344. [Google Scholar] [CrossRef]
- Bessegato, G.G.; Guaraldo, T.T.; de Brito, J.F.; Brugnera, M.F.; Zanoni, M.V.B. Achievements and Trends in Photoelectrocatalysis: From Environmental to Energy Applications. Electrocatalysis 2015, 6, 415–441. [Google Scholar] [CrossRef]
- Augugliaro, V.; Camera-Roda, G.; Loddo, V.; Palmisano, G.; Palmisano, L.; Soria, J.; Yurdakal, S. Heterogeneous Photocatalysis and Photoelectrocatalysis: From Unselective Abatement of Noxious Species to Selective Production of High-Value Chemicals. J. Phys. Chem. Lett. 2015, 6, 1968–1981. [Google Scholar] [CrossRef]
- Liu, X.; Chu, P.K.; Ding, C. Surface modification of titanium, titanium alloys, and related materials for biomedical applications. Mater. Sci. Eng. R Reports 2004, 47, 49–121. [Google Scholar] [CrossRef]
- Lugovskoy, A.; Lugovskoy, S. Production of hydroxyapatite layers on the plasma electrolytically oxidized surface of titanium alloys. Mater. Sci. Eng. C 2014, 43, 527–532. [Google Scholar] [CrossRef]
- Rafieerad, A.R.; Ashra, M.R.; Mahmoodian, R.; Bushroa, A.R. Surface characterization and corrosion behavior of calcium phosphate-base composite layer on titanium and its alloys via plasma electrolytic oxidation: A review paper. Mater. Sci. Eng. C 2015, 57, 397–413. [Google Scholar] [CrossRef]
- Akatsu, T.; Yamada, Y.; Hoshikawa, Y.; Onoki, T.; Shinoda, Y.; Wakai, F. Multifunctional porous titanium oxide coating with apatite forming ability and photocatalytic activity on a titanium substrate formed by plasma electrolytic oxidation. Mater. Sci. Eng. C 2013, 33, 4871–4875. [Google Scholar] [CrossRef] [PubMed]
- Umebayashi, T.; Yamaki, T.; Yamamoto, S.; Miyashita, A.; Tanaka, S.; Sumita, T.; Asai, K. Sulfur-doping of rutile-titanium dioxide by ion implantation: Photocurrent spectroscopy and first-principles band calculation studies. J. Appl. Phys. 2003, 93, 5156–5160. [Google Scholar] [CrossRef]
- Lee, J.H.; Youn-Il, J.; Kim, Y.J.; Kim, I.K.; Jang, K.W.; Oh, H.J. Photocatalytic characteristics of boron and nitrogen doped titania film synthesized by micro-arc oxidation. Ceram. Int. 2015, 41, 11899–11907. [Google Scholar] [CrossRef]
- Yao, Z.; Jia, F.; Tian, S.; Li, C.; Jiang, Z.; Bai, X. Microporous Ni-Doped TiO2 film photocatalyst by plasma electrolytic oxidation. ACS Appl. Mater. Interfaces 2010, 2, 2617–2622. [Google Scholar] [CrossRef]
- Yao, Z.; Jia, F.; Jiang, Y.; Li, C.; Jiang, Z.; Bai, X. Photocatalytic reduction of potassium chromate by Zn-doped TiO2/Ti film catalyst. Appl. Surf. Sci. 2010, 256, 1793–1797. [Google Scholar] [CrossRef]
- Wan, L.; Li, J.; Feng, J.; Sun, W. Photocatalysts of Cr doped TiO2 film prepared by micro arc oxidation. Chin. J. Chem. Phys. 2008, 21, 487. [Google Scholar] [CrossRef]
- Stojadinović, S.; Radić, N.; Grbić, B.; Maletić, S.; Stefanov, P.; Pačevski, A.; Vasilić, R. Structural, photoluminescent and photocatalytic properties of TiO2:Eu3+ coatings formed by plasma electrolytic oxidation. Appl. Surf. Sci. 2016, 370, 218–228. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Zuliani, A.; Ceresoli, D.; Martinazzo, R.; Selli, E. Exploiting the Photonic crystal properties of TiO2 nanotube arrays to enhance photocatalytic hydrogen production. ACS Catal. 2016, 6, 1345–1353. [Google Scholar] [CrossRef]
- Wan, L.; Li, J.F.; Feng, J.Y.; Sun, W.; Mao, Z.Q. Anatase TiO2 films with 2.2 eV band gap prepared by micro-arc oxidation. Mater. Sci. Eng. B Solid State Mater. Adv. Technol. 2007, 139, 216–220. [Google Scholar] [CrossRef]
