Transition Metal Catalyzed Azidation Reactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
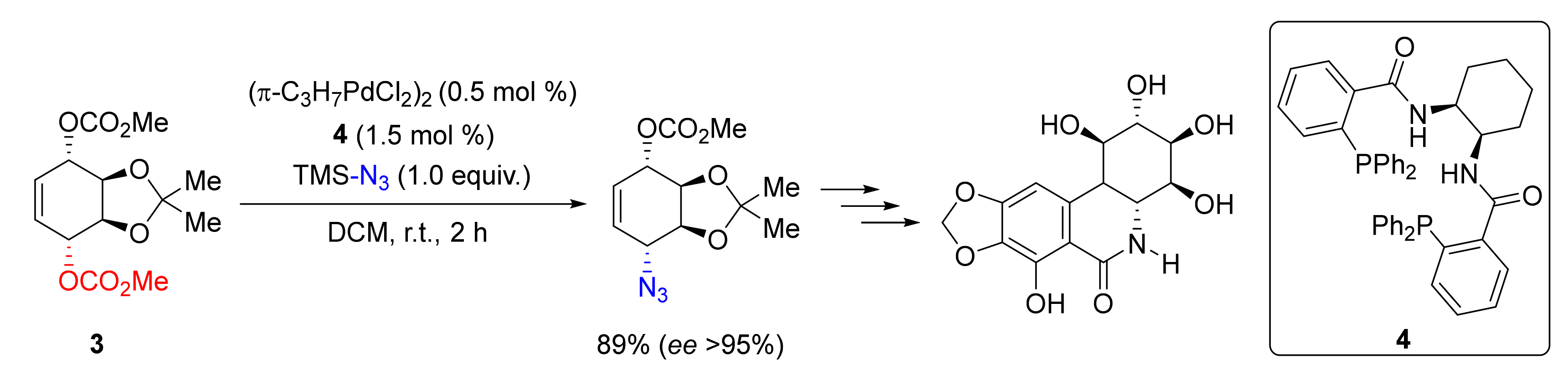
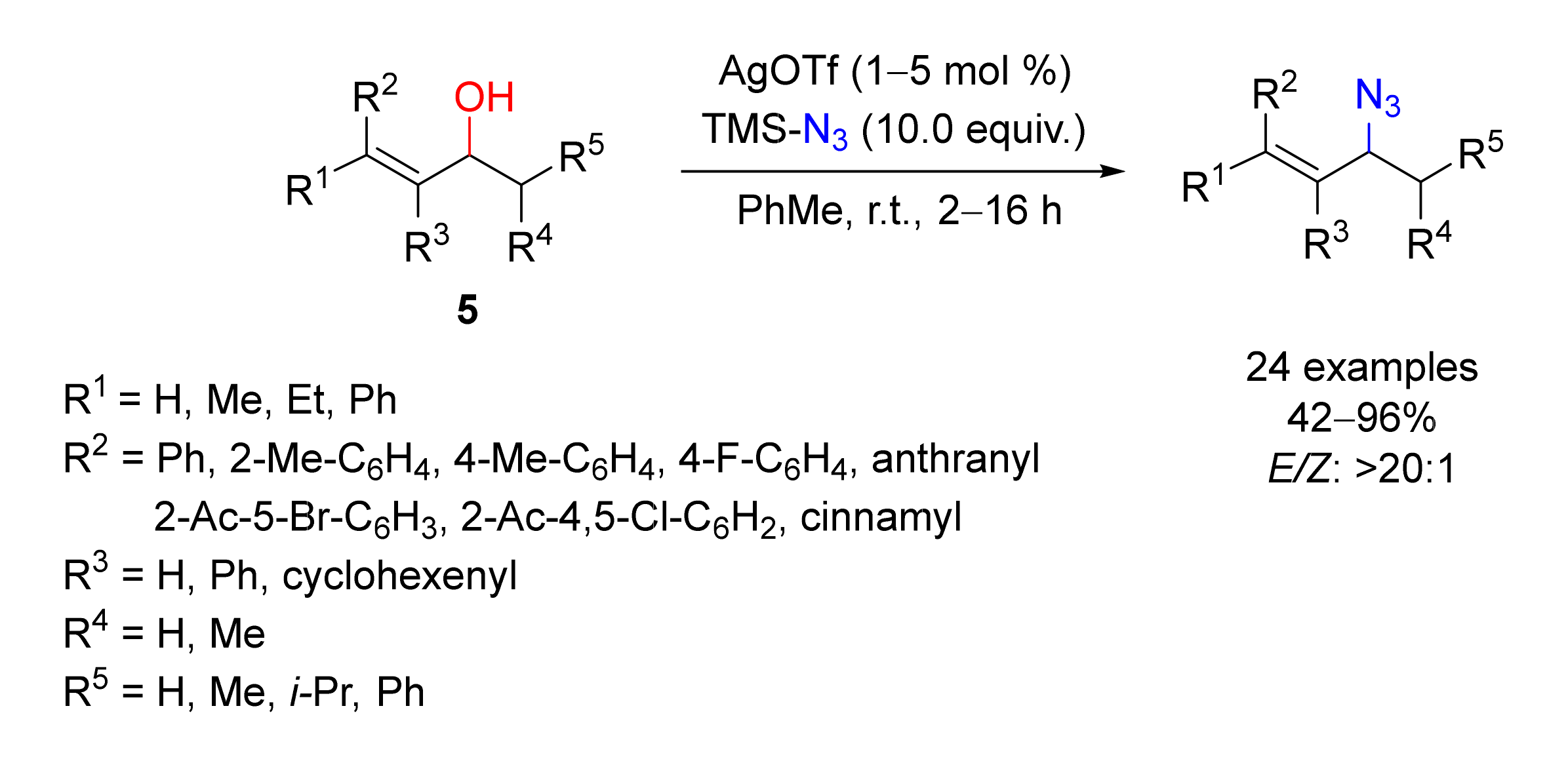
