Abstract
In this review, we discuss the use of titanium complexes bearing either bridged diphenolate or calix[n]arene (n = 4, 6, 8) ligation, in the formation of plastics from α-olefins or via the ring opening polymerization (ROP) of cyclic esters. The syntheses, molecular structures and catalytic behaviour of these systems are discussed, as well as where possible, the properties of the resultant polymers.
1. Introduction
Global demand for plastics remains high in both newly expanding as well as established economies, which stems from the favourable characteristics, including cost, associated with many petrochemical derived polymers. This availability/affordability, as well as the high ‘tune ability’ of their mechanical properties allows for incredible versatility, and as such polyolefin-based plastics are ubiquitous and play a central role in modern day society. Nevertheless, excessive and uncontrolled use of these materials for single-use applications (eg. packaging) and their still somewhat limited recyclability come at a cost to the environment. As well as changes in recent social behaviour, one possible route towards tackling the environmental issues is to limit the application of polyolefin based single-use plastics, and develop more biodegradable alternatives. Ideally, such alternative polymers should retain the favourable characteristics of known plastics, whilst not taking centuries to degrade (or readily fragmenting into microplastics). The challenge is not easy, and will require intense research efforts. One promising route to biodegradable polymers is via the ring opening polymerization (ROP) of cyclic esters. Both the polymerization of polyolefins and the ROP of cyclic esters has benefited from catalyst development and the ability of coordination chemistry to manipulate the ligands at the metal center of the catalyst to both optimize catalytic activity and to tune polymer (or oligomer) properties. Early transition metals, particularly those of group IV, have seen widespread use in the polyolefin area, and in more recent years have also been scoped for the ROP of cyclic esters. Chelating ligands can help to stabilize the catalytically active species and also offer the opportunity to bring metals into close proximity with the possibility of favourable cooperative effects. One type of chelate ligand that have seen application in a variety of catalytic processes are the diphenolates. This also led to exploration of macrocyclic phenols known as calix[n]arenes (Figure 1, right) given that these macrocycles can be visualized as larger cyclic versions of the aforementioned diphenolates (Figure 1, Type II) [1,2,3]. Such systems offer several advantages, for example the ligands employed are commercially available or can readily be synthesized in multigrams quantities. In addition, access to vast numbers of structural motifs is possible via their ready functionalization; such modifications allow for the control of parameters such as thermal stability and/or solubility. In turn, the inherent chirality of some of these ligands and, consequently, of their corresponding metal complexes can be particularly advantageous in the case of stereoselective reactions. In such a scenario, the cavity of the calix[n]arenes is ideal for selective substrate recognition [1]. All of these characteristics can be crucial in terms of catalysis. This review focuses on the use of both titanium diphenolates and titanocalix[n]arenes, the first examples of the latter were characterized back in the 1980s [4]. For the sake of clarity, titanium complexes bearing ligands derived from type I phenols (Figure 1, left) are not discussed here, since they have been extensively treated elsewhere [5].

Figure 1.
Phenol-based pro-ligands [1,2,3,4,5].
2. Olefin Polymerization
2.1. Titanium-Diphenolate Complexes
In 1989, Kakugo et al. reported the synthesis of titanium complexes (1 and 2, Figure 2) bearing a sulfur-bridged diphenolate ligand [6]. These compounds, activated by methylaluminoxane (MAO, Scheme 1) where tested in the polymerization of ethylene and of propylene (Table 1).

Figure 2.
Titanium complexes bearing sulfur- and methylene-bridged diphenolates [6,7].

Scheme 1.
Simplified mechanism for the Ti-catalyzed olefin polymerization.

Table 1.
Ethylene and propylene polymerization catalyzed by 1 and 2/MAO systems [6].
The chloride derivative 1 proved to be slightly better performing that its alkoxide congener 2 in both transformations. In fact, a slightly higher activity was exhibited by the former in ethylene polymerization (228 and 188 for 1 and 2, respectively, Table 1, entries 1 and 2) while an important increase (ca. 2-fold) was observed in the reaction involving propylene (Table 1, entries 3 and 4). High molecular weights and narrow polydispersities were achieved in all cases. Notably, both complexes were shown to be more active than [Ti(OiPr)4] (Table 1, entry 5). In terms of microstructure, 13C NMR spectroscopic analyses on the PP obtained in the presence of complex 1/MAO systems highlighted poor regio- and stereoregularity.
Subsequently, complex 1 along with its benzyl derivative 3 and their methylene-bridged analogues 4 and 5 activated by MAO were employed in the polymerization of ethylene by Orpen et al. (Table 2) [7]. Also in this case, complex 1 proved to be more active than the benzyl complex 3. However, higher molecular weight and better control were achieved with the latter catalyst (Table 2, entries 1 and 2). The rather broad polydispersities suggested the formation of more than one active species when MAO is used as co-catalyst. Better performance of the chloride pre-catalyst over the benzyl congener was observed also in the case of complexes 4 and 5 (Table 2, entries 3 and 4). Interestingly, sulfur-bridged compounds were shown to be more active than their methylene-bridged analogues (cf. Table 2 entry 1 vs. entry 3). Complexes 1 and 3 were also tested for the polymerization of 1-hexene. With both systems, atactic but regioregular poly-1-hexene was obtained. The polymers isolated exhibited high molecular weights and narrow polydispersities (7800 vs. 19,000 Da and 1.78 vs. 1.71 for 1 and 3, respectively). Eventually, the polymerization of 1,3-butadiene was achieved in the presence of the 1/MAO system. The microstructure of the polymer was found to be 65% 1,4-cis, 20% 1,4-trans and 15% 1,2-vinyl, as determined by 13C-NMR spectroscopic analyses. Interestingly, both 1 and 3 were found to be inactive when EtAlCl2 was used as co-catalyst instead of MAO.
Table 2.
Ethylene polymerization catalyzed by 1, 3, 4 and 5/MAO systems [7].
In order to further investigate the effect of the bridging unit on the efficiency of the catalyst, some tellurium-bridged diphenols and their corresponding titanium complexes were synthesized by Nakamure et al. (Figure 3 and Figure 4) [8]. All complexes activated by MAO were tested in the polymerization of ethylene (Table 3).

Figure 3.
Tellurium-bridged diphenolate titanium complexes [8].

