Environmental Reactions of Air-Quality Protection on Eco-Friendly Iron-Based Catalysts

 ,
,  ,
, 

Abstract
1. Introduction
2. Results
2.1. Catalytic Results
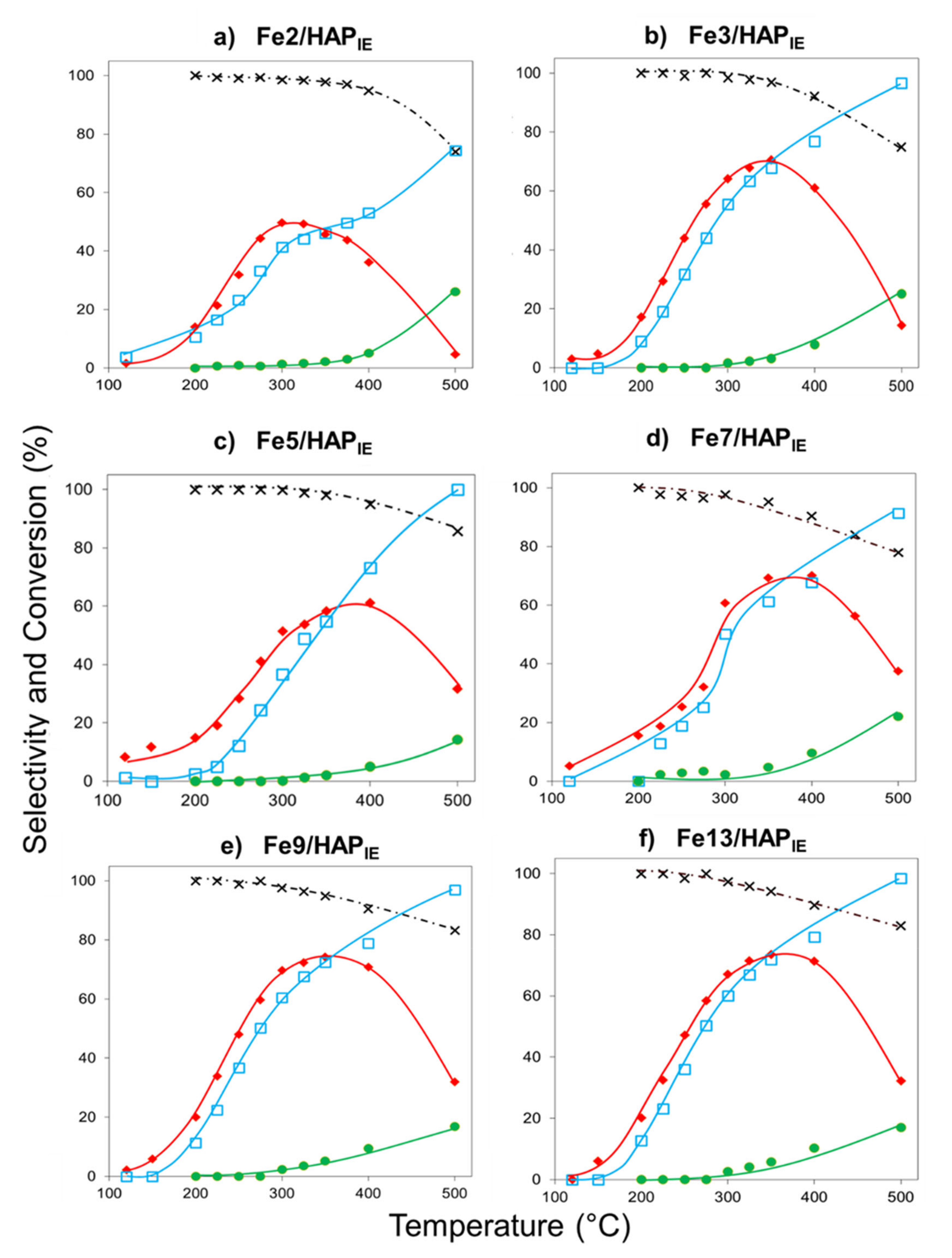
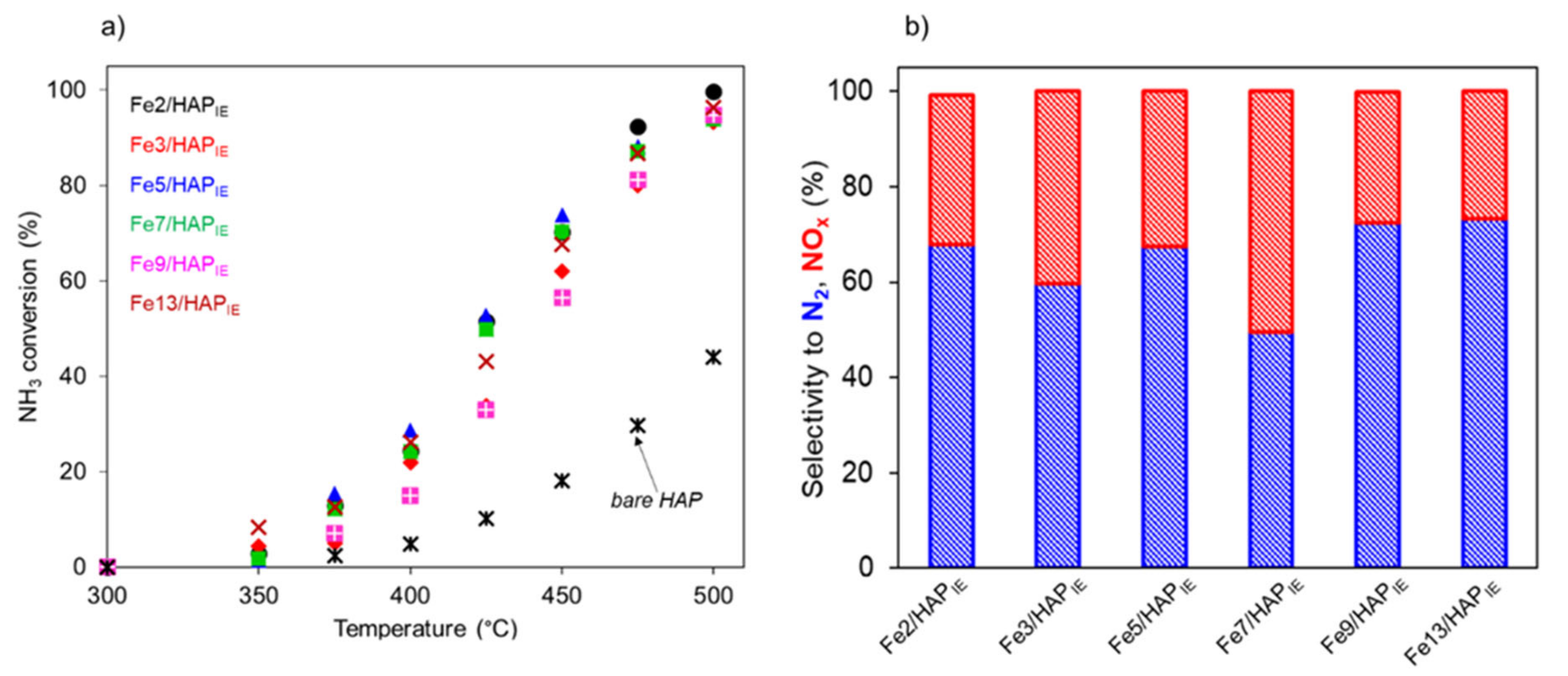
2.1.1. Selective Catalytic Reduction of NO by NH3 (NH3-SCR)
2.1.2. Selective Catalytic Oxidation of NH3 (NH3-SCO)
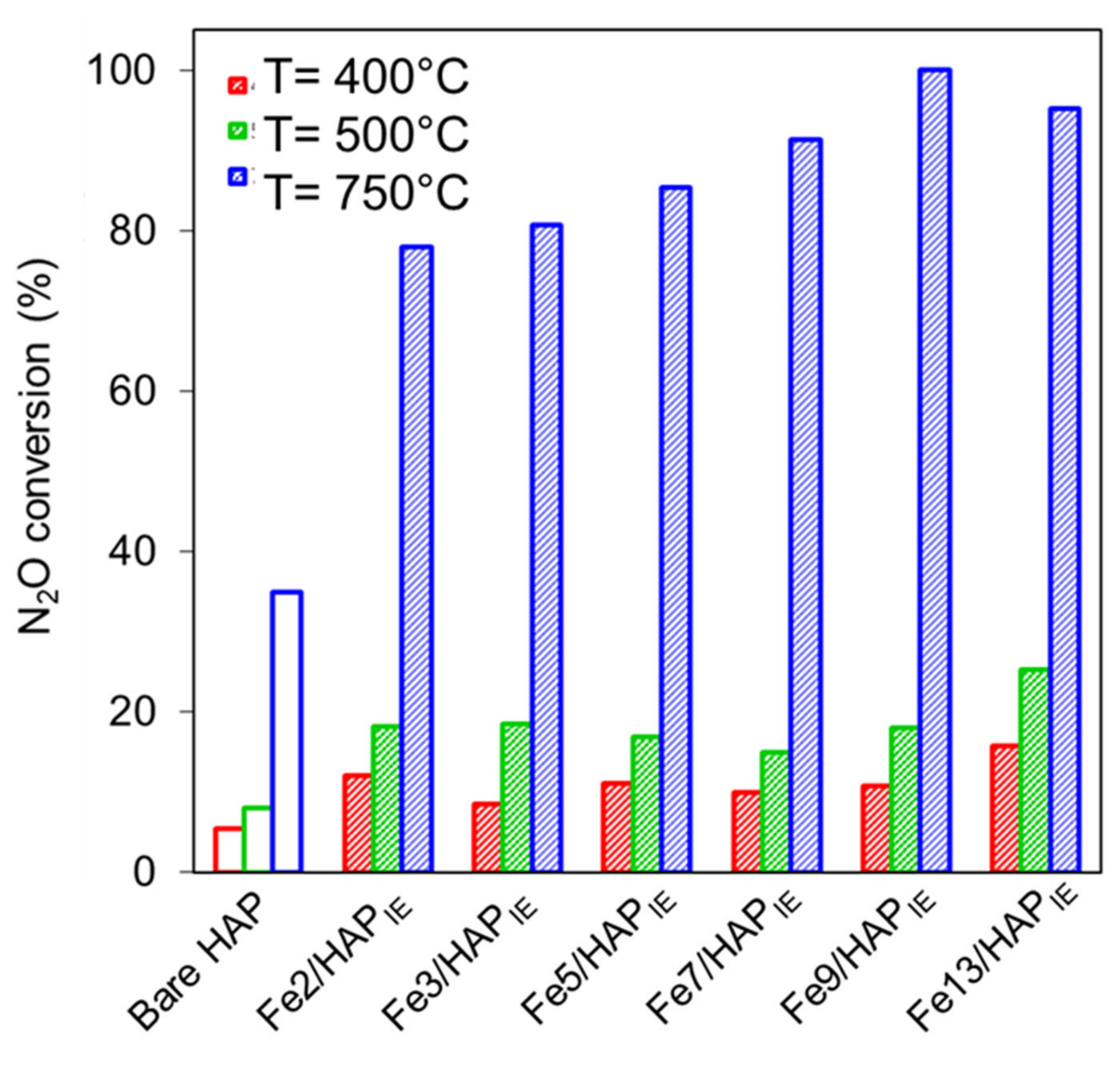
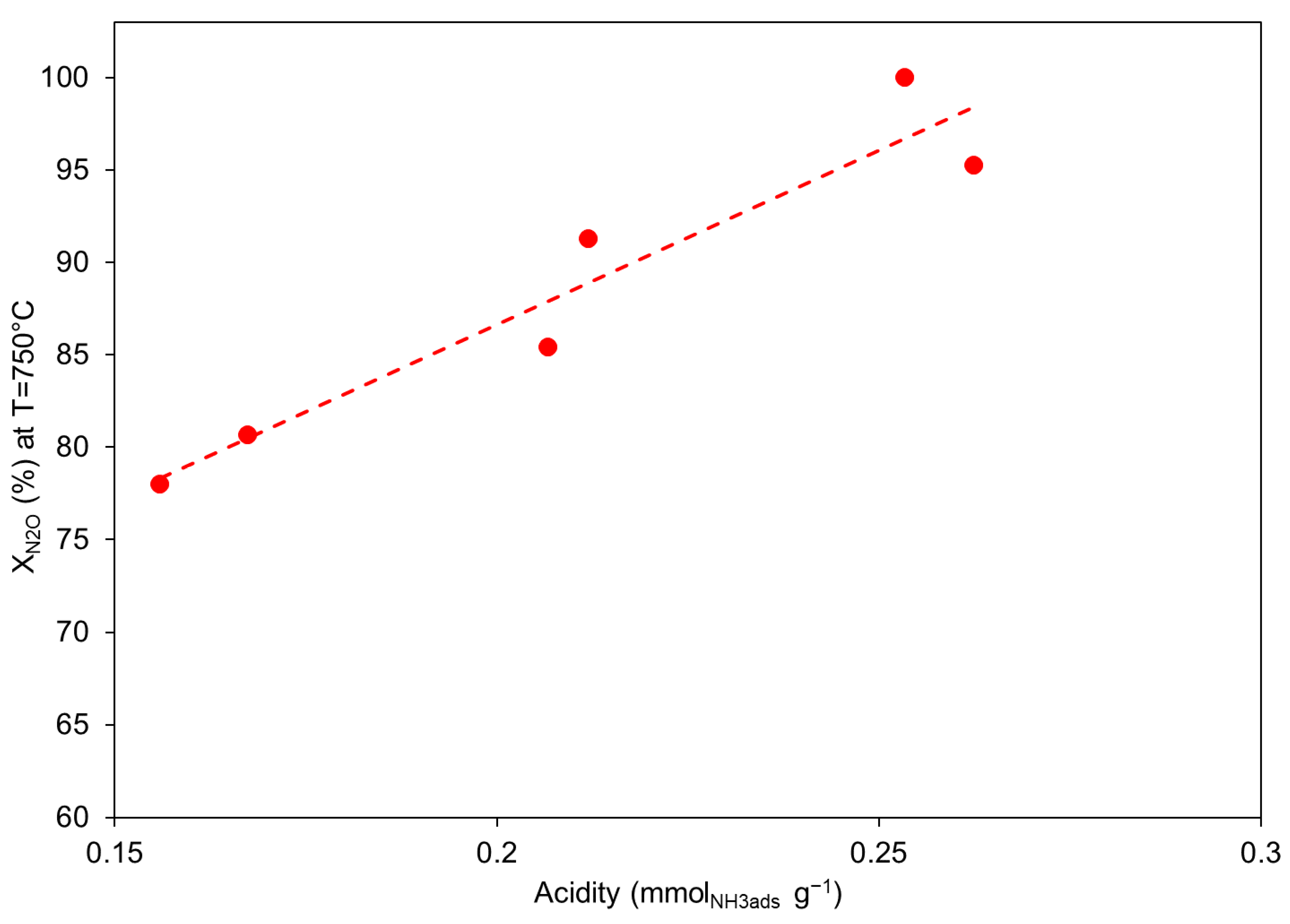
2.1.3. N2O Decomposition
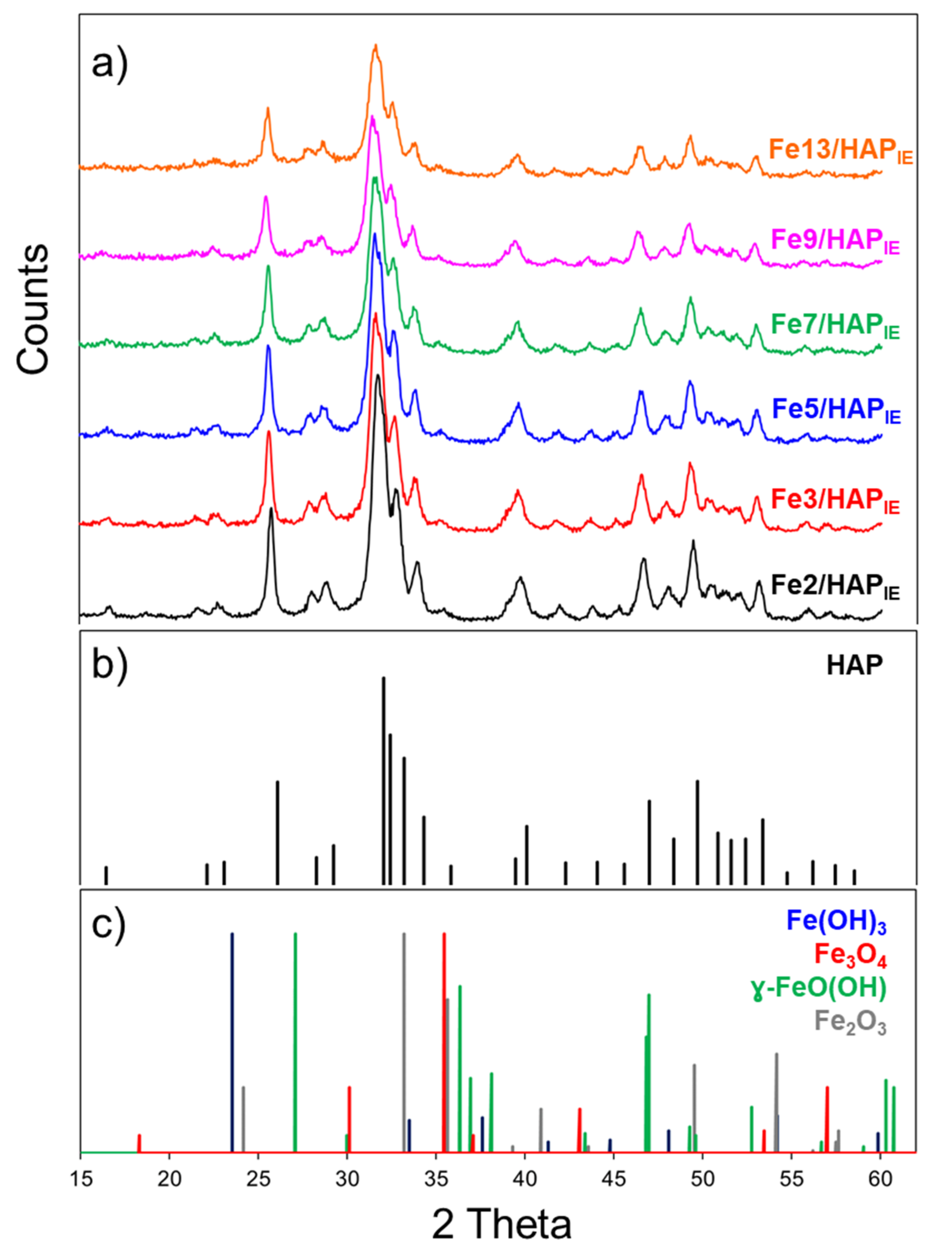
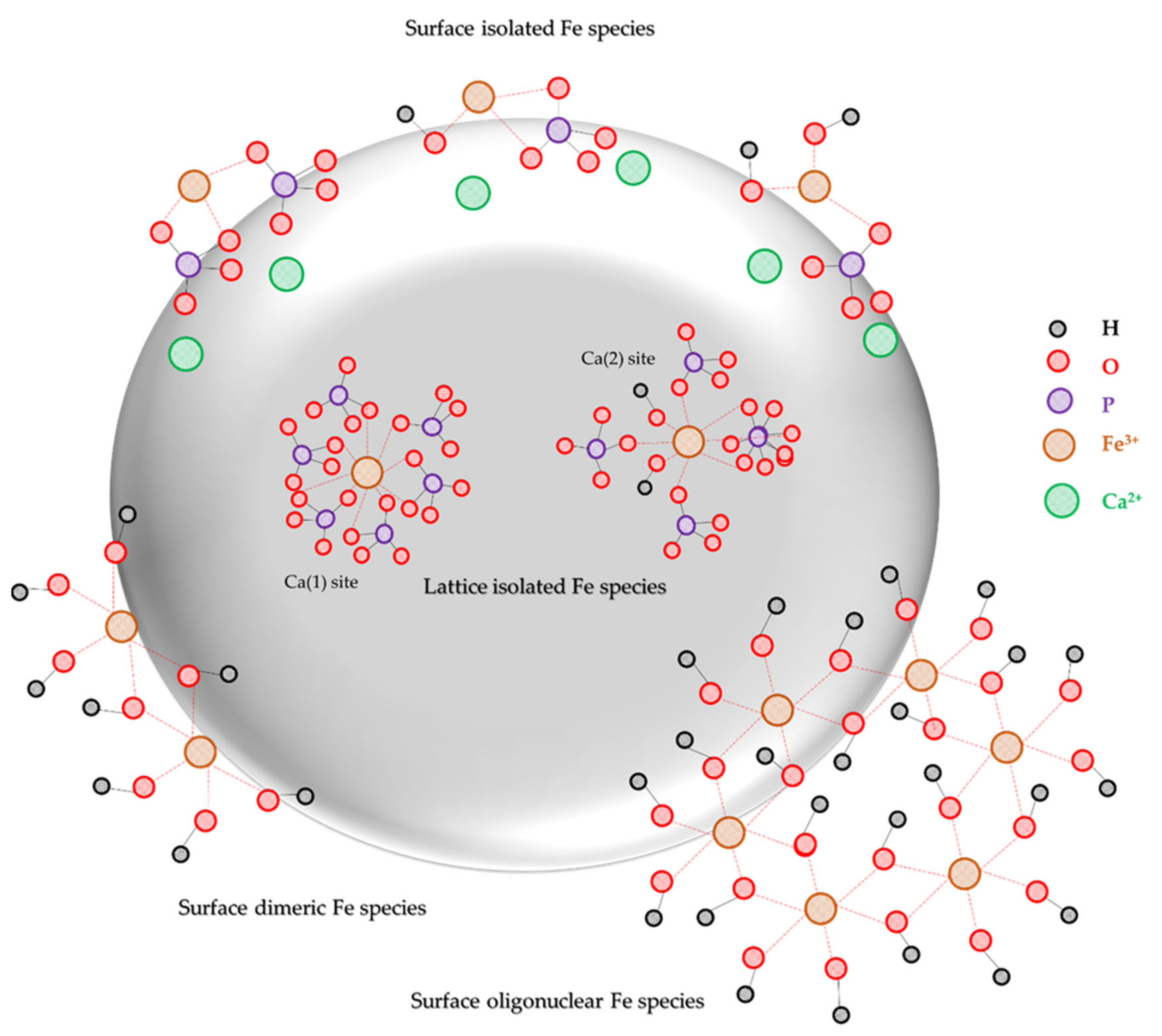
2.2. Catalyst Characterization
3. Discussion
4. Materials and Methods
4.1. Materials and Catalyst Preparation
4.2. Characterization
4.2.1. N2 Adsorption–Desorption Isotherms at −196 °C
4.2.2. X-ray Powder Diffraction (XRPD)
4.2.3. NH3 Adsorption
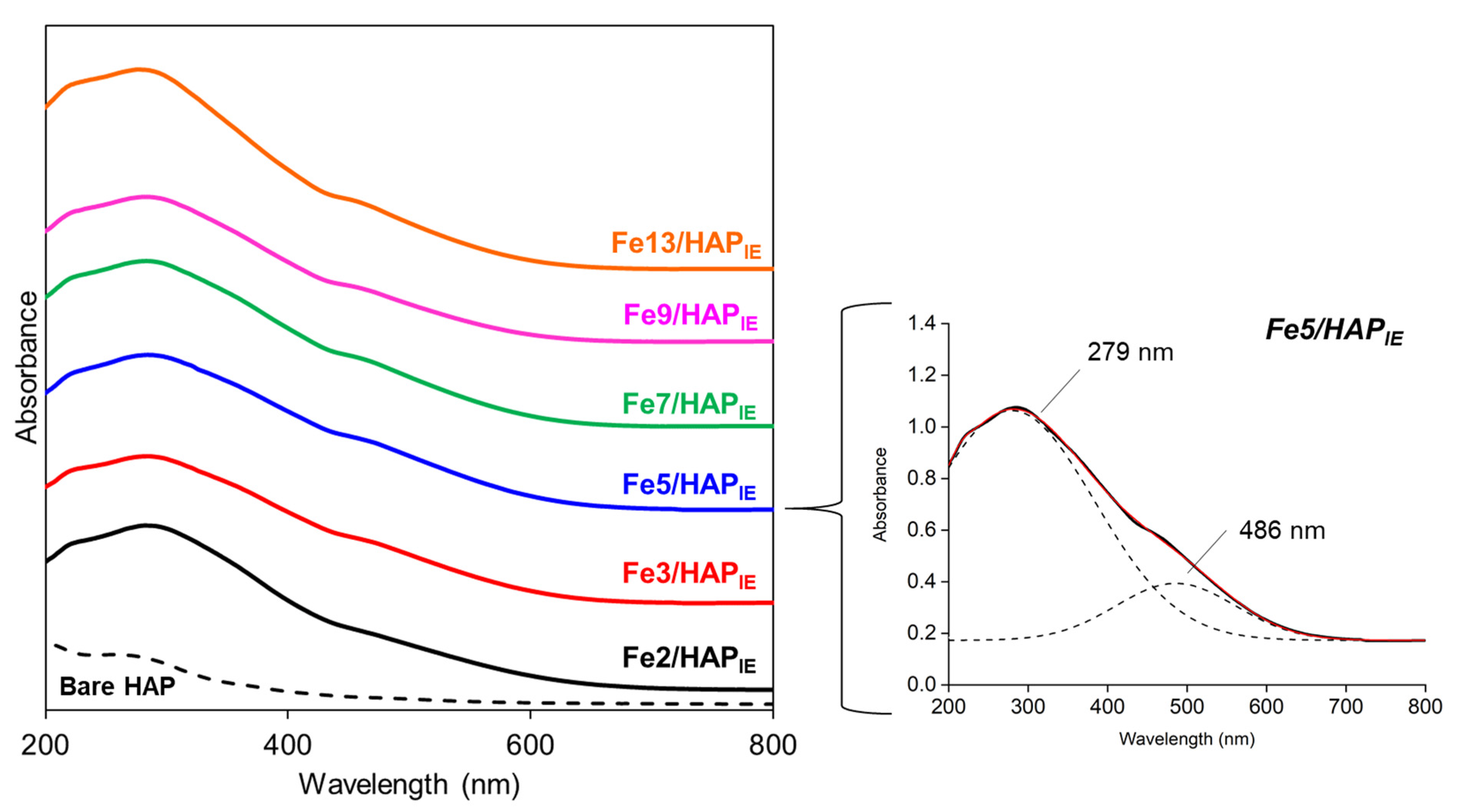
4.2.4. UV Diffuse Reflectance Spectroscopy (UV-DRS)
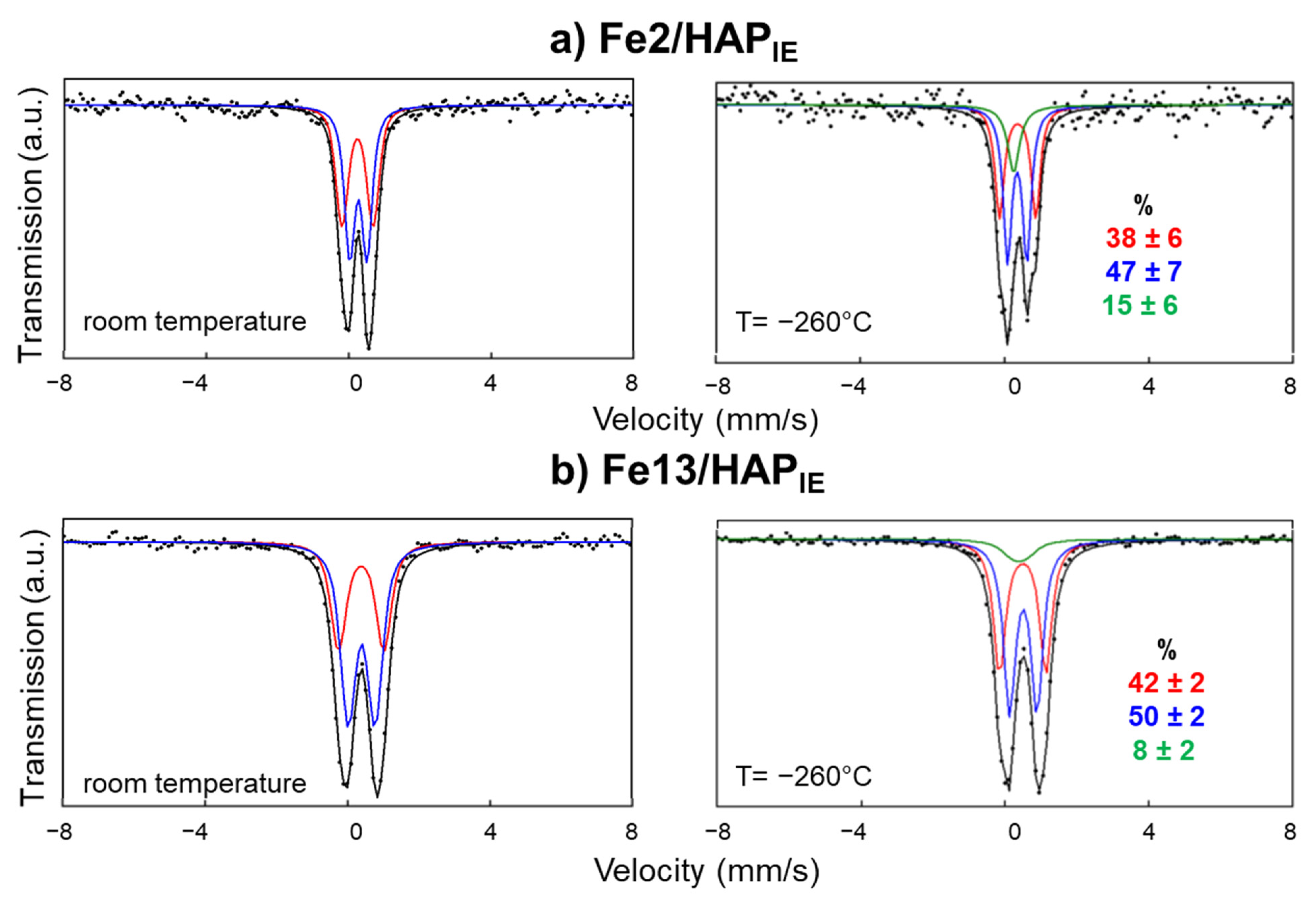
4.2.5. Mössbauer Spectroscopy
4.2.6. Catalytic Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Zhao, J.; Lee, J.-M. Conventional and New Materials for Selective Catalytic Reduction (SCR) of NOx. ChemCatChem 2018, 10, 1499–1511. [Google Scholar] [CrossRef]
- Bosch, H.; Janssen, F. Catalytic Reduction of Nitrogen Oxides: A Review on the Fundamentals and Technology; Elsevier: Amsterdam, The Netherlands, 1988; Volume 7. [Google Scholar]
- Damma, D.; Ettireddy, P.; Reddy, B.; Smirniotis, P. A Review of Low Temperature NH3-SCR for Removal of NOx. Catalysts 2019, 9, 349. [Google Scholar] [CrossRef]
- Yan, Z.; Shan, W.; Shi, X.; He, G.; Lian, Z.; Yu, Y.; Shan, Y.; Liu, J.; He, H. The way to enhance the thermal stability of V2O5-based catalysts for NH3-SCR. Catal. Today 2019. [Google Scholar] [CrossRef]