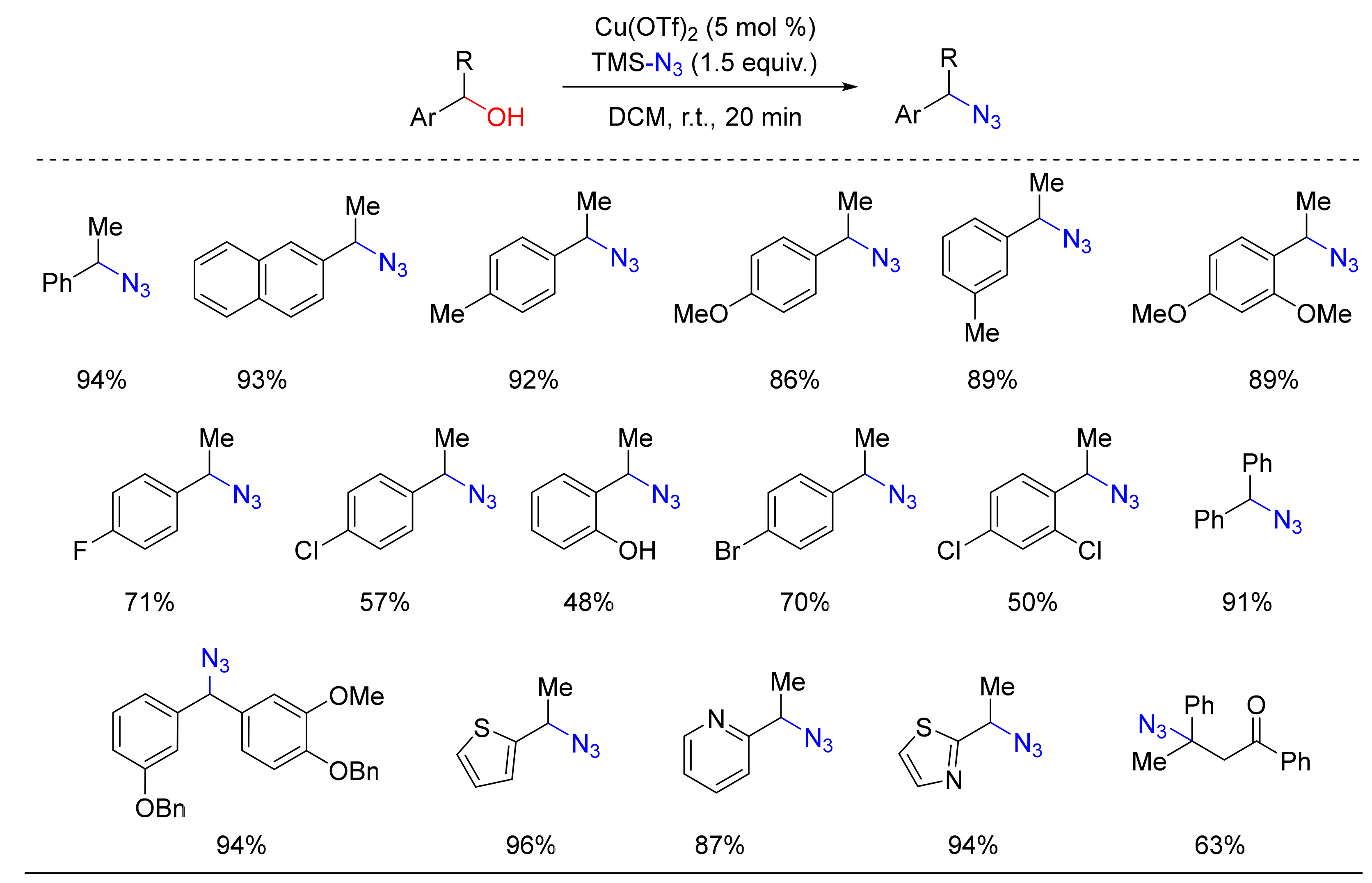
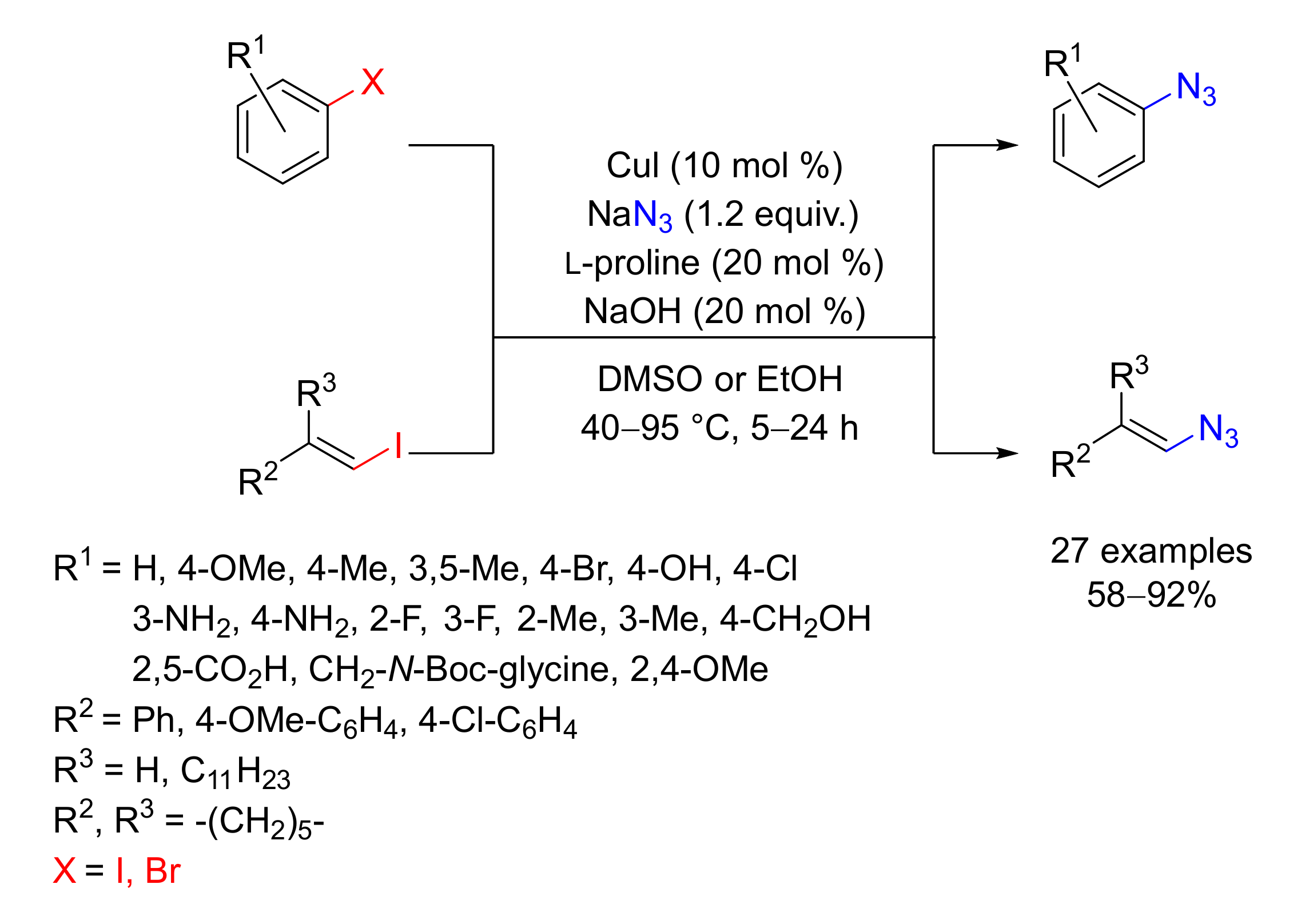