Figure 4.
Molecular structures of complexes 6 (left), 8 (center) and 10 (right) [8].
Table 3.
Ethylene polymerization catalyzed by 1, 4, 6 -10/MAO systems [8].
The chloride complex 6 bearing bulky substituents proved to be very active (Table 3, entry 1). Interestingly, its alkoxide analogue 8 was ca. 5-times less performing (Table 3, entry 5), whilst the less sterically hindered compound 7 was almost inactive (Table 3, entry 4). Notably, good activity was exhibited by the Cp-containing complex 9, while its Cp* analogue 10 was shown to be less performing (130 and 2.3 kgPE × molTi−1 × atm−1 × h−1, Table 3, entries 6 and 7 for 9 and 10, respectively). Such different reactivity was explained by considering the steric bulk around the Ti center exerted by the Cp* ligand. Complexes 1 and 4 were also tested under the same reaction conditions (Table 3, entries 8 and 9, respectively). Complex 4 was ca. two orders of magnitude less performing than 1. Notably, the activity of the sulfur-containing catalyst was three times higher than that of the tellurium-based complex. This evidence was inconsistent with theoretical studies previously performed in order to predict the activity of these systems [9]. This was explained in terms of stability of the catalytically active species. In fact, the 6/MAO system was shown to completely deactivate within 1 h, probably due to Al-Te exchange reactions. The poor performance of catalyst 4 was explained considering the higher flexibility of the sulfur and tellurium-containing ligands over the more rigid methylene-bridged congener.
Subsequently, Trivedi et al. reported the synthesis of titanium-isopropoxide complexes bearing various substituted methylene-bridged diphenols- (11–13) and binaphthols (14–15) (Figure 5) [10,11]. In a preliminary investigation, all complexes activated by ethylaluminum sequichloride (EASC) were tested in the polymerization of ethylene (Table 4). All systems proved to be active, following the trend 13 > 15 > 11 > 14 > 12. Low molecular weight waxes were obtained in all cases, with Mw values spanning from 770 to 3400 Da. Good control was observed in all cases, with polydispersity values found in the range 1.3-1.9. Notably, under the same reaction conditions the metallocene catalyst Cp2TiCl2 was found to be inactive.
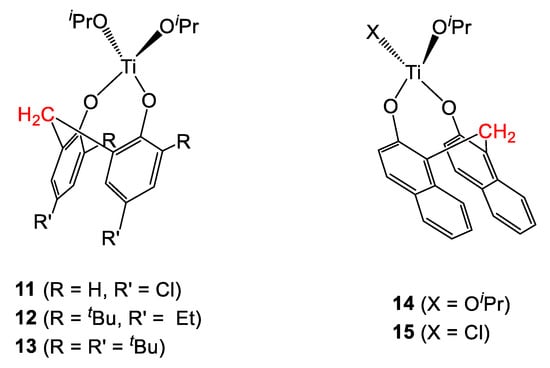
Figure 5.
Titanium complexes bearing methylene-bridged diphenol and binaphthol derived ligands [10,11].
Table 4.
Ethylene polymerization catalyzed by 11-15/EASC systems [10,11].
Furthermore, the effect of the reaction conditions on the performance of complex 15 in the polymerization of ethylene was investigated (Table 5) [11]. A drop of activity was observed when EASC was replaced by other Al-based co-catalysts (Table 5, entries 1–5) Lower activities were also observed by reducing the temperature (Table 5, entries 6 and 7). On the other hand, a significant improvement of productivity was achieved by increasing the monomer pressure (Table 5, entries 8 and 9). The nature of the reaction medium was also considered, and the catalytic system was inactive in the case of the reaction carried out in hexane (Table 5, entry 10), while enhanced activity was observed when the standard solvent (toluene) was replaced by chlorobenzene (Table 5, entry 11).
Table 5.
Effect of the reaction conditions on the ethylene polymerization promoted by 15 [11].
In all cases, low molecular weights and narrow polydispersities were observed (900–3200 Da and 1.3–1.7, respectively). Melting temperatures lower than that observed for HDPE and LDPE were detected (111–120 °C). Noteworthy, all polymers exhibited a degree of crystallinity in the range of 80–88%, as determined by X-ray diffraction analyses.
2.2. Titanocalix[n]arene Complexes
The first reports of the use of titanocalix[n]arenes for the polymerization of α-olefins dates back to 1999, when Ladipo et al. synthesized novel titanium-based complexes 16–22 bearing proximally bridged p-tert-butylcalix[4]arene ligands (Figure 6) [12]. When activated by MAO, these compounds exhibited moderated activity in the polymerization of ethylene (Table 6), but were inactive towards the polymerization of higher olefins such as propylene and 1-hexene.

Figure 6.
Titanium-calix[4]arene complexes disclosed by Ladipo et al. [12].
Table 6.
Polymerization of ethylene catalyzed by 16–22/MAO systems [12].
Frediani, Sémeril et al. employed the 1,3-di-n-propyloxycalix[4]arene complex 23 (Figure 7 and Figure 8), activated by MAO, to afford ultrahigh molecular weight polyethylene [13]. The influence of the temperature as well as the [ethylene]/[Ti] ratio on the productivity of the polymerization were studied (Table 7). Indeed, an improvement of catalytic activity was obtained by either increasing the temperature (Table 7, entries 1–6) or by employing higher monomer concentrations (Table 7, entries 7–9).

Figure 7.
Proximal and distal titanocalix[4]arene complexes [13,14,15].

Figure 8.
Molecular structure of 23 [13].
Table 7.
Polymerization of ethylene catalyzed by 23/MAO [13].
1H-NMR spectroscopic studies identified the active species as a titanium methyl cation. A similar species was observed by Proto et al. in their study concerning the polymerization of ethylene catalyzed by 24/MAO systems [14].
Subsequently, Taoufik, Bonnamour et al. extended these studies and investigated the effect of varying the 1,3-dialkyloxy R groups (for R = methyl, ethyl, n-propyl and i-butyl) on the catalytic activity of complexes 24–27 (Figure 9) during ethylene polymerization [15]. In the same study, a couple of 1,2-dialkoxycalix[4]arene derivatives were also studied such as the previously encountered 19 containing a chelating siloxide SiMe2 and the methyloxy 28. Depleted calix[4]arene complexes 29-31 bearing 1,2- or 1,3-titanium dichloride motifs were also investigated (Table 8). Whilst the behavior of the 1,2- and 1,3-systems was different, it was determined that within each series increasing the number of alkyloxy groups present was detrimental to the catalytic activity. Moderate activity was exhibited by complex 19 containing the chelating 1,2-siloxide moiety (Table 8, entry 1). Interestingly, low productivity values spanning from 14 to 21 kgPE. molcat-1 h−1, were achieved in the presence of complexes 24–27, regardless of the chain length of the 1,3-alkoxide substituents (Table 3, entries 2–5). The highest productivity was obtained in the presence of the 1,2-depleted complex 29 (Table 8, entries 7) while significantly lower activity was observed in the case of its 1,3-analogue 30 (Table 8, entry 8). In all cases, high number average molecular weights were determined. Noteworthy, the highest values (spanning from 2.3 to 3.2 × 106 g mol−1) were observed for the PEs synthesized in the presence of complexes 24–27. These numbers were found to be consistent with the range observed for Ultra High Molecular Weight (UHMW) PE. On the other hand, lower Mn values consistent with High Density (HD) PE were achieved in the presence of the depleted complexes 19 and 29 (1.7 and 1.4 × 106 g mol−1, respectively).

Figure 9.
Molecular structures of complexes 25 (left), 26 (center) and 27 (right) [15].
Table 8.
Polymerization of ethylene catalysed by 19- and 24–30/MAO systems [15].
In order to explain such differences, it was proposed that the increased protection around the metal center in complexes 24–27 would limit chain transfer reactions, enabling the growth of longer PE chains. Such a statement was supported by the trend of the molecular weight distributions. In fact, broad polydispersities were observed in the case of less hindered complexes 19 and 29 (4.4 and 5.4, respectively), while narrower values (spanning from 2.7 to 3.0) were achieved in the case of the more protected congeners 24–27.
A number of other less well-defined titanium calix[n]arenes have also been employed for ethylene polymerization [16,17,18,19]. In this regard, Li et al. reported the synthesis of titanocalixarene complexes by treating at −40 °C in dichloromethane p-tert-butylcalix[n]arene (n = 4, 6 and 8) with different amount of [TiCl4] (Scheme 2) [16] The formation of Ti-O bonds was determined via 1H- NMR spectroscopy, by monitoring the disappearance of the diagnostic OH signal normally observed at low field. Elemental analyses performed on each sample suggested the formation of di-, tri- and tetranuclear complexes for 32, 33 and 34, respectively.