- Li, J.; Chang, H.; Ma, L.; Hao, J.; Yang, R.T. Low-temperature selective catalytic reduction of NOx with NH3 over metal oxide and zeolite catalysts—A review. Catal. Today 2011, 175, 147–156. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, L.; Yang, R.T. Activity, propene poisoning resistance and hydrothermal stability of copper exchanged chabazite-like zeolite catalysts for SCR of NO with ammonia in comparison to Cu/ZSM-5. Appl. Catal. A Gen. 2012, 427–428, 24–34. [Google Scholar] [CrossRef]
- Klimczak, M.; Kern, P.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts-Part I: V2O5-WO3/TiO2 catalysts. Appl. Catal. B Environ. 2010, 95, 39–47. [Google Scholar] [CrossRef]
- Kern, P.; Klimczak, M.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts. Part II: Fe–zeolite catalysts. Appl. Catal. B Environ. 2010, 95, 48–56. [Google Scholar] [CrossRef]
- Lai, J.K.; Wachs, I.E. A Perspective on the Selective Catalytic Reduction (SCR) of NO with NH3 by Supported V2O5-WO3/TiO2 Catalysts. ACS Catal. 2018, 8, 6537–6551. [Google Scholar] [CrossRef]
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The State of the Art in Selective Catalytic Reduction of NOx by ammonia Using Metal-Exchanged Zeolite Catalysts. Catal. Rev. 2008, 50, 492–531. [Google Scholar] [CrossRef]
- Ibrahim, M.; Labaki, M.; Giraudon, J.-M.; Lamonier, J.-F. Hydroxyapatite, a multifunctional material for air, water and soil pollution control: A review. J. Hazard. Mater. 2020, 383, 121139. [Google Scholar] [CrossRef]