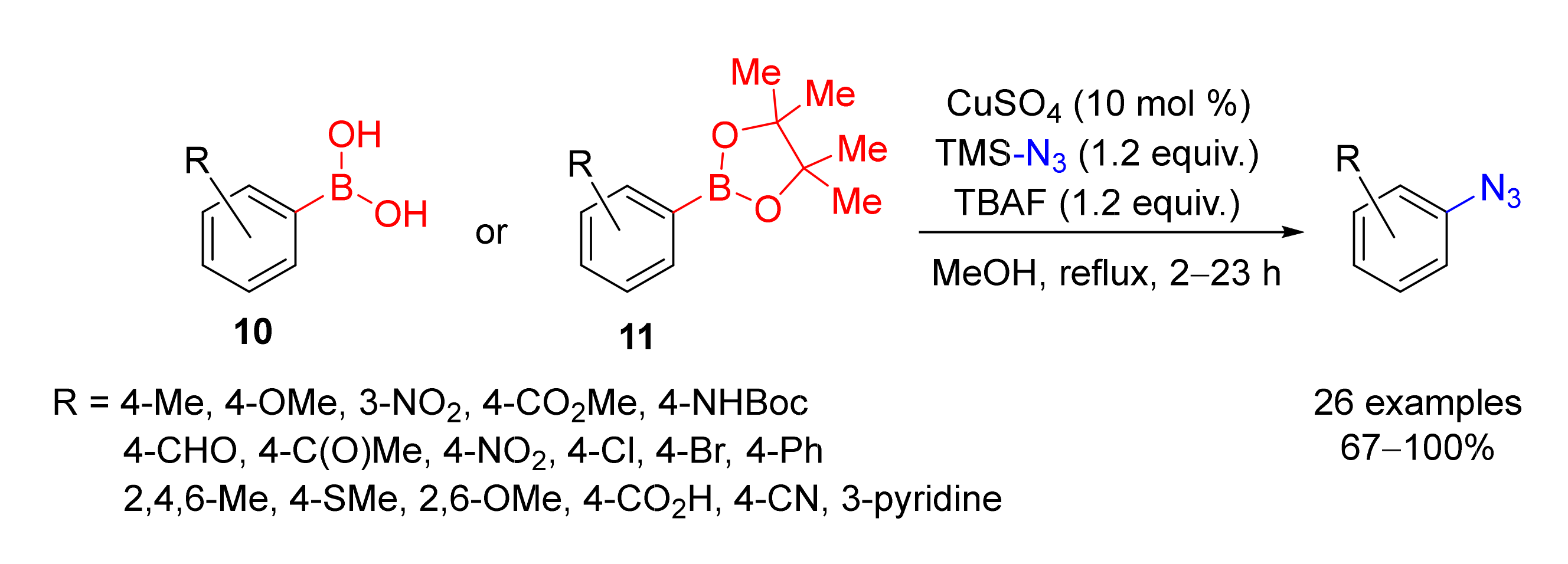
2. Azidation of Substrates Bearing a Leaving Group