Scheme 2.
Synthesis of multinuclear titanocalix[n]arene (n = 4, 6, 8) complexes [16].
Such complexes, activated by MAO, were employed as catalysts in the polymerization of ethylene (Table 9). All catalysts exhibited moderate activity affording high molecular weight PEs (1.2 – 5.6 × 106 g mol−1). The trend of productivity was found to be 32 < 33 < 34 suggesting that larger rings allow for a better Ti-monomer interaction. No influence of calixarene size was observed on the molecular weights of the polymers synthesized.
Table 9.
Polymerization of ethylene catalyzed by 32–34/MAO systems [16].
The polymerization of styrene was also investigated (Table 10). The activity of the catalytic systems was found to be the same as observed for ethylene polymerization. Interestingly, high syndiotacticity was achieved in the presence of 32 and 33 (Table 10, entries 1 and 2, 90.6 and 92.3%, respectively), while atactic PS was isolated by using the larger analogue 34 (Table 10, entry 3).
Table 10.
Polymerization of styrene catalyzed by 32–34/MAO systems [16].
Subsequently, Chen et al. studied in detail the effect of the reaction conditions on ethylene polymerization catalysed by 32/AlR3 systems (Table 11) [17]. By using Al(iBu)3 as co-catalyst, a positive effect on the productivity of the polymerization was observed by progressively increasing the Al/Ti ratio (Table 11, entries 1–4). Indeed, no conversion was obtained with an Al/Ti ratio of 10, while moderate activity was achieved by slightly increasing the amount of co-catalyst (Table 11, entries 1 and 2). Double the amount of polymer was collected by moving from an Al/Ti ratio of 15 to 20 (Table 11, entry 3), while only a slight improvement was achieved by doubling the ratio (Table 11, entry 4). A progressive drop of the polymer melting point was detected when increasing the amount of co-catalyst, possibly due to chain-transfer processes. The productivity of the process was shown to be dependent also on the polymerization temperature. Indeed, when performing the reaction at 35 or 60 °C good activities were observed (Table 11, entries 7 and 8). On the other hand, at 0 and 100 °C the catalyst exhibited only moderate activity (Table 11, entries 5 and 9). This was explained assuming the incomplete activation of the catalyst at low temperature and partial decomposition of the catalytic species under more forcing conditions. This assumption was confirmed by prolonging the reaction time (4 h instead of 1 h). In fact, while the amount of collected polymer doubled in the case of the reaction performed at 0 °C, no improvement was observed in the test carried out at higher temperature (Table 11, entries 6 and 10). Finally, the effect of different Al-alkyl co-activators was studied (Table 11, entries 11–14). Interestingly, the efficiency of the process was shown to follow the trend: AlEt3 > Al(iBu)3 > AlEt2Cl> Al(iBu)2Cl.
Table 11.
Effect of the reaction conditions on the ethylene polymerization catalyzed by 32/AlR3 systems [17].
Examples of ethylene polymerization catalyzed by titanocalix[n]arene complexes are also found in the patent literature [18,19]. Diaz-Barrios et al. patented the preparation of an additive by treating p-tert-butylcalix[8]arene with a hundred-fold excess of trimethylaluminum. This species was then reacted with [TiCl4] in the presence of magnesium dichloride affording a supported catalyst with a titanium content of 7.7%. Such a compound was further employed in the polymerization of ethylene affording a polymer with a melt flow index of 0.76 g/10 min and bulk density of 0.27 g/mL.
Titanium-based complexes bearing thiacalix[4]arenes are less explored than their conventional calix[4]arenes counterparts. In 2002, Morohashi, Miyano et al. disclosed the synthesis of the first dinuclear titanium-thiacalix[4]arene complex (Scheme 3) [20].

Scheme 3.
Synthesis of Ti-thiacalix[4]arene complexes 36 and 37 [20].
By treating p-tert-butylthiacalix[4]arene 35 with [TiCl4], the cone- and the 1,2-semi-alternate complexes 36 (Figure 10) and 37 were obtained. The isolation of the two species was possible due to their differing solubility in dichloromethane. Lately, Capacchione et al. investigated the efficiency of these compounds, activated by MAO, in the polymerization of ethylene (Table 12) [21]. Both complexes exhibited a maximum of activity at 25 °C (Table 12, entries 2 and 8, for 36 and 37, respectively, while a drop of productivity was observed at both lower (0 °C) and higher (50 °C) temperatures. This was ascribed to the incomplete activation of the catalyst in the former case and to its partial decomposition in the latter. Complex 37 exhibited significant lower activity compared to the cone isomer, probably due to the steric effect exerted by the p-tert-butyl substituent of the calixarene moiety. High melting points, spanning from 133 to 143 °C, were observed in all cases, suggesting the production of highly linear PEs. This assumption was also confirmed by 13C-NMR spectroscopy.

Figure 10.
Molecular structure of 36 [21].
Table 12.
Polymerization of ethylene catalysed by 36 and 37/MAO systems [21].
3. Ring Opening Polymerization (ROP)
3.1. Ti-diphenolate complexes
In the early 2000s, Takeuchi et al. investigated the ring opening polymerization (ROP) of ε-caprolactone (ε-CL) and other oxocyclic monomers, namely propylene oxide (PO) and oxetane (OX), catalyzed by complexes 38–43 (Figure 11) [22,23]. The isopropoxy adducts 39 and 42 proved to be active in the ROP of ε-CL, affording high molecular weight polymers with good control. The degree of polymerization was found to be ca. half of the expected value, suggesting the growth of two polymer chains for each Ti center. Neither of the two catalysts was active in the ROP of propylene oxide. Similarly, no polymerization was observed in the presence of the more Lewis acidic chloro-derivatives 38 and 41. However, 1H-NMR spectroscopy analysis on the crude reaction mixture suggested the presence of the ring opening product 1-chloro-2-propanol. This was thought to proceed from the formation of a chlorotitanium monoalcoholate species followed by its protonolysis by the water present in the deuterated solvent. [22] Subsequently, the same group disclosed the synthesis of ε-CL/OX (X = Cl or iPr) block copolymers catalyzed by the chlorotitanium monotriflate complexes 40 and 43. [23] In this scenario, the cationic polymerization of OX is followed, after quenching of the electrophilic growing end, by the anionic ROP of ε-caprolactone (Scheme 4).

Figure 11.
Diphenolate complexes employed in ROP of oxocyclic monomers [22,23].