- Campisi, S.; Evangelisti, C.; Postole, G.; Gervasini, A. Combination of interfacial reduction of hexavalent chromium and trivalent chromium immobilization on tin-functionalized hydroxyapatite materials. Appl. Surf. Sci. 2021, 539, 148227. [Google Scholar] [CrossRef]
- Campisi, S.; Castellano, C.; Gervasini, A. Tailoring the structural and morphological properties of hydroxyapatite materials to enhance the capture efficiency towards copper(II) and lead(II) ions. New J. Chem. 2018, 42, 4520–4530. [Google Scholar] [CrossRef]
- Agbeboh, N.I.; Oladele, I.O.; Daramola, O.O.; Adediran, A.A.; Olasukanmi, O.O.; Tanimola, M.O. Environmentally sustainable processes for the synthesis of hydroxyapatite. Heliyon 2020, 6, e03765. [Google Scholar] [CrossRef] [PubMed]
- Zoubeir, L.; Adeline, S.; Laurent, C.S.; Yoann, C.; Truc, H.T.; Benoît, L.G.; Federico, A. The use of the Novosol process for the treatment of polluted marine sediment. J. Hazard. Mater. 2007, 148, 606–612. [Google Scholar] [CrossRef]
- Pretto, M.; Costa, A.L.; Landi, E.; Tampieri, A.; Galassi, C. Dispersing behavior of hydroxyapatite powders produced by wet-chemical synthesis. J. Am. Ceram. Soc. 2003, 86, 1534–1539. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Celotti, G.C.; Mattioli-Belmonte, M.; Logroscino, G. Synthetic Biomimetic Nanostructured Hydroxyapatite. Key Eng. Mater. 2005, 284–286, 949–952. [Google Scholar] [CrossRef]
- Silvester, L.; Lamonier, J.-F.; Vannier, R.-N.; Lamonier, C.; Capron, M.; Mamede, A.-S.; Pourpoint, F.; Gervasini, A.; Dumeignil, F. Structural, textural and acid–base properties of carbonate-containing hydroxyapatites. J. Mater. Chem. A 2014, 2, 11073–11090. [Google Scholar] [CrossRef]