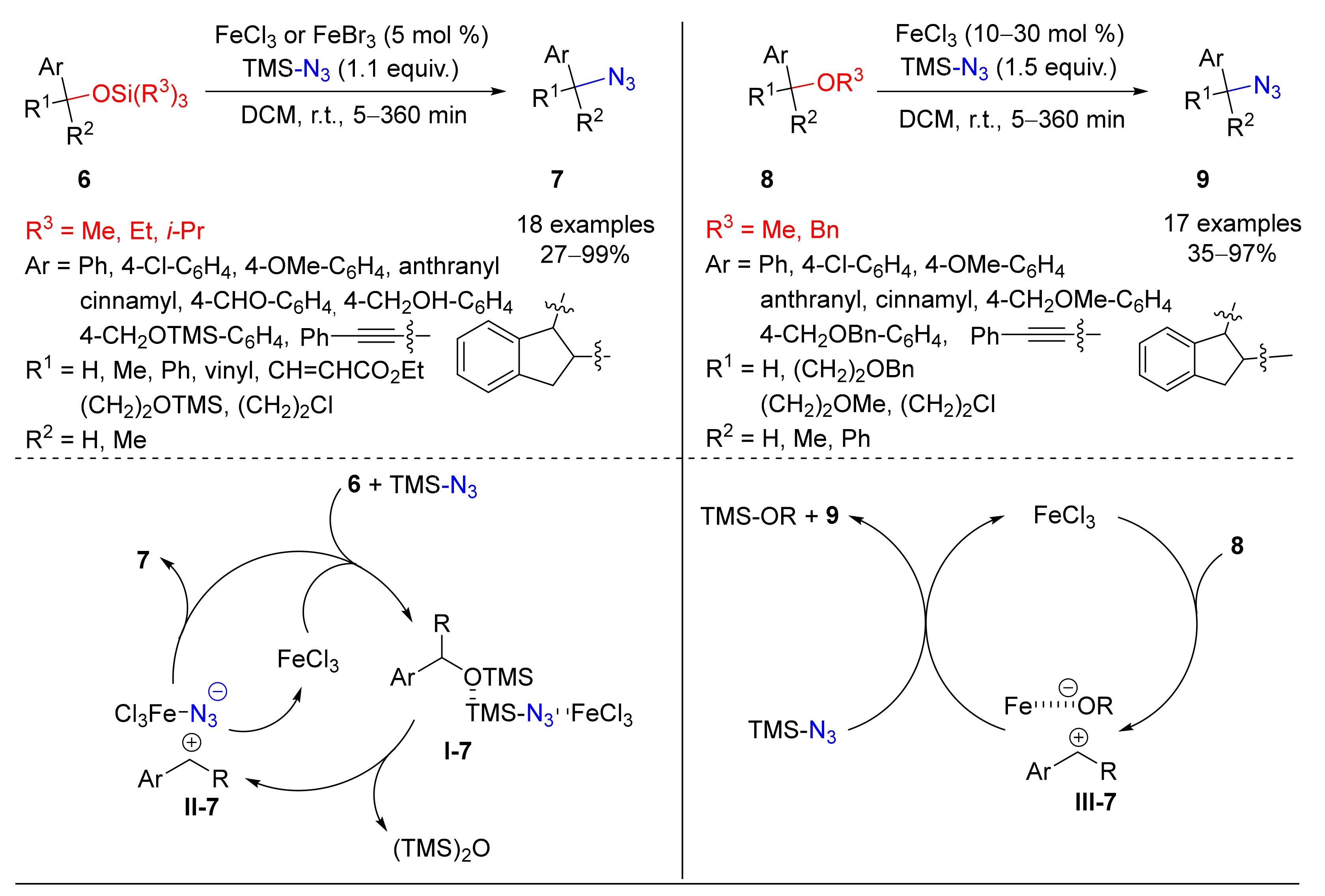
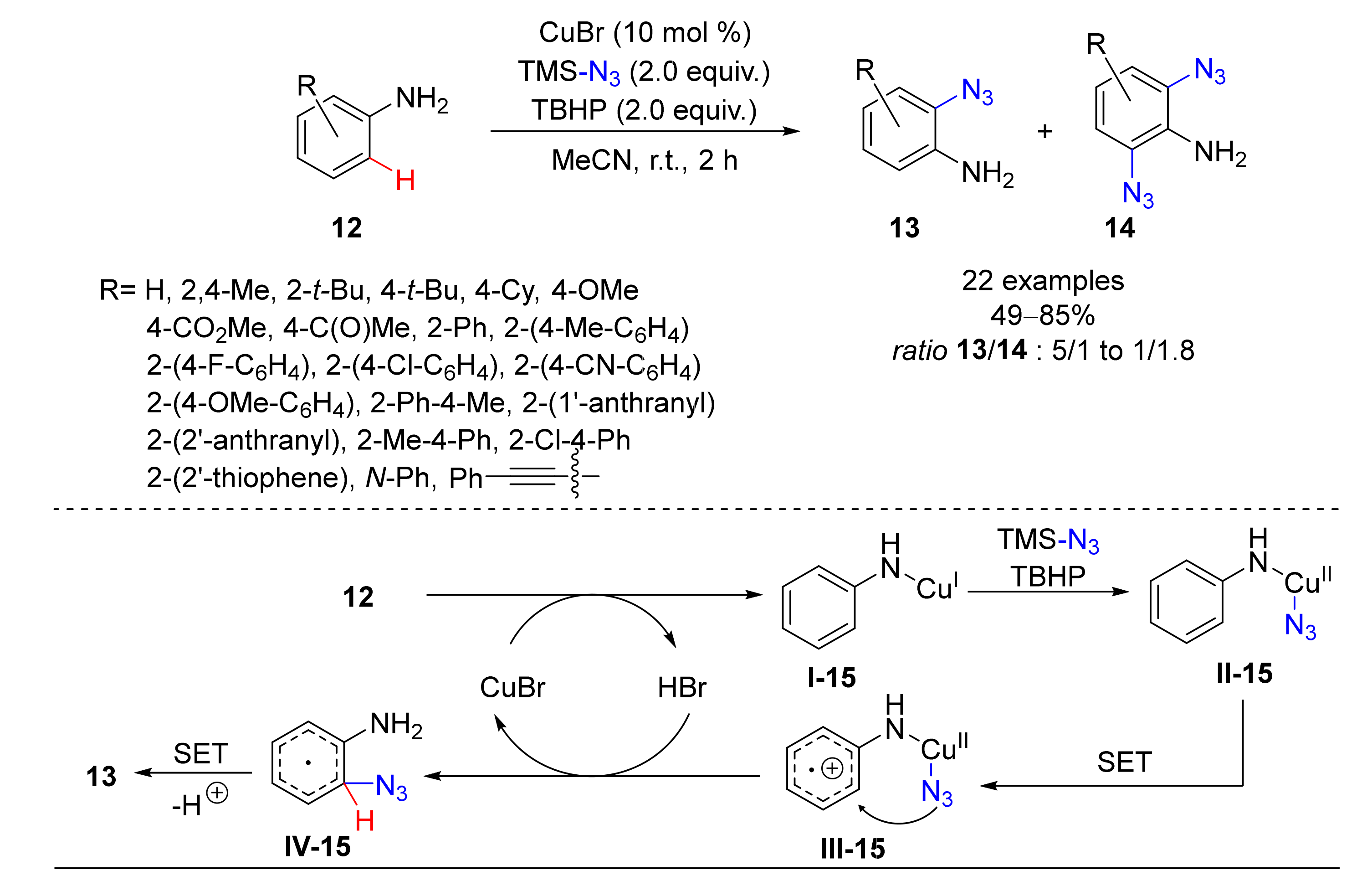