- Franz, D.S.; Perego, O.; Marchese, M.B. Photoelectrochemical AOPs Based on Nanotubular Titanium Diox-Ide, Ecotechnologies for Wastewater Treatment; ECOSTP: Karnataka, India, 2014. [Google Scholar]
- Quintero, D.; Galvis, O.; Calderón, J.A.; Castaño, J.G.; Echeverría, F. Effect of electrochemical parameters on the formation of anodic films on commercially pure titanium by plasma electrolytic oxidation. Surf. Coat. Technol. 2014, 258, 1223–1231. [Google Scholar] [CrossRef]
- Matykina, E.; Berkani, A.; Skeldon, P.; Thompson, G.E. Real-time imaging of coating growth during plasma electrolytic oxidation of titanium. Electrochim. Acta 2007, 53, 1987–1994. [Google Scholar] [CrossRef]
- Friedemann, A.E.R.; Thiel, K.; Haßlinger, U.; Ritter, M.; Gesing, T.M.; Plagemann, P. Investigations into the structure of PEO-layers for understanding of layer formation. Appl. Surf. Sci. 2018, 443, 467–474. [Google Scholar] [CrossRef]
- Hussein, R.O.; Nie, X.; Northwood, D.O.; Yerokhin, A.; Matthews, A. Spectroscopic study of electrolytic plasma and discharging behaviour during the plasma electrolytic oxidation (PEO) process. J. Phys. D Appl. Phys. 2010, 43, 105203. [Google Scholar] [CrossRef]
- Topalian, Z.; Niklasson, G.A.; Granqvist, C.G.; Österlund, L. Spectroscopic study of the photofixation of SO2 on anatase TiO2 thin films and their oleophobic properties. ACS Appl. Mater. Interfaces 2012, 4, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Ebraheem, S.; El-Saied, A. Band Gap Determination from Diffuse Reflectance Measurements of Irradiated Lead Borate Glass System Doped with TiO2 by Using Diffuse Reflectance Technique. Mater. Sci. Appl. 2013, 4, 324–329. [Google Scholar]
- Murphy, A.B. Band-gap determination from diffuse reflectance measurements of semiconductor films, and application to photoelectrochemical water-splitting. Sol. Energy Mater. Sol. Cells 2007, 91, 1326–1337. [Google Scholar] [CrossRef]
- Spurr, R.A.; Myers, H. Quantitative Analysis of Anatase-Rutile Mixtures with an X-Ray Diffractometer. Anal. Chem. 1957, 29, 760–762. [Google Scholar] [CrossRef]
- Berti, G.; Calloni, A.; Brambilla, A.; Bussetti, G.; Duò, L.; Ciccacci, F. Direct observation of spin-resolved full and empty electron states in ferromagnetic surfaces. Rev. Sci. Instrum 2014, 85, 73901. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franz, S.; Arab, H.; Lucotti, A.; Castiglioni, C.; Vicenzo, A.; Morini, F.; Bestetti, M. Exploiting Direct Current Plasma Electrolytic Oxidation to Boost Photoelectrocatalysis. Catalysts 2020, 10, 325. https://doi.org/10.3390/catal10030325
Franz S, Arab H, Lucotti A, Castiglioni C, Vicenzo A, Morini F, Bestetti M. Exploiting Direct Current Plasma Electrolytic Oxidation to Boost Photoelectrocatalysis. Catalysts. 2020; 10(3):325. https://doi.org/10.3390/catal10030325
Chicago/Turabian StyleFranz, Silvia, Hamed Arab, Andrea Lucotti, Chiara Castiglioni, Antonello Vicenzo, Federico Morini, and Massimiliano Bestetti. 2020. "Exploiting Direct Current Plasma Electrolytic Oxidation to Boost Photoelectrocatalysis" Catalysts 10, no. 3: 325. https://doi.org/10.3390/catal10030325
APA StyleFranz, S., Arab, H., Lucotti, A., Castiglioni, C., Vicenzo, A., Morini, F., & Bestetti, M. (2020). Exploiting Direct Current Plasma Electrolytic Oxidation to Boost Photoelectrocatalysis. Catalysts, 10(3), 325. https://doi.org/10.3390/catal10030325