Scheme 4.
Block polymerization of oxetane and ε-caprolactone catalyzed by 40 and 43 [23].
Based on the promising results achieved in the polymerization of ethylene [6,7,8], Harada et al. investigated the use of complexes 1, 5, 6, 8 and 10 in the ROP of ε-CL (Table 13) with a view to obtaining a biodegradable polymer [24]. The compound bearing the sulfur-bridged ligand (1) proved to be active both in the presence and in the absence of solvent (Table 13, entries 1–3). In fact, almost complete conversion was achieved in 6 h in all cases. The highest Mn was obtained in toluene, while the lowest in anisole; the control of the process was shown to follow the opposite trend. Under solvent-free conditions, the Te-bridged catalysts 6 exhibited low activity. In fact, only moderate conversion was achieved after 12 h at 100 °C (Table 13, entry 4). By running the reaction in toluene at 50 °C, no reaction was observed, while 88% conversion was achieved by increasing the temperature (Table 13, entry 5 and 6, respectively). Similar outcomes were obtained by replacing toluene with anisole or dioxane in the presence of a lower catalyst loading (Table 13, entries 7 and 8). Similarly to the previous case, the highest Mn was observed in the reaction in toluene, while the polydispersities were found in a narrow range (1.06–1.20). The isopropoxy analogue of catalyst 6 (namely 8) was also shown to promote the reaction albeit requiring longer reaction times (12 h instead of 6 h, Table 13, entry 9). The Cp- and Cp* analogues 9 and 10 were inactive (Table 13, entries 10 and 11). The Te-bridged complexes were also tested in the ROP of δ-valerolactone (δ-VL). Moderate conversions were achieved in the presence of complex 6 both in toluene and under solvent-free conditions (Table 13, entries 12 and 13), while its congener 8 was shown to be active only in the absence of solvent (Table 13, entries 14 and 15). Catalyst 6 was inactive in the ROP of β-propiolactone (β-PL) (Table 13, entry 16), while exhibiting interesting activity in the polymerization of L-lactide (L-LA). In fact, almost complete conversion was achieved after 48 at 100 °C in anisole, providing a polymer with narrow polydispersity (Table 13, entry 18). The nature of solvent was shown to influence also the structure of the isolated polymers. In fact, the formation of cyclic oligomers arising from backbiting processes was observed in the tests performed in toluene. On the other hand, linear polymers were obtained when anisole and dioxane were employed, suggesting the living nature of the process occurring in such reaction media. This was confirmed by performing a diblock copolymerization of L-LA and ε-CL in the presence of 6 in anisole. The pre-polymerization of L-LA was first carried out, followed by progressive addition of the co-monomer. GPC analyses highlighted the linear increase of the copolymer Mn consistent with the amount of co-monomer employed.
Table 13.
ROP of cyclic esters catalyzed by titanium-diphenolate complexes [24].
Lately, the same group disclosed the synthesis and catalytic activity of diethylamido complexes bearing sulfur- or methylene-bridged ligands 44 and 45 (Figure 12) [25]. Concerning the polymerization of ε-CL, both catalysts prove to be active. However, the reaction proceeded faster in the case of the CH2-derivative. In fact, while ca. 90% conversion was achieved in 8 h with 45, longer reaction times were required in the presence of its sulfur-containing analogue (Table 14, entries 2 and 4). Moreover, the molecular weight of the polymer synthesized with 45 was found to be ca. three times higher than that obtained in the presence of analogue 44 (56 and 19 kDa, respectively). 1H-NMR spectroscopy analyses on the polymers highlighted the presence of Et2N-terminal groups, suggesting that the initiation step of the process would be the formal migratory insertion of the monomer in the Ti-NEt2 bond generating a titanium-alkoxide species. The S-containing complex was also employed in the ROP of L-Lactide (Table 14, entries 5 and 6). High conversion (90%) was achieved after 5 days at 100 °C in toluene affording a polymer with high molecular weight and rather narrow polydispersity (20.4 kDA and 1.23, respectively). The Mn value increased linearly versus the polymer yield, suggesting the occurrence of a living process.

Figure 12.
Diethylamido-derivatives of S- and CH2-bridged diphenolate titanium complexes [25].
Table 14.
ROP of cyclic esters catalyzed by Ti-diphenolate complexes 47 and 48 [26].
Subsequently, Sobota et al. synthesized two novel complexes by treating the pro-ligand 46 with a titanium alkoxide precursor (Scheme 5) [26]. Interestingly, a dimeric species was obtained when [Ti(OEt)4] was employed, while a monomeric complex was isolated in the case of the reaction involving [Ti(OiPr)4]. This was explained by considering the higher steric hindrance of the latter alkoxide ligand over the former. Both complexes were active in the ROP of L- and r-LA, affording polymers with high molecular weight and narrow polydispersity (18.0 kDa and 1.05, respectively). Nevertheless, higher rates were achieved with the monomeric species 48. In fact, 98% conversion was achieved in 1 h while longer reaction times (2.5 h) were required by its dimeric analogue to reach similar values.
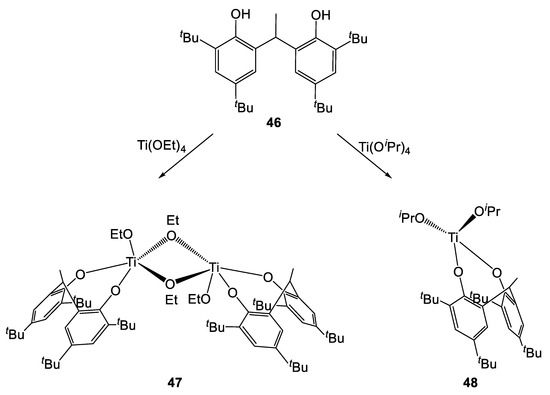
Scheme 5.
Synthesis of complexes 47 and 48 [26].
The Mn increased linearly to the monomer conversion, indicating the living nature of the polymerization. The analysis of the polymer end-groups indicated the presence of terminal ethoxy- or isopropoxy groups, suggesting that the initiation steps involves the insertion of the monomer into the titanium-alkoxo bond. Finally, the NMR spectroscopic analysis of the microstructure of the polymer obtained from r-LA highlighted the preference of complexes 47 and 48 for heteroatactic addition.
A series mixed titanium-M (where M = Li, Na, Zn and Mg) heterobimetallic complexes having various substituents on the methylene bridge of the diphenolate ligand were prepared by Lin et al. (Figure 13) [27]. All complexes were tested in the ROP of L-lactide, and their activity was compared to that of the monometallic complex 48 (Table 15). All complexes were shown to be highly active, with good control of molecular weight and narrow molecular weight distributions. Notably, all Mn values were found to be close to the calculated ones. Similar activity values were found for the monometallic complex 48 and for those containing Li and Na (Table 15, entries 1–3). Notably, the rate of the reaction increased in the presence of Ti-Zn bimetallic species 51–53 and 57 (Table 15, entries 4–7 and 13, respectively). Interestingly, a drop of activity was observed when their Mg-containing congeners (complexes 54–56) were employed (Table 15, entries 8–12). In fact, in order to achieve complete conversion of the monomer, higher temperatures and longer reaction time were required (50 or 70 °C instead of 30 °C and 1–3.5 h instead of 30 min, respectively). The higher activity of the zinc-containing complexes was explained considering the higher charge density of Mg compared to that of Zn, leading to stronger M-OR bonds which are thus less prone to monomer insertion. Overall, Mg- and Zn-Ti bimetallic complexes were shown to be better performing that their Li- and Na analogues, as well as than the monometallic catalyst 48. Within the series, the activity followed the trend 56 > 55 > 54 for Mg-Ti species and 53 > 52 > 51 > 57 for the Ti-Zn congeners. This suggested a positive effect conferred by the presence of electron-donating substituent at the ortho positions of the ligand.

Figure 13.
Diphenolate-supported mixed heterobimetallic Ti complexes [27].
Table 15.
ROP of L-LA catalyzed by bimetallic Ti-diphenolate complexes 48–57 [27].
Very recently, Chen et al. disclosed the synthesis of a number of dinuclear titanium complexes supported by diphenolate ligands having various substituents on the methyne bridge (Figure 14) [28]. The influence of the nature of the substituents on the ROP of ε-CL and L-LA was investigated (Table 16). In the first case, complete conversion of the monomer was achieved within 2-4 h in the presence of all catalysts (Table 16, entries 1–5). The molecular weights determined by GPC were also found to be close to the calculated values.

Figure 14.
Dinuclear titanium complexes supported by substituted diphenolate ligands [28].
Table 16.
ROP of ε-CL and L-LA catalyzed by bimetallic titanium complexes 58–62 [28].
However, slightly broader polydispersities spanning from 1.3 to 1.5 were observed in the case of catalysts 60–62. This was attributed to undesired transesterification reactions. Concerning L-LA, higher temperature and longer reaction times were required (Table 16, entries 6–10). The methyl-substituted complex 59 outperformed its congeners while the methoxyanisole-derivative 62 was shown to be the least performing (Table 16, entries 7 and 10, respectively). Compared to the ROP of ε-CL, better control was achieved, with molecular weight distributions found in a narrow range (1.06–1.20). Kinetic studies indicated that the rate of the reaction is in all cases first order dependent on the monomer concentration. The activity trend for ε-CL was found to be 58 > 59 > 60 > 61 > 62 and 59 > 58 > 60 > 61 > 62 for L-LA, suggesting the increasing in the size of the substituent (R) at the bridge has a detrimental effect on the polymerization rate, possible due to higher steric bulk around the metal center.
We have recently synthesized the chlorotitanium diphenolate complex 63, by treatment of the proligand 46 with an equimolar amount of [TiCl4] (Scheme 6) [29].