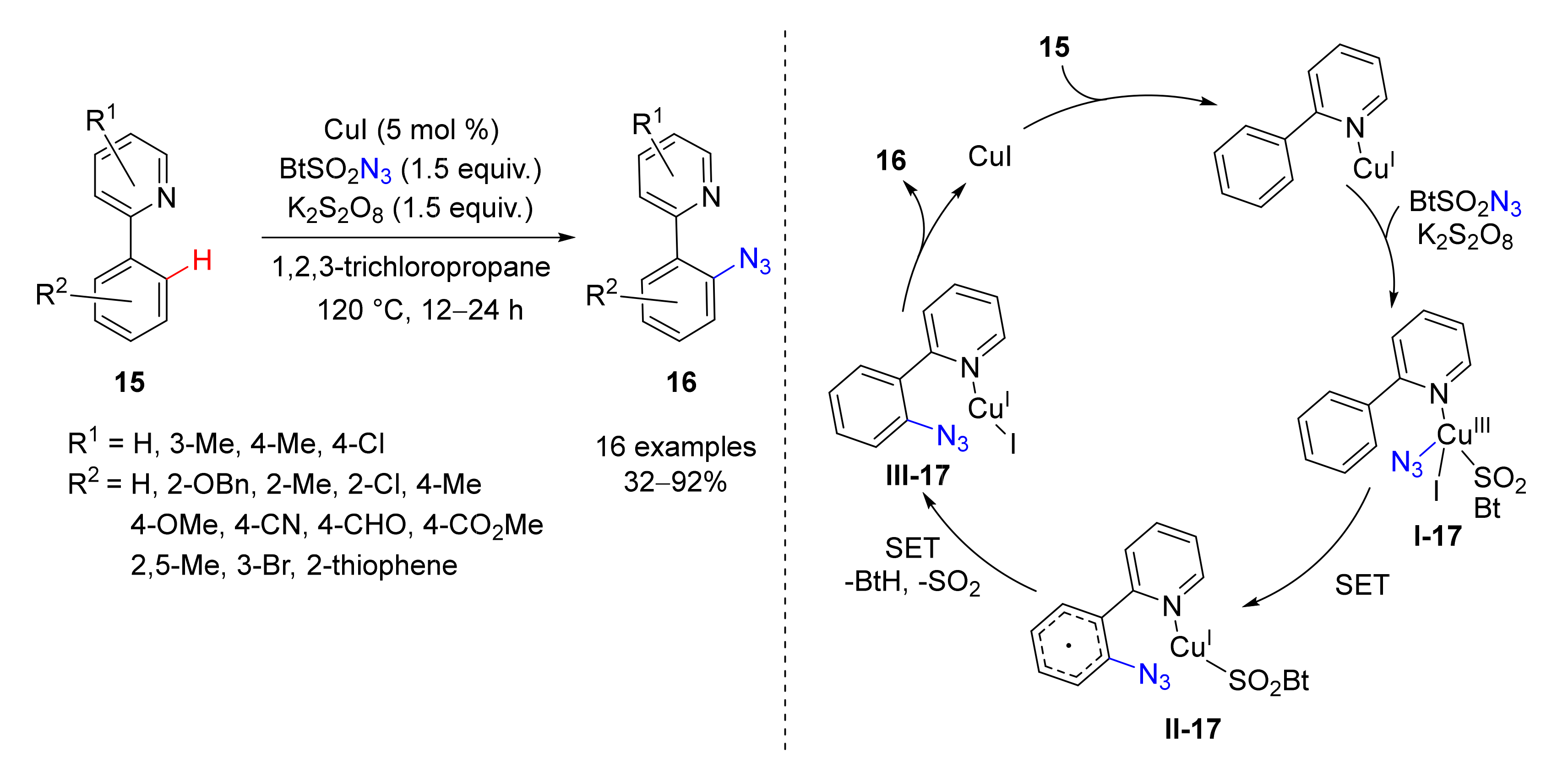
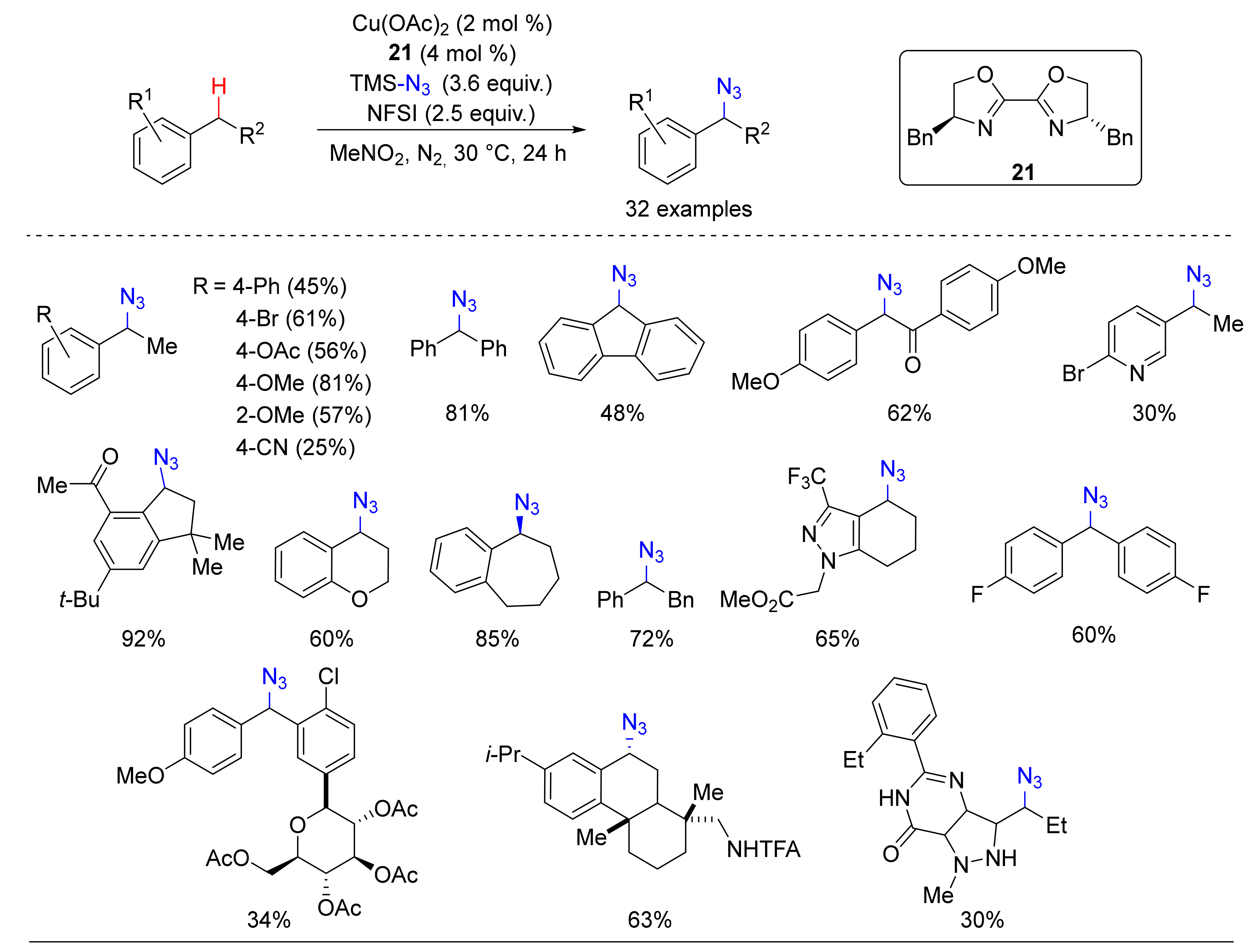