- Chlala, D.; Labaki, M.; Giraudon, J.-M.; Gardoll, O.; Denicourt-Nowicki, A.; Roucoux, A.; Lamonier, J.-F. Toluene total oxidation over Pd and Au nanoparticles supported on hydroxyapatite. Comptes Rendus Chim. 2016, 19, 525–537. [Google Scholar] [CrossRef]
- Chlala, D.; Giraudon, J.-M.; Nuns, N.; Lancelot, C.; Vannier, R.-N.; Labaki, M.; Lamonier, J.-F. Active Mn species well dispersed on Ca2+ enriched apatite for total oxidation of toluene. Appl. Catal. B Environ. 2016, 184, 87–95. [Google Scholar] [CrossRef]
- Boukha, Z.; Kacimi, M.; Ziyad, M.; Ensuque, A.; Bozon-Verduraz, F. Comparative study of catalytic activity of Pd loaded hydroxyapatite and fluoroapatite in butan-2-ol conversion and methane oxidation. J. Mol. Catal. A Chem. 2007, 270, 205–213. [Google Scholar] [CrossRef]
- Boukha, Z.; González-Prior, J.; de Rivas, B.; González-Velasco, J.R.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I. Synthesis, characterisation and behaviour of Co/hydroxyapatite catalysts in the oxidation of 1,2-dichloroethane. Appl. Catal. B Environ. 2016, 190, 125–136. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Crocker, M.; Zhang, Y.; Zhu, X.; Shi, C. Understanding on the origins of hydroxyapatite stabilized gold nanoparticles as high-efficiency catalysts for formaldehyde and benzene oxidation. Catal. Commun. 2015, 59, 195–200. [Google Scholar] [CrossRef]
- Kumar, P.A.; Reddy, M.P.; Ju, L.K.; Phil, H.H. Novel Silver Loaded Hydroxyapatite Catalyst for the Selective Catalytic Reduction of NOx by Propene. Catal. Lett. 2008, 126, 78–83. [Google Scholar] [CrossRef]
- Tounsi, H.; Djemal, S.; Petitto, C.; Delahay, G. Copper loaded hydroxyapatite catalyst for selective catalytic reduction of nitric oxide with ammonia. Appl. Catal. B Environ. 2011, 107, 158–163. [Google Scholar] [CrossRef]
