The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation



Abstract
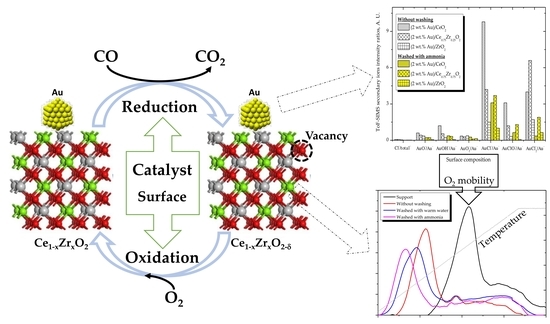
{kind=link}
{kind=link}
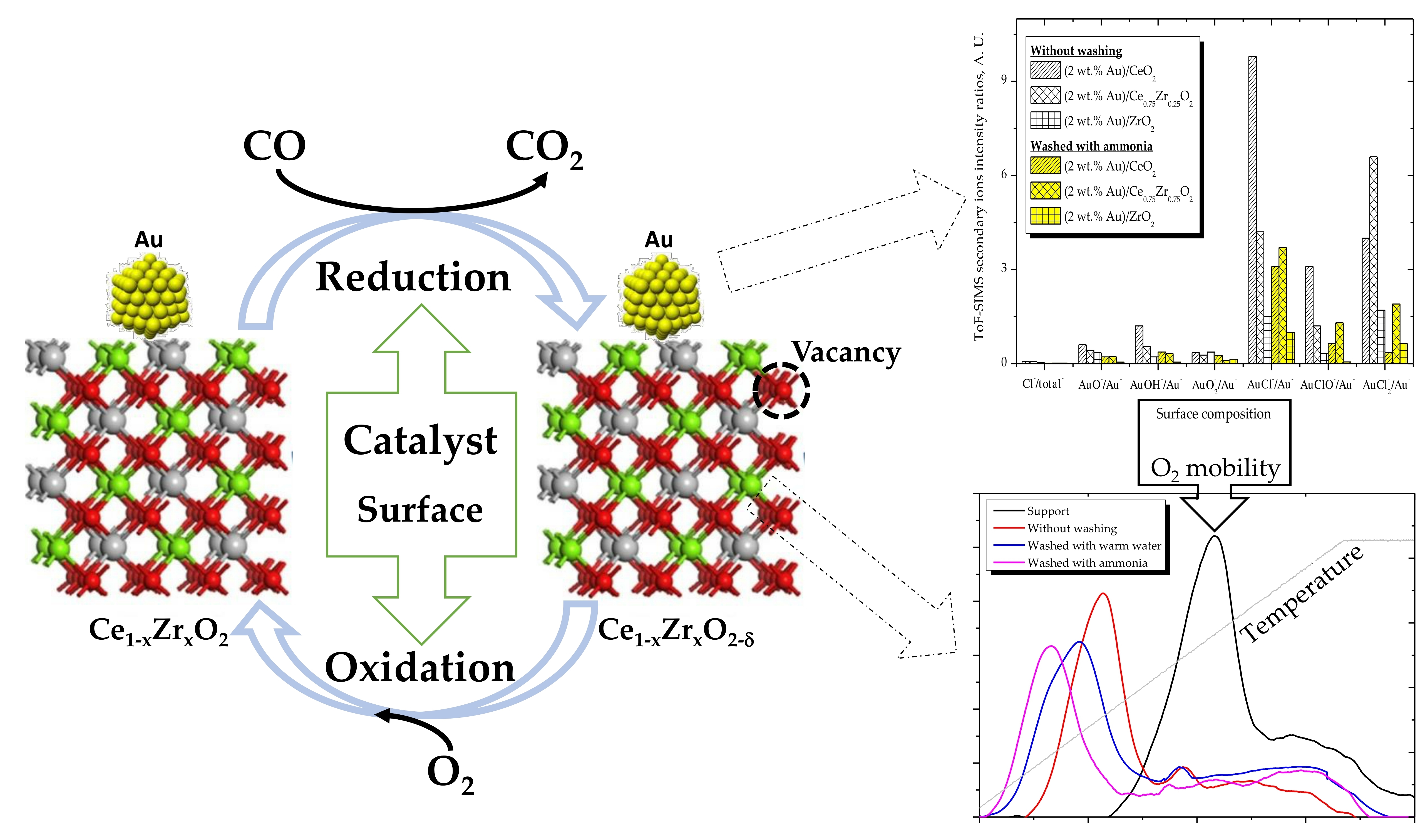
{kind=link}
{kind=link}
{kind=link}
{kind=link}
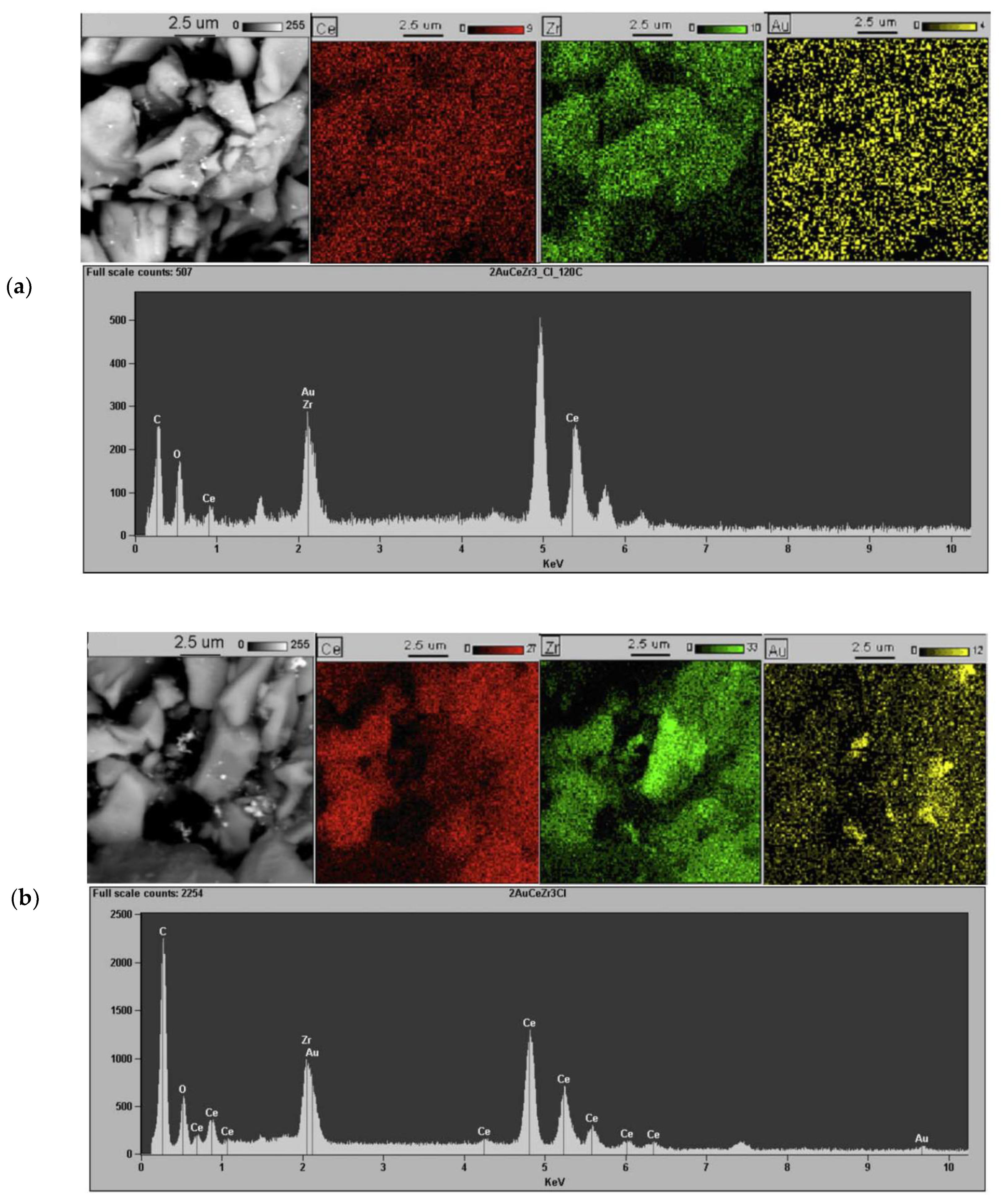
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Factors Affecting CO Oxidation Reaction
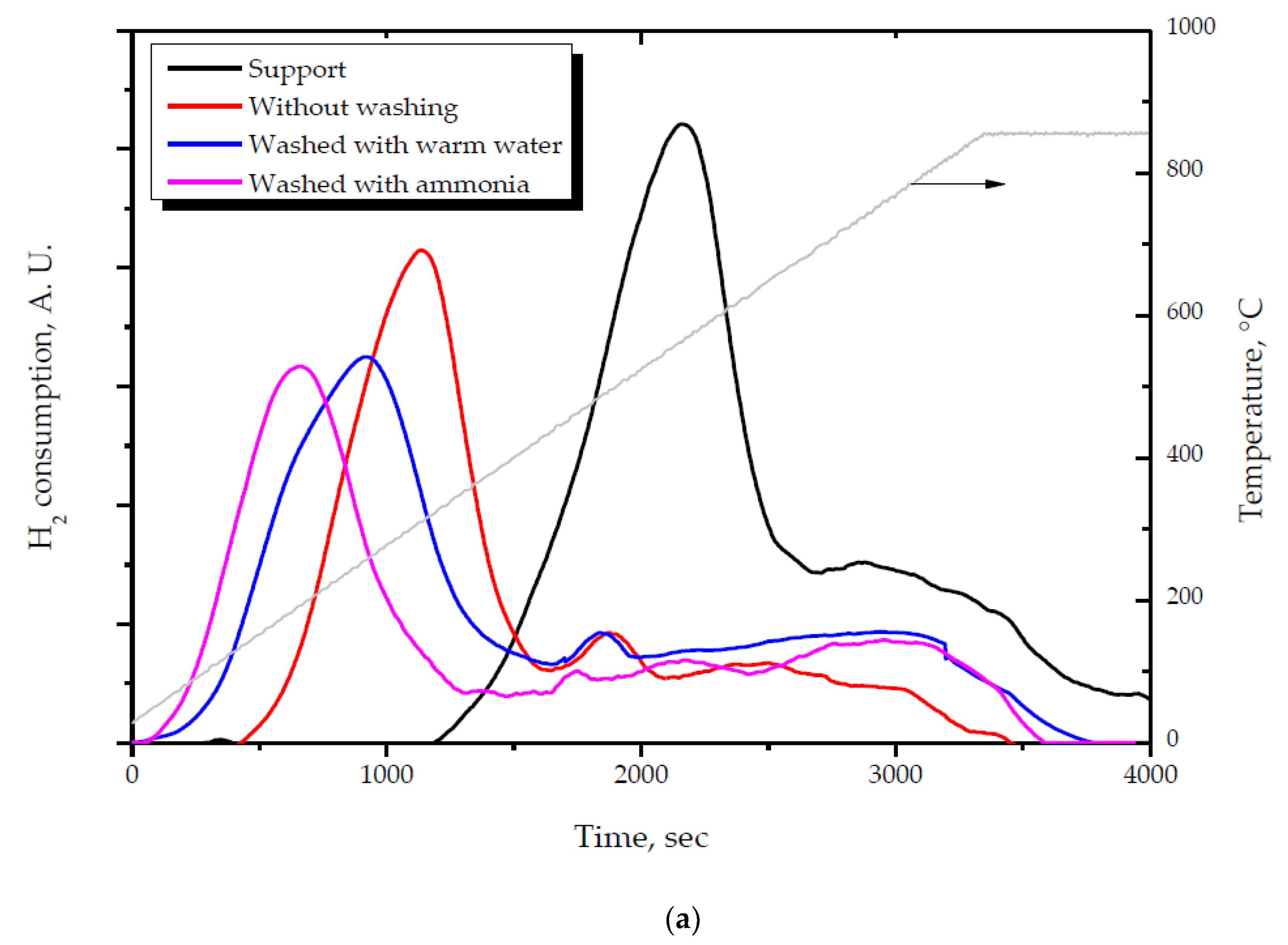
2.1. Effect of the Preparation Conditions and the Catalyst Properties
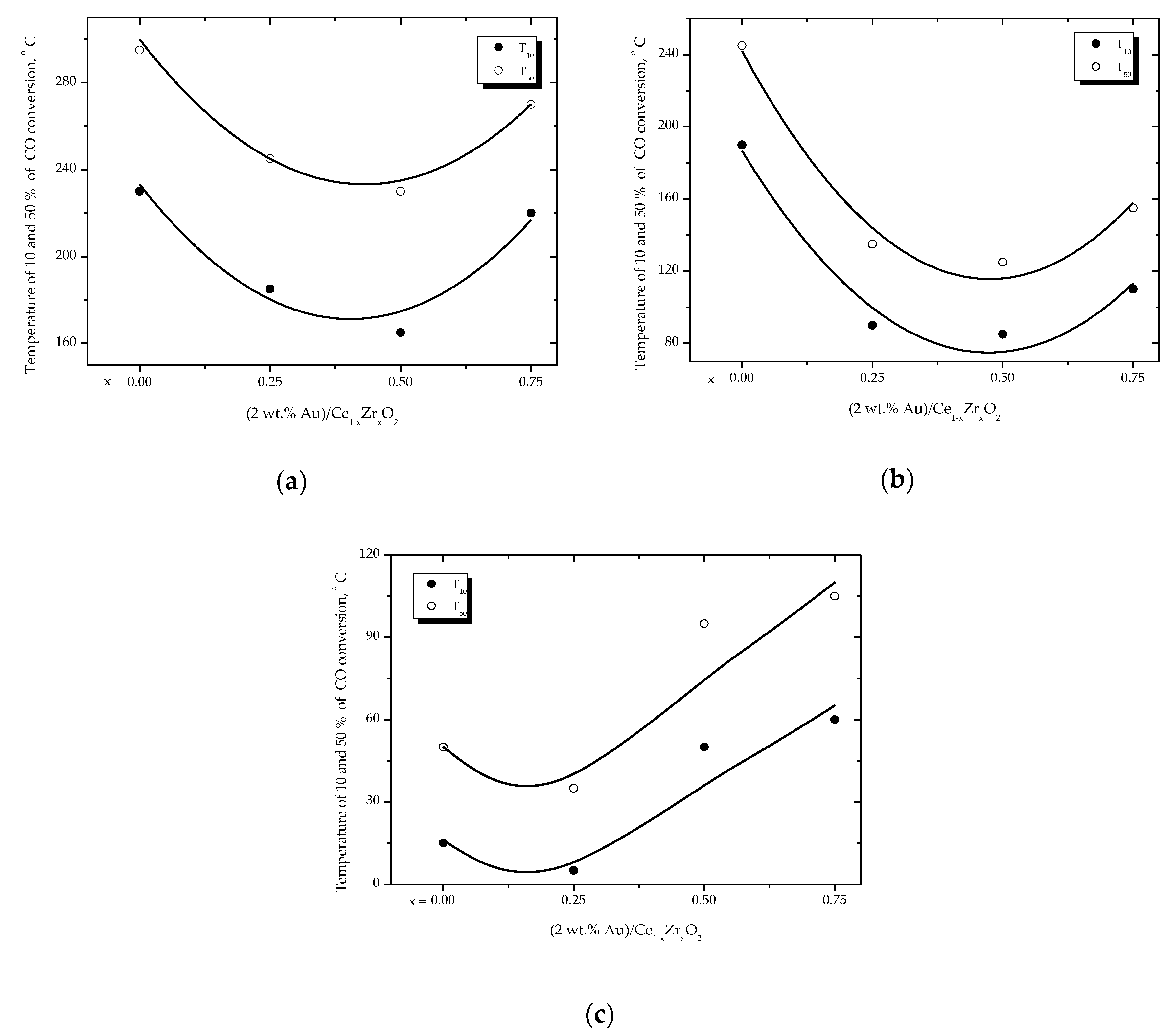
2.2. Effect of the Support Composition (Ce/Zr Molar Ratio)
2.3. Influence of the Pretreatment Atmosphere
3. Materials and Methods
3.1. Synthesis of Catalysts
3.2. Characterization Methods
4. Conclusions
- High activity of (2 wt. % Au)/Ce1−xZrxO2 catalysts in low-temperature CO oxidation is displayed by Au nanoparticles with an optimum size of ca. 4 nm. In the presence of residual