Scheme 6.
Synthesis of the chlorotitanium diphenolate complex 63 [29].
The complex, activated by BnOH, was tested in the ROP of several cyclic esters (Table 17). By conducting the reaction at 80 °C over 24 h, ca. 50 and 35 % conversion was achieved for ε-CL and δ-VL (Table 17, entries 1 and 2, respectively). Higher activity was exhibited in the case of r-LA at 130 °C. In fact, 80% conversion was observed after 24 h (Table 17, entry 3). Due to possible intramolecular transesterification reactions, Mn lower than the calculated values were obtained in all cases.
Table 17.
ROP of cyclic esters catalyzed by 63 [29].
3.2. Titanocalix[n]arene complexes
Reports using titanocalix[n]arenes in the ROP of cyclic esters are scant. Frediani, Sémeril et al. employed the nitro-containing complex 64 (Figure 15 and Figure 16) under solvent-free conditions, for the well-controlled ROP of L-lactide (Table 17) [30].

Figure 15.
Titanocalix[4]arenes 64 tested in the ROP of L-lactide [30].

Figure 16.
Molecular structure of 64 [30].
By increasing the monomer/catalyst ratio, an enhancement of the productivity and higher molecular weights were obtained (Table 18, entries 1–4). A beneficial effect on the catalyst activity was also achieved by using an alcohol as co-catalyst (Table 18, entry 5). Nevertheless, a drop of the Mn, allegedly caused by alcohol-promoted chain transfer processes, was observed. Number average molecular weights lower than the expected values were obtained in all cases. This was explained by considering intramolecular transesterification reactions taking place during the propagation step. In fact, the complex could be considered as a dual-site catalyst, since two polymer chains could independently grow on the same titanium center. The occurrence of transesterification reactions was confirmed by MALDI-TOF analyses. Concerning the microstructure of the polymers, highly isotactic PLA was obtained in all cases, as observed by NMR spectroscopy.
Table 18.
Polymerization of L-lactide catalyzed by 64 [30].
Subsequently, the same research group employed the previously reported complex [1,3-L(O)2(OnPr)2TiCl2] (23) for the ROP of rac-lactide (r-LA) [31]. The thermally induced reaction was first investigated (Table 19). As per catalyst 64, the activity of complex 23 as well as the molecular weight of the polymers were found to be directly proportional to the monomer/Ti ratio. Relatively narrow polydispersity values (1.2–1.3) highlighted the good control of the process. NMR spectroscopic analyses suggested a partial isotactic-stereoblock microstructure of the polymers formed.
Table 19.
Thermally induced polymerization of r-lactide catalyzed by 23 [31].
The ROP of r-lactide promoted by microwave irradiation was also explored (Table 20). It was found that an induction time is required by the polymerization. Indeed, only 3% conversion was achieved after 20 min (Table 20, entry 1). However, the conversion increased with the reaction time, reaching 88% after 80 min (Table 20, entry 4). The activity achieved its peak after 60 min (Table 20, entry 3), while longer reaction times led to polymer decomposition.
Table 20.
Microwave-assisted polymerization of r-lactide catalyzed by 23 [31].
Recently, we have reported the isolation of acetonitrile solvates of the known titanocalix[4]arene complexes 24, 26 (bearing 1,3-methoxy- and propoxy- substituents, respectively) as well as new solvate forms of their analogues 65 and 66 (1,3-pentoxy- and monomethoxy derivative, respectively, Figure 16) [29]. A chloro-bridged compound {[TiL(O)3(OR)]2(μ-Cl)2}.(R = n-decyl) (67) was also synthesized and characterized by X-Ray diffraction studies on single crystals (Figure 17 and Figure 18).

Figure 17.
Titanium calix[4]arene complexes 65 and 66 (isolated as MeCN solvates) [29].