- Campisi, S.; Galloni, M.G.; Marchetti, S.G.; Auroux, A.; Postole, G.; Gervasini, A. Functionalized Iron Hydroxyapatite as Eco-friendly Catalyst for NH3-SCR Reaction: Activity and Role of Iron Speciation on the Surface. ChemCatChem 2020, 12, 1676–1690. [Google Scholar] [CrossRef]
- Campisi, S.; Galloni, M.G.; Bossola, F.; Gervasini, A. Comparative performance of copper and iron functionalized hydroxyapatite catalysts in NH3-SCR. Catal. Commun. 2019, 123, 79–85. [Google Scholar] [CrossRef]
- Schiavoni, M.; Campisi, S.; Carniti, P.; Gervasini, A.; Delplanche, T. Focus on the catalytic performances of Cu-functionalized hydroxyapatites in NH3-SCR reaction. Appl. Catal. A Gen. 2018, 563, 43–53. [Google Scholar] [CrossRef]
- Ferri, M.; Campisi, S.; Scavini, M.; Evangelisti, C.; Carniti, P.; Gervasini, A. In-depth study of the mechanism of heavy metal trapping on the surface of hydroxyapatite. Appl. Surf. Sci. 2019, 475, 397–409. [Google Scholar] [CrossRef]
- Sakhno, Y.; Bertinetti, L.; Iafisco, M.; Tampieri, A.; Roveri, N.; Martra, G. Surface hydration and cationic sites of nanohydroxyapatites with amorphous or crystalline surfaces: A comparative study. J. Phys. Chem. C 2010, 114, 16640–16648. [Google Scholar] [CrossRef]
- Nikolenko, N.V.; Esajenko, E.E. Surface properties of synthetic calcium hydroxyapatite. Adsorpt. Sci. Technol. 2005, 23, 543–553. [Google Scholar] [CrossRef]
- Misra, D.N. Adsorption on hydroxyapatite: Role of hydrogen bonding and interphase coupling. Langmuir 1988, 4, 953–958. [Google Scholar] [CrossRef]
- Sotomayor, F.J.; Cychosz, K.A.; Thommes, M. Characterization of Micro/Mesoporous Materials by Physisorption: Concepts and Case Studies. Acc. Mater. Surf. Res. 2018, 3, 34–50. [Google Scholar]