chlorine, Au agglomeration during the calcination process occurs. Washing of the catalysts with ammonia effectively prevents Au sintering. The smallest Au nanoparticles and the highest activity in CO oxidation presented catalysts washed with ammonia.
- The electronic properties of nanosized Au can be modified by the oxide support. The structure and properties of oxide support define the nature of Au-support interactions. High oxygen mobility at low temperatures is critical for high activity. The catalytic performance of (2 wt. % Au)/Ce1−xZrxO2 contacts strongly depends on the support’s redox properties. Oxygen vacancies on the support surface are centers for oxygen molecule activation.
- Concerning Au active species: (i) Two active forms of Au can be responsible for high activity in CO oxidation: Au0 and Au3+. The Au0 is crucial for CO adsorption. The Au3+ species, in conjunction with Au0, are essential for the activation of surface hydroxyl groups. Next, the activated OH groups can react with the adsorbed CO to form carboxylate species. However, a high amount of Aux+ or Au° decreases the activity. In fact, the appropriate ratio of Aux+ and Au° is critical to obtain the highest catalytic activity in CO oxidation; (ii) The adsorption and activation of O2 take place on the support oxygen vacancies; (iii) The moisture can both enhance the dissociation of O2 molecules adsorbed on the oxygen vacancies and modify the electronic state of gold. Next, the activated oxygen can rapidly react with CO adsorbed on the metallic Au0; (iv) High CO oxidation yields require the presence of water-derived species on the catalyst surface. Thus, the moisture adsorbed on the catalyst surface, rather than its content in the feed stream, is responsible for the creation of the catalytic performance.
- Regarding the reaction mechanism: The Au-assisted MvK model was suggested as more relevant to describe the kinetics of CO oxidation over Ce1−xZrxO2 and (2 wt. % Au)/Ce1−xZrxO2 catalysts. Consequently, the principal role of Au in CO oxidation over (2 wt. % Au)/Ce1−xZrxO2 catalysts can be related to the promotion in the transformation process of reversibly adsorbed or inactive surface oxygen into irreversibly adsorbed species active in CO oxidation. Additionally, at oxygen-lean conditions, the activity seems to be dependent on the ability of the oxide to release first the surface and then bulk oxygen. Thus, considering that extra oxygen is available from the catalyst surface bulk, it was possible to properly fit the observed catalyst activity. Finally, the proposed fitting procedure, based on MvK expression, can be considered as successful for all conditions tested for the CO oxidation reaction.