Figure 18.
Structure of {[TiL(O)3(OR)]2(μ-Cl)2}.(R = n-decyl) 67 [29].
All complexes were tested as catalysts for the ROP of cyclic esters (Table 21) in the presence of benzyl alcohol. In the case of ε-caprolactone, low to moderate conversions were achieved in the presence of 24, 26 and 65 (32, 33 and 68%, respectively, Table 21 entries 1–3), while complete conversion was obtained when the monomethoxy complex 66 and its dimeric decyl-congener 67 were employed (Table 21, entries 4 and 6, respectively). In all cases, the molecular weights obtained were lower than the expected values, probably due to undesired transesterification reactions. Compared to the reactions catalyzed by 24, 26 and 65, higher Mn were achieved in the presence of 66 and 67.
Table 21.
ROP of cyclic esters catalyzed by 24, 26, 65–67 [29].
However, better control was exhibited by the complexes 24, 26 and 65 (PDI 1.1 vs. 1.5, cf. Table 21 entries 1 and 3). The analysis of the terminal groups of the polymer isolated in the presence of 66 was performed by 1H-NMR spectroscopy. As expected, the presence of the diagnostic resonances for the BnO- and –CH2OH terminations were detected. Further investigations using MALDI-TOF spectrometry confirmed these results. Nevertheless, a second distribution compatible with cyclic species was also identified. Noteworthy, all catalysts proved also to be active in air, allowing for the complete conversion of the monomer within 24 h at 130 °C (Table 21, entries 7–11). Compared to the runs performed at 80 °C, broader polydispersities were observed in all cases, highlighting the loss of control at these high reaction temperatures. Further structural investigation by MALDI-TOF spectrometry highlighted the presence of only cyclic species for low Mn (20–30 repeating units), while a second distribution accountable to hydroxyl-terminated linear polymer was also detect for higher fractions (>35 repeating units). The formation of such terminations was attributed to the insertion of the first monomer unit in a Ti-OH species arising from the reaction of the pre-catalyst with adventitious water.
The complexes proved to be efficient catalysts also for the ROP of δ-valerolactone (Table 21, entries 12–16). Similarly to the previous case, 66 and 67 were shown to be better performing than their dichloride counterparts. In fact, moderate conversions (ca. 50%) were achieved with the chlorides while ca. 80% conversion was obtained in the presence of 66/67. However, in spite of the higher conversions, a loss of control was also observed (PDIs spanning 1.45 to 1.60). Complex 66 was active also under aerobic conditions at 130 °C (Table 21, entry 17).
Compared to the reaction performed under inert atmosphere (Table 21, entry 15), lower conversion was achieved (62 and 81%, respectively). The ROP of r-lactide was also investigated (Table 21, entries 18–23), and all catalysts were active at 130 °C with near complete conversion observed in the presence of 24, 26, 65 and 66 (Table 21, entries 18–21). Surprisingly, only 65% conversion was obtained when using 67 (Table 21, entry 22). Noteworthily, complete conversion was reached when using 66 under air (Table 21, entry 23). The microstructure of the polymers was studied by means of 2D J-resolved 1H-NMR spectroscopy [32]. While complexes 24 and 26 afforded almost atactic polymers (Pr 0.46 and 0.42, respectively), isotactic materials were isolated in the case of systems 65–67.
Complex 66 was also employed in ε-CL/δ-VL co-polymerization leading to a high molecular weight copolymer (Mn > 23,000). NMR spectroscopic studies showed the complete conversion of both monomers suggesting the formation of a copolymer with a 1:1 CL/VL ratio. The analysis of the CL-CL, VL-VL, CL-VL and VL-CL dyads by 13C-NMR spectroscopy allowed for the determination of the number-average sequence length that was found to be 2.22 and 1.85 for CL and VL, respectively. These values suggested the formation of random-type co-polymers. Similarly, ε-CL/r-LA copolymerization was performed, and NMR spectroscopic analyses on the material produced highlighted that only 65% of ε-caprolactone had been converted, while full consumption of r-lactide was achieved. Differing reactivity of the two co-monomers in ε-CL/r-LA copolymerization has also been reported elsewhere [33,34,35]. The co-polymer compositions were investigated by 13C-NMR spectroscopy. By studying the triad distributions in the carbonyl range of the spectrum, the LA/CL ratio was found to be 75:35 and the average sequence length was 3.04 and 2.42 for CL and LA, respectively. Notably, both reactions were performed in air without significant loss of activity and with no effect on the structural features of the isolated copolymers. The monochloride titanocalix[4]arene was tested also in the ROP of the large monomer ω-pentadecalactone [36,37,38,39,40]. However, only 50% conversion was achieved after 24 h at 130 °C when the reaction was performed under an inert atmosphere. Even lower conversion (11%) was observed under air when employing the same reaction conditions.
Furthermore, complex 66 was supported on silica by following a slightly modified reported procedure [41], and tested in the ROP of all monomers previously investigated (Table 22). The catalyst efficiently promoted the polymerization of ε-CL, and complete conversion was observed after 24 h at 130 °C, regardless of the use of benzyl alcohol (Table 22, entries 1 and 2).
Table 22.
Polymerization of cyclic esters catalyzed by Si-supported-66 [29].
Notably, higher molecular mass and narrower polydispersity was achieved in the absence of the initiator. Moderate conversions were observed in the tests involving δ-VL and r-LA (Table 22, entries 3 and 4). Interestingly, the supported complex was not active in the ROP of the larger monomer ω-pentadecalactone (Table 22, entry 7).
Finally, the activity of the diphenolate complex 63 in the ROP of various cyclic esters was compared to that of the titanocalixarene species 64 and 66 (Table 23); the parent diphenol can be visualized as a ‘half-calix[4]arene’ molecule. In all cases, the monochloride catalyst 66 outperformed the other systems, both in terms of conversion and polymer Mn (Table 23, entries 3, 6, 9). Nevertheless, narrower polydispersities were achieved in the presence of 63 and 64.
Table 23.
Comparison between the activities of various Ti-based ROP catalysts [29].
Noteworthily, the diphenolate species was found to be inactive in the ROP of the larger pentadecalactone (Table 23, entry 10), while the calixarene-based complexes 64 and 66 afforded polymers in low and moderate conversion (Table 23, entries 11 and 12, respectively).
Interestingly, 63 and 64 turned out to be inactive for the ROP of ε-CL under solvent-free conditions, while the monochloride complex 66 allowed for ca. 50% conversion over 24 h at 130 °C. The inactivity of 63 and 64 was explained by considering the inefficient activation of the dichloride species in the absence of the solvent.
Concerning Ti-based complexes bearing larger calix[n]arene ligands, McIntosh et al. recently reported preliminary the synthesis and catalytic activity in the ROP of r-LA of complex [Ti4L2(O)8(OnPr)8(THF)2] (where L2(OH)8 = p-tert-butylcalix[8]arene, 68, Figure 19 and Figure 20) [42]. Preliminary polymerization tests were performed in toluene at 130 °C with a monomer/Ti ratio of 100:1, allowing for the complete conversion of the monomer after 16 h. Mass spectrometry revealed that each terminal polymer chain was terminated by an nPrO- group. Molecular weights lower than the expected ones were observed, suggesting the occurrence of transesterification reactions. Interestingly, the presence of cyclic oligomers was highlighted by NMR spectroscopy.

Figure 19.
Tetranuclear titanium complex 68 synthesized by McIntosh et al. [42].