- Schiavoni, M.; Campisi, S.; Gervasini, A. Effect of Cu deposition method on silico aluminophosphate catalysts in NH3-SCR and NH3-SCO reactions. Appl. Catal. A Gen. 2017, 543, 162–172. [Google Scholar] [CrossRef]
- Campisi, S.; Palliggiano, S.; Gervasini, A.; Evangelisti, C. Finely Iron-Dispersed Particles on β Zeolite from Solvated Iron Atoms: Promising Catalysts for NH3-SCO. J. Phys. Chem. C 2019, 123, 11723–11733. [Google Scholar] [CrossRef]
- Madia, G.; Koebel, M.; Elsener, M.; Wokaun, A. Side Reactions in the Selective Catalytic Reduction of NOx with Various NO2 Fractions. Ind. Eng. Chem. Res. 2002, 41, 4008–4015. [Google Scholar] [CrossRef]
- Li, Y.; TeckNam, C.; PingOoi, C. Iron(III) and manganese(II) substituted hydroxyapatite nanoparticles: Characterization and cytotoxicity analysis. J. Phys. Conf. Ser. 2009, 187. [Google Scholar] [CrossRef]
- Khachani, M.; Kacimi, M.; Ensuque, A.; Piquemal, J.-Y.; Connan, C.; Bozon-Verduraz, F.; Ziyad, M. Iron–calcium–hydroxyapatite catalysts: Iron speciation and comparative performances in butan-2-ol conversion and propane oxidative dehydrogenation. Appl. Catal. A Gen. 2010, 388, 113–123. [Google Scholar] [CrossRef]
- Kandori, K.; Toshima, S.; Wakamura, M.; Fukusumi, M.; Morisada, Y. Effects of Modification of Calcium Hydroxyapatites by Trivalent Metal Ions on the Protein Adsorption Behavior. J. Phys. Chem. B 2010, 114, 2399–2404. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. Chapter 7 Characterization. In The Iron Oxides: Structure, Properties, Reactions, Occurrences, and Uses; Cornell, R.M., Schwertmann, U., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 1996; pp. 139–183. [Google Scholar]