Author Contributions
Funding
Conflicts of Interest
References
- Lykaki, M.; Stefa, S.; Carabineiro, S.A.C.; Pandis, P.K.; Stathopoulos, V.N.; Konsolakis, M. Facet-Dependent Reactivity of Fe2O3/CeO2 Nanocomposites: Effect of Ceria Morphology on CO Oxidation. Catalysts 2019, 9, 371. [Google Scholar] [CrossRef]
- Soliman, N.K. Factors Affecting CO Oxidation Reaction over Nanosized Materials: A Review. J. Mater. Res. Technol. 2019, 8, 2395–2407. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Gold-catalyzed oxidation of carbon monoxide. Cat. Rev. Sci. Eng. 1999, 41, 319. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7937. [Google Scholar] [CrossRef]
- Du, M.; Sun, D.; Yang, H.; Huang, J.; Jing, X.; Odoom-Wubah, T.; Wang, H.; Jia, L.; Li, Q. Influence of Au particle size on Au/TiO2 catalysts for CO oxidation. J. Phys. Chem. C 2014, 118, 19150–19157. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared of coprecipitation for low-temperature oxidation of hydrogen and carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Haruta, M.; Date, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A Gen. 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Zhou, A.; Wang, J.; Wang, H.; Li, H.; Wang, J.; Shen, M. Effect of active oxygen on the performance of Pt/CeO2 catalysts for CO oxidation. J. Rare Earths. 2018, 36, 257–264. [Google Scholar] [CrossRef]
- Haruta, M. Size- and Support-Dependency in the Catalysis of Gold. Catal. Today 1996, 36, 153–166. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, L.; Chen, M.; Lv, C.; Lian, X.; Wu, C.; Yang, B.; Miao, Z.; Wang, F.; Hu, X. CO Oxidation over Metal Oxide (La2O3, Fe2O3, PrO2, Sm2O3, and MnO2) Doped CuO-Based Catalysts Supported on Mesoporous Ce0.8Zr0.2O2 with Intensified Low-Temperature Activity. Catalysts 2019, 9, 724. [Google Scholar] [CrossRef]
- Bansmann, J.; Abdel-Mageed, A.M.; Chen, S.; Fauth, C.; Häring, T.; Kucerová, G.; Wang, Y.; Behm, R.J. Chemical and Electronic Changes of the CeO2 Support during CO Oxidation on Au/CeO2 Catalysts: Time-Resolved Operando XAS at the Ce LIII Edge. Catalysts 2019, 9, 785. [Google Scholar] [CrossRef]
- Sui, C.; Xing, L.; Cai, X.; Wang, Y.; Zhou, Q.; Li, M. Co-Supported CeO2 Nanoparticles for CO Catalytic Oxidation: Effects of Different Synthesis Methods on Catalytic Performance. Catalysts 2020, 10, 243. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Tanaka, S.; Jikihara, Y.; Nakayama, T.; Bando, Y.; Haruta, M. Room Temperature Carbon Monoxide Oxidation Based on Two-dimensional Gold-loaded Mesoporous Iron Oxide Nanoflakes. Chem. Commun. 2018, 54, 8514–8517. [Google Scholar] [CrossRef] [PubMed]
- Zanella, R.; Giorgio, S.; Henry, C.R.; Louis, C. Alternative methods for the preparation of gold nanoparticles supported on TiO2. J. Phys. Chem. B 2002, 106, 7634–7642. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. The influence of the preparation methods on the catalytic activity of platinum and gold supported on TiO2 for CO oxidation. Catal. Lett. 1997, 44, 83–87. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Kiener, C.; Wögerbauer, C.; Baiker, A. Preparation of supported gold catalysts for low-temperature CO oxidation via “size-controlled” gold colloids. J. Catal. 1999, 181, 223–232. [Google Scholar] [CrossRef]
- Ivanova, S.; Petit, C.; Pitchon, V. A new preparation method for the formation of gold nanoparticles on an oxide support. Appl. Catal. A Gen. 2004, 267, 191–201. [Google Scholar] [CrossRef]
- Dobrosz, I.; Jiratova, K.; Pitchon, V.; Rynkowski, J.M. Effect of the preparation of supported gold particles on the catalytic activity in CO oxidation reaction. J. Mol. Catal. A Chem. 2005, 234, 187–197. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Dobrosz-Gómez, I. Ceria-Zirconia Supported Gold Catalysts. Annales Universitatis Mariae Curie-Skłodowska. Ann. UMCS Chem. 2009, 64, 197–217. [Google Scholar]
- Trovarelli, A.; Fornasiero, P. (Eds.) Catalysis by Ceria and Related Materials, 2nd ed.; Imperial College Press: London, UK, 2013. [Google Scholar]
- Dobrosz-Gómez, I.; Kocemba, I.; Rynkowski, J.M. Au/Ce1-xZrxO2 as effective catalysts for low-temperature CO oxidation. Appl. Catal. B Environ. 2008, 83, 240–255. [Google Scholar] [CrossRef]