Figure 20.
Molecular structure of 60 [42].
4. Conclusions
In this review, we have described the use of titanium complexes bearing bridged-diphenolate or calix[n]arene ligands as pre-catalysts for either α-olefin polymerization or ROP of cyclic esters. It was shown that the structure of the complex can dramatically influence the outcome of the transformation, both in terms of activity of the catalytic system and the properties of the polymer obtained. A comparison between the performances of selected catalysts in the ethylene polymerization is shown in Table 24. In the case of diphenolate-containing complexes, both the nature of the bridging unit and the labile ligands proved to affect the polymerization process; for the former aspect, the catalyst activity was shown to follow the trend S > CH2 > Te (1, 2 > 4 > 6, 8). This could be due to the higher flexibility of the system provided by the S-bridging unit compared to that of the CH2-group; the lower activity of the Te-containing congener was explained considering the possible Te-AlMAO exchange leading to early catalyst decomposition. Notably, systems containing Ti-Cl proved to be better performing than Ti-OR congeners. In fact, both in the case of S- and Te-bridged systems, the activity of the chloride derivatives are ca. 2-fold higher than that of their iso-propoxy analogues (1 > 2 and 6 > 8, respectively). This could be ascribed to the faster activation of the chloride pre-catalysts. However, in the presence of the propoxide analogues, the molecular weights of the isolated polymers were higher, albeit with slightly broader polydispersities, suggesting the formation of multiple active species which were allegedly more robust than those generate from their Cl-congeners. On the other hand, titanocalix[n]arenes proved to be less active than diphenolate systems by 10-15 times to several orders of magnitude. The accessibility of the metal center proved to be crucial for the catalyst activity. In fact, the 1,2-depleted complex 29 was shown to be better performing than dialkoxy titanocalix[4]arenes 28 and 24. The size of the calixarene also proved to affect the catalyst performance. In fact, the activity for titanocalix[n]arenes was found to follow the trend n = 8 > n = 6 > n = 4 (34, 33 and 32, respectively), which perhaps reflects the increased conformational flexibility of the larger ring system. Finally, systems having thiacalixarene ligands (namely 36 and 37) exhibited activities similar to those achieved in the presence of 1,2-dialkoxy calixarenes. Nevertheless, the broad and bimodal molecular weight distributions for such thiacalixarene systems suggest an uncontrolled process and/or the formation of multiple active sites.
Table 24.
Comparison between titanium catalysts for ethylene polymerization.
Similar conclusions could be drawn by comparing the activity of titanium diphenolate catalysts in the ROP of cyclic esters (Table 25). S-bridged complexes were shown to be better performing than Te-containing analogues (1 > 6 and 8). Unlike for ethylene polymerization, methylene-bridged complexes outperformed the other systems in the ROP of ε-CL (45 > 44), suggesting that a lower bridge flexibility is beneficial for the ROP process. With respect to the polymerization of L-lactide, bimetallic species (49–51 and 54) proved to be better performing than monometallic analogues.
Table 25.
Comparison between titanium diphenolate catalysts in the ROP of ε-CL and L-LA.
In particular, the highest activity was shown by the mixed heterobimetallic Ti-Zn complex. This was ascribed to a cooperative effect between the two metal centers. The higher activity of the Zn-adduct complexes was attributed to the lower charge density of Zn compared to that of the other metals investigated (ie. Mg), leading to weaker M-OR bonds, which are more prone to form M-lactide adducts via monomer insertion. Bulky substituents at the bridging unit of the diphenolate ligand were also shown to negatively influence the performance of the catalytic systems (58 > 62), possibly because of reduced accessibility at the metal center.
Reports concerning the use of titanocalix[n]arenes as catalysts for the ROP of cyclic ester are still scarce. After the seminal work by Frediani et al., the research field has remained widely unexplored. We have recently disclosed the synthesis of new complexes based on calix[4]arenes which exhibited interesting activity in the ROP of the most common cyclic esters such as ε-CL, δ-VL and r-LA, as well as the more challenging substrate ω-pentadecalactone. The highest activities were observed in the presence of complexes 64 and 66, suggesting that an electron-withdrawing group (NO2) at the lower rim or a labile ligand (MeCN) at the metal center are beneficial for the ROP process. Moreover, these species proved to be better performing that their biphenolate counterparts, highlighting the positive effect of the calixarene moiety.
Whilst the results described in this review indicate that titanocalix[4]arenes and related species can perform well on a laboratory scale, their potential to deliver useful polymeric materials on a bulk scale is the next challenge. In the case of biodegradable polymers, future studies will also need to evaluate the impact and fate of any chemicals released upon decomposition. Such studies are on-going in our laboratories.
Author Contributions
Conceptualization, C.R. and O.S.; methodology, C.R. and O.S; formal analysis, C.R. and O.S.; investigation, C.R. and O.S.; resources, C.R. and O.S.; writing—original draft preparation, C.R. and O.S; writing—review and editing, C.R. and O.S.; supervision, C.R..; project administration, C.R.; funding acquisition, C.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by UKRI Creative Circular Plastic grant (EP/S025537/1).
Acknowledgments
This work was financially supported by UKRI Creative Circular Plastic grant (EP/S025537/1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Homden, D.H.; Redshaw, C. The Use of Calixarenes in Metal-Based Catalysis Chem. Review 2008, 108, 5086–5130. [Google Scholar]
- Li, Y.; Zhao, K.-Q.; Redshaw, C.; Martínez Ortega, B.A.; Nuñez, A.Y.; Hanna, T.A. Patai’s Chemistry of Functional Groups. Coordination Chemistry and Applications of Phenolic Calixarene–metal Complexes; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Amor, F.; Fokken, S.; Kleinhenn, T.; Spaniol, T.P.; Okuda, J. Mono(cyclopentadienyl)titanium complexes containing a sulfide-bridged bis(phenolato) ligand. Molecular structure of Ti{2,2′-S(OC6H2-4-Me-6-tBu)2}(η5-C5H5)Cl. J. Organomet. Chem. 2001, 621, 3–9. [Google Scholar] [CrossRef]
- Olmstead, M.M.; Sigel, G.; Hope, H.; Xu, X.; Power, P.P. Metallocalixarenes: Syntheses and X-Ray crystal structures of titanium(IV), iron(III), and cobalt(II) complexes of p-tert-butylcalix[4]arene. J. Am. Chem. Soc. 1985, 107, 8087–8091. [Google Scholar] [CrossRef]
- Kissin, Y.V.; Mink, R.I.; Brandolini, A.J.; Nowlin, T.E. AlR2Cl/MgR2 Combinations as Universal Cocatalysts for Ziegler–Natta, Metallocene, and Post-Metallocene Catalysts. J. Polym. Sci. Pol. Chem 2009, 47, 3271–3285. [Google Scholar] [CrossRef]
- Miyatake, T.; Mizunuma, K.; Seki, Y.; Kakugo, M. 2,2’-Thiobis(6-tert-butyl-4-methylphenoxy)titanium or zirconium complex-methylalumoxane catalysts for polymerization of olefins. Makromol. Chem., Rapid Commun. 1989, 10, 349–352. [Google Scholar] [CrossRef]
- van der Linden, A.; Schaverien, C.J.; Meijboon, N.; Canter, C.; Orpen, A.G. Polymerization of a-Olefins and Butadiene and Catalytic Cyclotrimerization of 1-Alkynes by a New Class of Group IV Catalysts. Control of Molecular Weight and Polymer Microstructure via Ligand Tuning in Sterically Hindered Chelating Phenoxide T.itanium and Zirconium Species. J. Am. Chem. Soc. 1995, 117, 3008–3021. [Google Scholar] [CrossRef]
- Nakayama, Y.; Watanabe, K.; Ueyama, N.; Nakamura, A.; Harada, A.; Okuda, J. Titanium Complexes Having Chelating Diaryloxo Ligands Bridged by Tellurium and Their Catalytic Behavior in the Polymerization of Ethylene. Organometallics 2000, 19, 2498–2503. [Google Scholar] [CrossRef]