- Long, G.J.; Grandjean, F. Mössbauer Spectroscopy Applied to Inorganic Chemistry; Modern Inorganic Chemistry; Springer: New York, NY, USA, 2013; ISBN 9781489922892. [Google Scholar]
- Long, G.J.; Grandjean, F. Mössbauer Spectroscopy Applied to Magnetism and Materials Science; Modern Inorganic Chemistry; Springer: New York, NY, USA, 2013; ISBN 9781489924094. [Google Scholar]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.Y.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, N.; Lei, Z.; Chen, B. Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts. Chem. Rev. 2016, 116, 3658–3721. [Google Scholar] [CrossRef]
- Kröcher, O.; Brandenberger, S. Active sites, deactivation and stabilization of Fe-ZSM-5 for the selective catalytic reduction (SCR) of NO with NH3. Chimia 2012, 66, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The determination of the activities of different iron species in Fe-ZSM-5 for SCR of NO by NH3. Appl. Catal. B Environ. 2010, 95, 348–357. [Google Scholar] [CrossRef]
- Chmielarz, L.; Jabłońska, M. Advances in selective catalytic oxidation of ammonia to dinitrogen: A review. RSC Adv. 2015, 5, 43408–43431. [Google Scholar] [CrossRef]
- Heyden, A.; Hansen, N.; Bell, A.T.; Keil, F.J. Nitrous oxide decomposition over Fe-ZSM-5 in the presence of nitric oxide: A comprehensive DFT study. J. Phys. Chem. B 2006, 110, 17096–17114. [Google Scholar] [CrossRef] [PubMed]
- Heyden, A.; Peters, B.; Bell, A.T.; Keil, F.J. Comprehensive DFT study of nitrous oxide decomposition over Fe-ZSM-5. J. Phys. Chem. B 2005, 109, 1857–1873. [Google Scholar] [CrossRef]