- Dobrosz-Gómez, I.; Gómez-García, M.Á.; Szynkowska, M.I.; Kocemba, I.; Rynkowski, J.M. Surface, structural and morphological characterization of nanocrystalline ceria–zirconia mixed oxides upon thermal aging. Catal. Today 2012, 191, 142–145. [Google Scholar] [CrossRef]
- Dobrosz, I.; Gómez-García, M.Á.; Kocemba, I.; Maniukiewicz, W.; Rynkowski, J.M. Characterization of Au/Ce1-xZrxO2 catalysts in CO oxidation. Polish J. Environ. Studies 2005, 14, 231–234. [Google Scholar]
- Dobrosz-Gómez, I.; Kocemba, I.; Rynkowski, J.M. Factors influencing structure and catalytic activity of Au/Ce1-xZrxO2 catalysts in CO oxidation. Appl. Catal. B Environ. 2009, 88, 83–97. [Google Scholar] [CrossRef]
- Daturi, M.; Binet, C.; Lavalley, J.C.; Blanchard, G. Surface FTIR investigations on CexZr1−xO2 system. Surf. Interface. Anal. 2000, 30, 273–277. [Google Scholar] [CrossRef]
- Su, X.; Yang, X.F.; Huang, Y.Q.; Liu, B.; Zhang, T. Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products. Acc. Chem. Res. 2019, 52, 656–664. [Google Scholar] [CrossRef]
- Wang, W.W.; Du, P.P.; Zou, S.H.; He, H.Y.; Wang, R.X.; Jin, Z.; Shi, S.; Huang, Y.Y.; Si, R.; Song, Q.S.; et al. Highly Dispersed Copper Oxide Clusters as Active Species in Copper-Ceria Catalyst for Preferential Oxidation of Carbon Monoxide. ACS Catal. 2015, 5, 2088–2099. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. A DFT Study on the Possibility of Using A Single Cu Atom Incorporated Nitrogen-Doped Graphene as A Promising and Highly Active Catalyst for Oxidation of CO. Int. J. Quant. Chem. 2019, 119, e25857. [Google Scholar] [CrossRef]
- Wang, H.; Shen, J.H.; Huang, J.F.; Xu, T.J.; Zhu, J.R.; Zhu, Y.H.; Li, C.Z. Atomically Dispersed Au Catalysts Supported on CeO2 Foam: Controllable Synthesis and CO Oxidation Reaction Mechanism. Nanoscale 2017, 9, 16817–16825. [Google Scholar] [CrossRef]
- Qiao, B.T.; Liu, J.X.; Wang, Y.G.; Lin, Q.Q.; Liu, X.Y.; Wang, A.Q.; Li, J.; Zhang, T.; Liu, J.Y. Highly Efficient Catalysis of Preferential Oxidation of CO in H2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249–6254. [Google Scholar] [CrossRef]
- Al Soubaihi, R.M.; Saoud, K.M.; Dutta, J. Critical Review of Low-Temperature CO Oxidation and Hysteresis Phenomenon on Heterogeneous Catalysts. Catalysts 2018, 8, 660. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.-G.; Li, J. Theoretical Investigations of Pt-CeO2 Single-Atom Catalyst for CO Oxidation. J. Phys. Chem. C. 2017, 121, 11281–11289. [Google Scholar] [CrossRef]
- Feng, Y.X.; Wang, Q.; Xiong, H.F.; Zhou, S.L.; Chen, X.; Hernandez, X.I.P.; Wang, Y.; Lin, S.; Datye, A.K.; Guo, H. Correlating DFT Calculations with CO Oxidation Reactivity on Ga-Doped Pt/CeO2 Single-Atom Catalysts. J. Phys. Chem. C 2018, 122, 22460–22468. [Google Scholar] [CrossRef]
- Kropp, T.; Lu, Z.; Li, Z.; Chin, Y.-H.C.; Mavrikakis, M. Anionic Single-Atom Catalysts for CO Oxidation: Support-Independent Activity at Low Temperatures. ACS Catal. 2019, 9, 1595–1604. [Google Scholar] [CrossRef]
- Yu, M.; Feng, Y.; Gao, L.; Lin, S. Phosphomolybdic Acid Supported Single-Metal-Atom Catalysis in CO Oxidation: First-Principles Calculations. Phys. Chem. Chem. Phys. 2018, 20, 20661–20668. [Google Scholar] [CrossRef]
- Wang, S.; Feng, Y.; Lin, S.; Guo, H. Phosphomolybdic Acid Supported Atomically Dispersed Transition Metal Atoms (M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au): Stable Single Atom Catalysts Studied by Density Functional Theory. RSC Adv. 2017, 7, 24925–24932. [Google Scholar] [CrossRef]
- Liu, X.; Sui, Y.H.; Duan, T.; Meng, C.G.; Han, Y. CO Oxidation Catalyzed by Pt-Embedded Graphene: A First-Principles Investigation. Phys. Chem. Chem. Phys. 2014, 16, 23584–23593. [Google Scholar] [CrossRef]
- Tang, Y.N.; Dai, X.Q.; Yang, Z.X.; Pan, L.J.; Chen, W.G.; Ma, D.W.; Lu, Z.S. Formation and Catalytic Activity of Pt Supported on Oxidized Graphene for the CO Oxidation Reaction. Phys. Chem. Chem. Phys. 2014, 16, 7887–7895. [Google Scholar] [CrossRef]
- Morgan, K.; Cole, K.J.; Goguet, A.; Hardacre, C.; Hutchings, G.J.; Maguire, N.; Shekhtman, S.O.; Taylor, S.H. TAP studies of CO oxidation over CuMnOX and Au/CuMnOX catalysts. J. Catal. 2010, 276, 38–48. [Google Scholar] [CrossRef]