- Froese, R.D.J.; Musaev, D.G.; Matsubara, T.; Morokuma, K. Theoretical Studies of Ethylene Polymerization Reactions Catalyzed by Zirconium and Titanium Chelating Alkoxide Complexes. J. Am. Chem. Soc. 1997, 119, 7190–7196. [Google Scholar] [CrossRef]
- Umare, P.S.; Rao, K.; Tembe, G.L.; Dhoble, D.A.; Trivedi, B. Controlled Synthesis of Low-Molecular-Weight Polyethylene Waxes by Titanium–Biphenolate–Ethylaluminum Sesquichloride Based Catalyst Systems. J. Appl. Polym. Sci. 2007, 104, 1531–1539. [Google Scholar] [CrossRef]
- Umare, P.S.; Rao, K.; Tembe, G.L.; Dhoble, D.A.; Trivedi, B. Polyethylene Waxes: Catalytic Synthesis by Ti-Biphenolates. J. Macromol. Sci. A. 2007, 44, 977–987. [Google Scholar] [CrossRef]
- Ozerova, O.V.; Rathb, N.P.; Ladipo, F.T. Synthesis, characterization, and reactivity of titanium(IV) complexes supported by proximally bridged p-tert-butylcalix[4]arene ligands J. Organomet. Chem. 1999, 586, 223–233. [Google Scholar] [CrossRef]
- Frediani, M.; Sémeril, D.; Comucci, A.; Bettucci, L.; Frediani, P.; Rosi, L.; Matt, D.; Toupet, L.; Kaminsky, W. Ultrahigh-Molecular-Weight Polyethylene by Using a Titanium Calix[4]arene Complex with High Thermal Stability under Polymerization Conditions. Macromol. Chem. Phys. 2007, 208, 938–945. [Google Scholar] [CrossRef]
- Capacchione, C.; Neri, P.; Proto, A. Polymerization of ethylene in the presence of 1,3-dimethoxy-p-But-calix[4]arene titanium dichloride. NMR evidence of the cationic titanium compound generated by methylalumoxane. Inorg. Chem. Commun. 2003, 6, 339–342. [Google Scholar] [CrossRef]
- Espinas, J.; Darbost, U.; Pelletier, J.; Jeanneau, E.; Duchamp, C.; Bayard, F.; Boyron, O.; Broyer, J.-P.; Thivolle-Cazat, J.; Basset, J.-M.; et al. Titanacalixarenes in Homogeneous Catalysis: Synthesis, Conformation and Catalytic Activity in Ethylene Polymerisation. Eur. J. Inorg. Chem. 2010, 1349–1359. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Y.S.; Xie, G.H. Synthesis of calixtitanium compounds and using for ethylene and styrene polymerizations. Acta Polym. Sin. 1998, 101–103. [Google Scholar]
- Chen, Y.; Zhang, Y.; Shen, Z.; Sun, W. Polymerization of ethylene with calix[4]titanium-Al(i-Bu)3 systems. Acta. Polym. Sin. 2000, 2, 239–241. [Google Scholar]
- Diaz-Barrios, A.; Liscano, J.; Trujillo, M.; Agrifoglio, G.; Matos, J.O. Assignee: Intevep S.A. U.S. Patent 5,767,034, 1998. [Google Scholar]
- Matos, J.O.; Diaz-Barrios, A.; Liscano, J.; Trujillo, M.; Agrifoglio, G. Assignee: Intevep S.A. European Patent EP1125951, 2001. [Google Scholar]
- Morohashi, N.; Hattori, T.; Yokomakura, K.; Kabutob, C.; Miyanoa, S. Dinuclear titanium(IV) complex of p-tert-butylthiacalix[4]arene as a novel bidentate Lewis acid catalyst. Tetrahedron Lett. 2002, 43, 7769–7772. [Google Scholar] [CrossRef]
- Proto, A.; Giugliano, F.; Capacchione, C. Ethylene polymerization promoted by dinuclear titanium p-tert-butylthiacalix[4]arene complexes. Eur. Polym. J. 2009, 45, 2138–2141. [Google Scholar] [CrossRef]
- Takeuchi, D.; Nakamura, T.; Aida, T. Bulky Titanium Bis(phenolate) Complexes as Novel Initiators for Living Anionic Polymerization of ε-Caprolactone. Macromolecules 2000, 33, 725–729. [Google Scholar] [CrossRef]
- Takeuchi, D.; Aida, T. Sequential Cationic and Anionic Polymerizations by Triflate Complexes of Bulky Titanium Bisphenolates: One-Pot Synthesis of Polyoxetane-Poly(E-caprolactone) Block Copolymer. Macromolecules 2000, 33, 4607–4609. [Google Scholar] [CrossRef]
- Takashima, Y.; Nakayama, Y.; Watanabe, H.; Itono, T.; Ueyama, N.; Nakamura, A.; Yasuda, H.; Harada, A.; Okuda, J. Polymerizations of Cyclic Esters Catalyzed by Titanium Complexes Having Chalcogen-Bridged Chelating Diaryloxo Ligands. Macromolecules 2002, 35, 7538–7544. [Google Scholar] [CrossRef]
- Takashima, Y.; Nakayama, Y.; Hirao, T.; Yasuda, H.; Harada, A. Bis(amido)titanium complexes having chelating diaryloxo ligands bridged by sulfur or methylene and their catalytic behaviors for ring-opening polymerization of cyclic esters. J. Organomet. Chem. 2004, 689, 612–619. [Google Scholar] [CrossRef]
- Ejfler, J.; Kobyłka, M.; Jerzykiewicz, L.B.; Sobota, P. Titanium complexes supported by bis(aryloxo) ligand: Structure and lactide polymerization activities. J. Mol. Catal. A: Chem. 2006, 257, 105–111. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Liu, M.-Y.; Sutar, A.K.; Lin, C.-C. Synthesis and Structural Studies of Heterobimetallic Alkoxide Complexes Supported by Bis(phenolate) Ligands: Efficient Catalysts for Ring-Opening Polymerization of L-Lactide. Inorg. Chem. 2010, 49, 665–674. [Google Scholar] [CrossRef]
- Jiang, M.-T.; Kosuru, S.R.; Lee, Y.H.; Lu, W.-Y.; Vandavasi, J.K.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y. Factors influencing catalytic behavior of titanium complexes bearing bisphenolate ligands toward ring-opening polymerization of L-lactide and ε-caprolactone. Express Polym. Lett. 2018, 12, 126–135. [Google Scholar] [CrossRef]
- Sun, Z.; Zhao, Y.; Santoro, O.; Elsegood, M.R.J.; Bedwell, E.V.; Zahra, K.; Walton, A.; Redshaw, C. Use of titanocalix[4]arenes in the ring opening polymerization of cyclic esters. Cat. Sci. & Tech 2020. in-press. [Google Scholar] [CrossRef]
- Frediani, M.; Sémeril, D.; Mariotti, A.; Rosi, L.; Frediani, P.; Rosi, L.; Matt, D.; Toupet, L. Ring Opening Polymerization of Lactide under Solvent-Free Conditions Catalyzed by a Chlorotitanium Calix[4]arene Complex. Macromol. Rapid Commun. 2008, 29, 1554–1560. [Google Scholar] [CrossRef]
- Frediani, M.; Sémeril, D.; Matt, D.; Rosi, L.; Frediani, P.; Rizzolo, F.; Papini, A.M. Ring-Opening Polymerisation of rac-Lactide Using a Calix[4]arene-Based Titanium (IV) Complex. Int. J. Polym. Sci. 2010, 6. [Google Scholar] [CrossRef]
- Walton, M.J.; Lancaster, S.J.; Redshaw, C. Highly selective and immortal magnesium calixarene complexes for the ring-opening polymerization of rac-lactide. ChemCatChem 2014, 6, 1892–1898. [Google Scholar] [CrossRef]
- Huang, Y.-T.; Wang, W.-C.; Hsu, C.-P.; Lu, W.-Y.; Chuang, W.-J.; Chiang, M.Y.; Laia, Y.-C.; Chen, H.-Y. The ring-opening polymerization of ε-caprolactone and L-lactide using aluminum complexes bearing benzothiazole ligands as catalysts. Polym. Chem. 2016, 7, 4367–4377. [Google Scholar] [CrossRef]
- Florcza, M.; Duda, A. Effect of the configuration of the active center on comonomer reactivities: The case of epsilon-caprolactone/L,L-lactide copolymerization. Angew. Chem., Int. Ed. 2008, 47, 9088–9091. [Google Scholar] [CrossRef]
- Duda, A.; Biela, T.; Libiszowski, J.; Penczek, S.; Dubois, P.; Mecerreyes, D.; Jérôme, R. Block and random copolymers of є-caprolactone. Polym. Degrad. Stab. 1998, 59, 215–222. [Google Scholar] [CrossRef]
- Zhong, Z.; Dijkstra, P.J.; Feijen, J. Controlled ring-opening polymerization of ω-pentadecalactone with yttrium isopropoxide as an initiator. Macromol. Chem. Phys. 2000, 201, 1329–1333. [Google Scholar] [CrossRef]
- Lebedev, B.; Yevstropov, A. Thermodynamic properties of polylactones. Makromol. Chem. 1984, 185, 1235–1253. [Google Scholar] [CrossRef]
- Naumann, S.; Scholten, P.B.V.; Wilson, J.A.; Dove, A.P. Dual Catalysis for Selective Ring-Opening Polymerization of Lactones: Evolution toward Simplicity. J. Am. Chem. Soc. 2015, 137, 14439–14445. [Google Scholar] [CrossRef]
- Jedliński, Z.; Juzwa, M.; Adamus, G.; Kowalczuk, M.; Montaudo, M. Anionic polymerization of pentadecanolide. A new route to a potentially biodegradable aliphatic polyester. Macromol. Chem. Phys. 1996, 197, 2923–2929. [Google Scholar] [CrossRef]
- Bouyahyi, M.; Duchateau, R. Metal-Based Catalysts for Controlled Ring-Opening Polymerization of Macrolactones: High Molecular Weight and Well-Defined Copolymer Architectures. Macromolecules 2014, 47, 517–524. [Google Scholar] [CrossRef]
- Grosso-Giordano, N.A.; Solovyov, A.; Hwang, S.; Katz, A. Effect of Coordination Environment in Grafted Single-Site Ti-SiO2 Olefin Epoxidation Catalysis. Top Catal. 2016, 59, 1110–1122. [Google Scholar] [CrossRef]
- Ryan, J.D.; Gagnon, K.J.; Teat, S.J.; McIntosh, R.D. Flexible macrocycles as versatile supports for catalytically active metal clusters. Chem. Commun. 2016, 52, 9071–9073. [Google Scholar] [CrossRef]


























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).