- Lagarec, K.; Rancourt, D.G. Recoil-Mössbauer Spectral Analysis Software for Windows; University of Ottawa: Ottawa, ON, Canada, 1998. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Fe Loading a (wt.%) | S.A. b (m2·g−1) | Pore Volume c (cm3·g−1) | Mean Pore Radius d (nm) | Acidity (μmolNH3(ads)·g−1) | Acidity Ratio e |
|---|---|---|---|---|---|---|
| HAP/500 f | - | 44 | 0.19 | 8.2 | 140.5 | - |
| Fe2/HAPIE | 2.07 | 67 | 0.21 | 6.9 | 155.9 ± 2.3 | 1.11 |
| Fe3/HAPIE | 3.02 | 64 | 0.22 | 7.4 | 167.4 ± 12 | 1.19 |
| Fe5/HAPIE | 4.77 | 66 | 0.20 | 6.4 | 206.7 ± 7.3 | 1.47 |
| Fe7/HAPIE | 6.83 | 76 | 0.24 | 6.4 | 212.0 ± 6.9 | 1.51 |
| Fe9/HAPIE | 8.62 | 78 | 0.25 | 6.1 | 253.4 ± 0.4 | 1.80 |
| Fe13/HAPIE | 12.93 | 82 | 0.30 | 5.6 | 262.4 ± 7.3 | 1.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galloni, M.G.; Campisi, S.; Marchetti, S.G.; Gervasini, A. Environmental Reactions of Air-Quality Protection on Eco-Friendly Iron-Based Catalysts. Catalysts 2020, 10, 1415. https://doi.org/10.3390/catal10121415
Galloni MG, Campisi S, Marchetti SG, Gervasini A. Environmental Reactions of Air-Quality Protection on Eco-Friendly Iron-Based Catalysts. Catalysts. 2020; 10(12):1415. https://doi.org/10.3390/catal10121415
Chicago/Turabian StyleGalloni, Melissa Greta, Sebastiano Campisi, Sergio Gustavo Marchetti, and Antonella Gervasini. 2020. "Environmental Reactions of Air-Quality Protection on Eco-Friendly Iron-Based Catalysts" Catalysts 10, no. 12: 1415. https://doi.org/10.3390/catal10121415
APA StyleGalloni, M. G., Campisi, S., Marchetti, S. G., & Gervasini, A. (2020). Environmental Reactions of Air-Quality Protection on Eco-Friendly Iron-Based Catalysts. Catalysts, 10(12), 1415. https://doi.org/10.3390/catal10121415