- Schlexer, P.; Widmann, D.; Behm, R.J.; Pacchioni, G. CO Oxidation on a Au/TiO2 Nanoparticle Catalyst via the Au-Assisted Mars–van Krevelen Mechanism. ACS Catal. 2018, 8, 6513–6525. [Google Scholar] [CrossRef]
- Saqlain, M.A.; Hussain, A.; Siddiq, M.; Leitao, A.A. A DFT+U study of the Mars van Krevelen Mechanism of CO Oxidation on Au/TiO2 catalysts. Appl. Catal. A 2016, 519, 27–33. [Google Scholar] [CrossRef]
- Duan, Z.; Henkelman, G. CO Oxidation at the Au/TiO2 Boundary: The Role of the Au/Ti5c Site. ACS Catal. 2015, 5, 1589. [Google Scholar] [CrossRef]
- Widmann, D.; Behm, R.J. Activation of Molecular Oxygen and the Nature of the Active Oxygen Species for CO Oxidation on Oxide Supported Au Catalysts. Acc. Chem. Res. 2014, 47, 740–749. [Google Scholar] [CrossRef]
- Dobrosz, I.; Kocemba, I.; Rynkowski, J.M. The Optimisation of Au/Ce0.75Zr0.25O2 Catalyst Preparation for CO Oxidation. Pol. J. Environ. Studies 2006, 15, 44. [Google Scholar]
- Dobrosz-Gómez, I.; Kocemba, I.; Rynkowski, J.M. Carbon Monoxide Oxidation over Au/Ce1-xZrxO2 Catalysts: Effects of Moisture Content in the Reactant Gas and Catalyst Pretreatment. Catal. Lett. 2009, 128, 297–306. [Google Scholar] [CrossRef]
- Dobrosz-Gómez, I.; Gómez-García, M.Á.; Rynkowski, J.M. The Role of Au-Support Interactions in Creation of Catalytic Performance of Au/Ce0.75Zr0.25O2 in CO Oxidation. Pol. J. Environ. Studies 2009, 18, 587–591. [Google Scholar]
- Dobrosz-Gómez, I.; Gómez-García, M.Á.; Rynkowski, J.M. CO Oxidation over Au/CeO2–ZrO2 Catalysts: The Effect of the Support Composition of the Au Support Interaction. Kinet. Catal. 2010, 51, 823–827. [Google Scholar] [CrossRef]
- Gómez-García, M.Á.; Gómez-Mendoza, N.A.; Dobrosz-Gómez, I.; Gil Pavas, E.; Rynkowski, J.M. Temperature-Scanning Method for the Kinetic Studies of CO Oxidation over Ceria–Zirconia Supported Gold Catalysts. Chem. Eng. J. 2015, 282, 20–28. [Google Scholar] [CrossRef]
- Force, C.; Belzunegui, J.P.; Sanz, J.; Martinez-Arias, A.; Soria, J. Influence of precursor salt on metal particle formation in Rh/CeO2 catalysts. J. Catal. 2001, 197, 192–199. [Google Scholar] [CrossRef]
- Ivanova, S.; Pitchon, V.; Zimmermann, Y.; Petit, C. Preparation of alumina supported gold catalysts: Influence of washing procedures, mechanism of particles size growth. Appl. Catal. A Gen. 2006, 298, 57–64. [Google Scholar] [CrossRef]
- Schulz, A.; Hargittai, M. Structural variations and bonding in gold halides: A quantum chemical study of monometric and dimetric gold monohalide and gold trihalide molecules, AuX, Au2X2, AuX3, and Au2X6 (X = F, Cl, Br, I). Chem. Eur. J. 2001, 7, 3657–3670. [Google Scholar] [CrossRef]
- Arena, F.; Famulari, P.; Trunfio, G.; Bonura, G.; Frusteri, F.; Spadaro, L. Physicochemical properties and reactivity of Au/CeO2 catalysts in total and selective oxidation of CO. Appl. Catal. B Environ. 2006, 66, 81–91. [Google Scholar] [CrossRef]
- Fu, L.; Wu, N.Q.; Yang, J.H.; Qu, F.; Johnson, D.L.; Kung, M.C.; Kung, H.H.; Dravid, V.P. Direct evidence of oxidized gold on supported gold catalysts. J. Phys. Chem. Lett. B 2005, 109, 3704–3706. [Google Scholar] [CrossRef]
- Cao, A.; Lu, R.; Veser, G. Stabilizing Metal Nanoparticles for Heterogeneous Catalysis. Phys. Chem. Chem. Phys. 2010, 12, 13499–13510. [Google Scholar] [CrossRef]
- Cuenya, B.R. Synthesis and Catalytic Properties of Metal Nanoparticles: Size, Shape, Support, Composition, and Oxidation State Effects. Thin Solid Films 2010, 518, 3127–3150. [Google Scholar] [CrossRef]
- Jóźwiak, W.K.; Kaczmarek, E.; Ignaczak, W. Activity requirements of gold catalysts Au/Fe2O3 in low-temperature CO oxidation. Pol. J. Environ. Studies 2005, 14, 127. [Google Scholar]
- Moreau, F.; Bond, G.C.; Taylor, A.O. Gold on Titania Catalysts for the Oxidation of Carbon Monoxide: Control of PH during Preparation with Various Gold Contents. J. Catal. 2005, 231, 105–114. [Google Scholar] [CrossRef]
- Zanella, R.; Delannoy, L.; Louis, C. Mechanism of Deposition of Gold Precursors onto TiO2 during the Preparation by Cation Adsorption and Deposition–Precipitation with NaOH and Urea. Appl. Catal. A 2005, 291, 62–72. [Google Scholar] [CrossRef]
- Grzybowska-Świerkosz, B. Nano-Au/oxide Support Catalysts in Oxidation Reactions: Provenance of Active Oxygen Species. Catal. Today 2006, 112, 3–7. [Google Scholar] [CrossRef]
- Guzmán, J.; Carrettin, S.; Corma, A. Spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef]
- Cunningham, D.A.H.; Vogel, W.; Haruta, M. Structural analysis of Au/TiO2 catalysts by debye function analysis. Catal. Lett. 1999, 63, 43–47. [Google Scholar] [CrossRef]
- Olea, M.; Tada, M.; Iwasawa, Y. TAP study on carbon monoxide oxidation over supported gold catalysts Au/Ti(OH)*4 and Au/Fe(OH)*3: Moisture effect. J. Catal. 2007, 248, 60–67. [Google Scholar] [CrossRef]
- Costello, C.K.; Yang, J.H.; Law, H.Y.; Wang, Y.; Lin, J.-N.; Marks, L.D.; Kung, M.C.; Kung, H.H. On the potential role of hydroxyl groups in CO oxidation over Au/Al2O3. Appl. Catal. A. Gen. 2003, 243, 15–24. [Google Scholar] [CrossRef]
- Ruszel, M.; Grzybowska, B.; Samson, K.; Gressel, I.; Klisinska, A. MIICr2O4-spinels as supports for Au nanoparticles in oxidation of CO. Catal. Today 2006, 112, 126–129. [Google Scholar] [CrossRef]
- Kang, Y.-M.; Wan, B.-Z. Gold and iron supported on Y-type zeolite for carbon monoxide oxidation. Catal. Today 1997, 35, 379–392. [Google Scholar] [CrossRef]
- Visco, A.M.; Neri, F.; Neri, G.; Donato, A.; Milone, C.; Galvagno, S.X. X-ray photoelectron spectroscopy of Au/Fe2O3 catalysts. Phys. Chem. Chem. Phys. 1999, 1, 2869–2873. [Google Scholar] [CrossRef]
- Pillai, U.R.; Deevi, S. Highly active gold-ceria catalyst for the room temperature oxidation of carbon monoxide. Appl. Catal. A Gen. 2006, 299, 266–273. [Google Scholar] [CrossRef]
- Abd El-Moemen, A.; Karpenko, A.; Denkwitz, Y.; Behm, R.J. Activity, stability and deactivation behavior of Au/CeO2 catalysts in the water gas shift reaction at increased reaction temperature (300 °C). J. Power Sources 2009, 190, 64–75. [Google Scholar] [CrossRef]
- Widmann, D.; Leppelt, R.; Behm, R.J. Activation of a Au/CeO2 catalyst for the CO oxidation reaction by surface oxygen removal/oxygen vacancy formation. J. Catal. 2007, 251, 437–442. [Google Scholar] [CrossRef]
- Wang, A.Q.; Hsieh, Y.P.; Chen, Y.F.; Mou, C.Y. Au–Ag alloy nanoparticle as catalyst for CO oxidation: Effect of Si/Al ratio of mesoporous support. J. Catal. 2006, 237, 197–206. [Google Scholar] [CrossRef]
- Qian, K.; Huang, W.; Fang, J.; Lv, S.; He, B.; Jiang, Z.; Wei, S. Low-temperature CO oxidation over Au/ZnO/SiO2 catalysts: Some mechanism insights. J. Catal. 2008, 255, 269–278. [Google Scholar] [CrossRef]
- del Río, E.; Collins, S.E.; Aguirre, A.; Chen, X.; Delgado, J.J.; Calvino, J.J.; Bernal, S. Reversible deactivation of a Au/Ce0.62Zr0.38O2 catalyst in CO oxidation: A systematic study of CO2-triggered carbonate inhibition. J. Catal. 2014, 316, 210–218. [Google Scholar] [CrossRef]
- Provendier, H.; Petit, C.; Schmitt, J.L.; Kiennemann, A.; Chaumont, C. Characterization of the solid solution La(Ni,Fe)O3 prepared via sol-gel related method using propionic acid. J. Mat. Sci. 1999, 34, 4121–4127. [Google Scholar] [CrossRef]
- Johnson, M.F.L.; Mooi, J. Cerium dioxide crystallite sizes by temperature-programmed reduction. J. Catal. 1987, 103, 502. [Google Scholar] [CrossRef]
- Johnson, M.F.L.; Mooi, J. Cerium dioxide crystallite sizes by temperature-programmed reduction. J. Catal. 1993, 140, 612. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrosz-Gómez, I.; Gómez-García, M.-Á.; Rynkowski, J.M. The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation. Catalysts 2020, 10, 1312. https://doi.org/10.3390/catal10111312
Dobrosz-Gómez I, Gómez-García M-Á, Rynkowski JM. The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation. Catalysts. 2020; 10(11):1312. https://doi.org/10.3390/catal10111312
Chicago/Turabian StyleDobrosz-Gómez, Izabela, Miguel-Ángel Gómez-García, and Jacek Michał Rynkowski. 2020. "The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation" Catalysts 10, no. 11: 1312. https://doi.org/10.3390/catal10111312
APA StyleDobrosz-Gómez, I., Gómez-García, M.-Á., & Rynkowski, J. M. (2020). The Origin of Au/Ce1-xZrxO2 Catalyst’s Active Sites in Low-Temperature CO Oxidation. Catalysts, 10(11), 1312. https://doi.org/10.3390/catal10111312