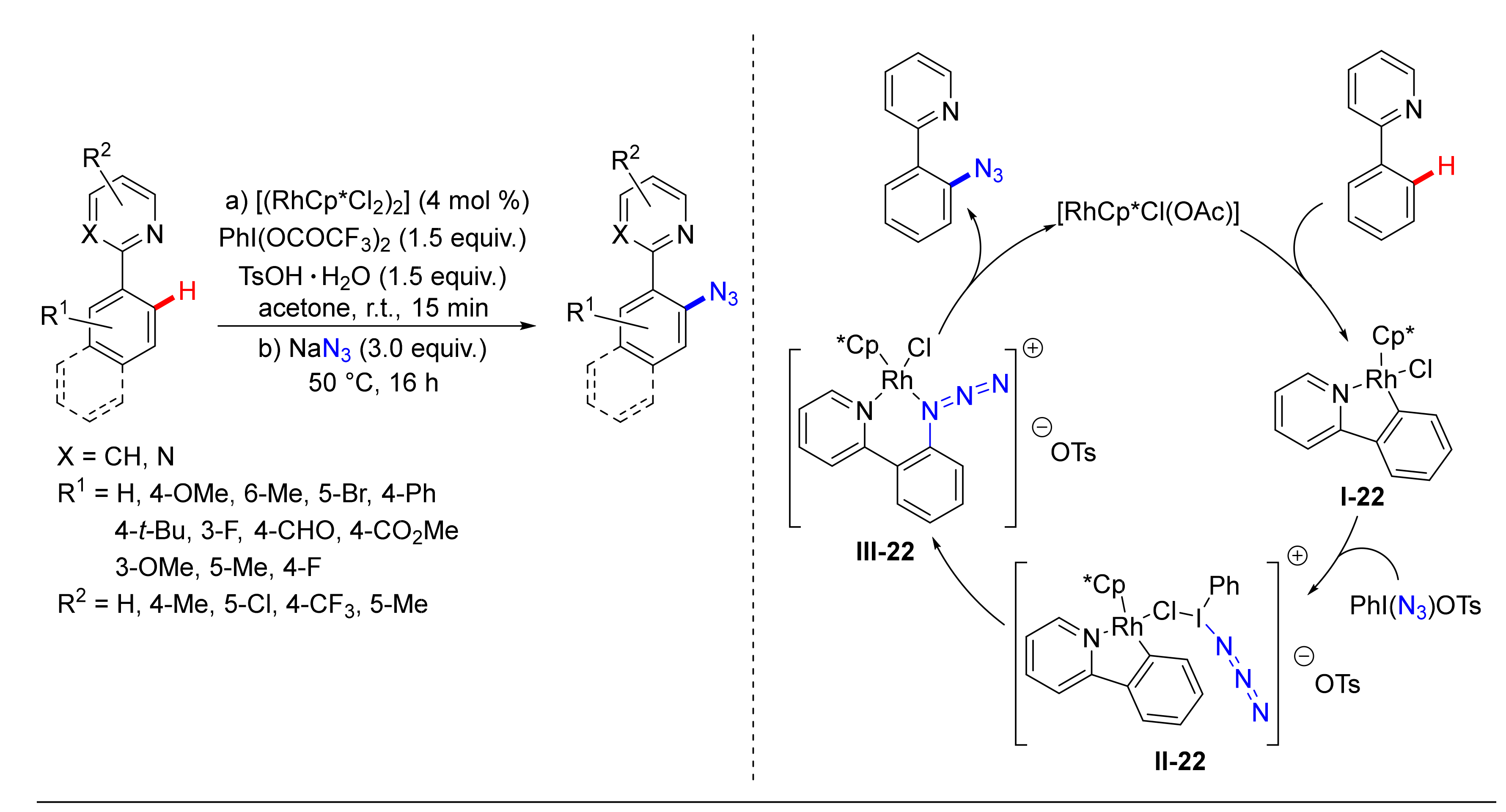
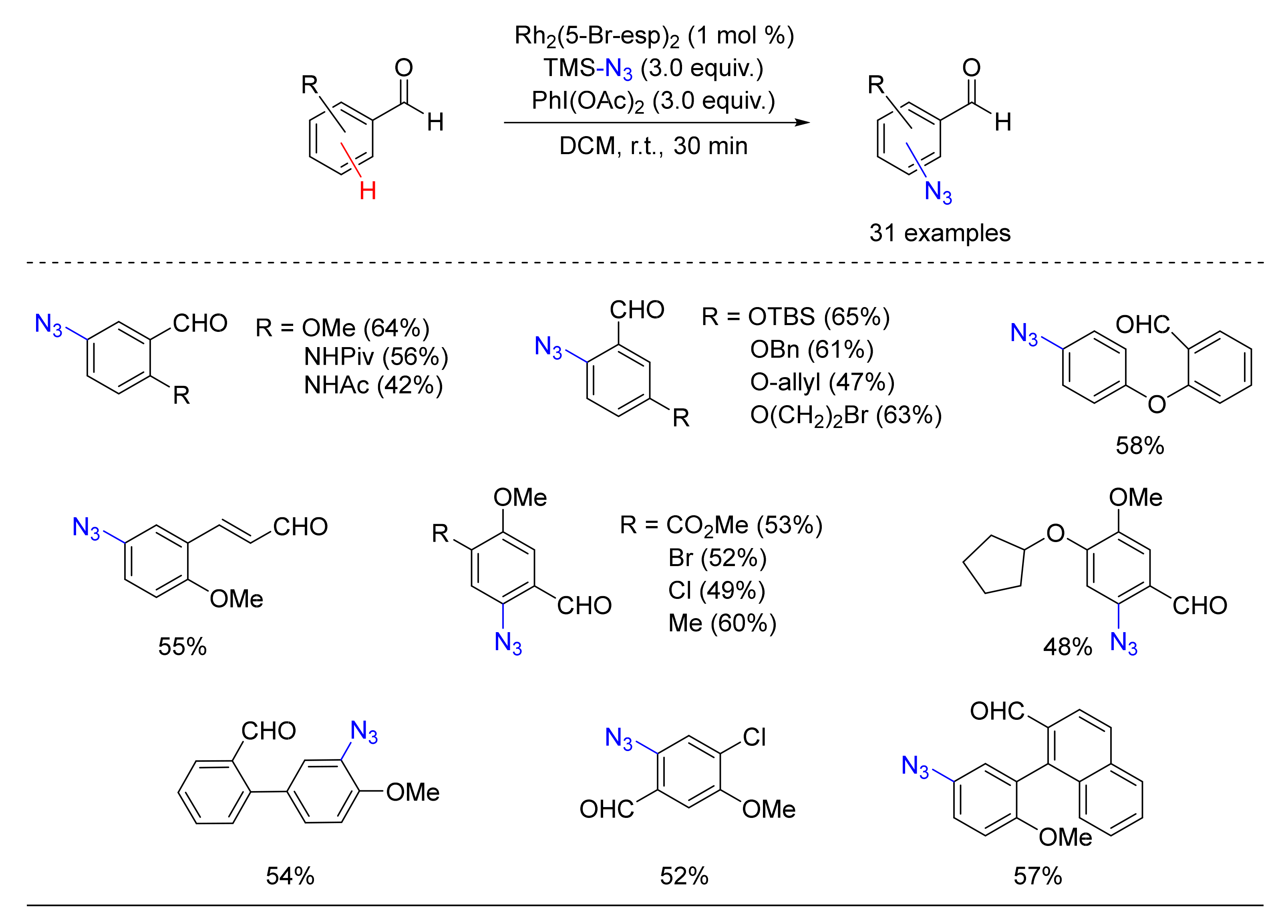
3. Azidation of C-H Bonds
4. Azidation Involving the Formation of More Than One Bond
4.1. Amination/Azidation
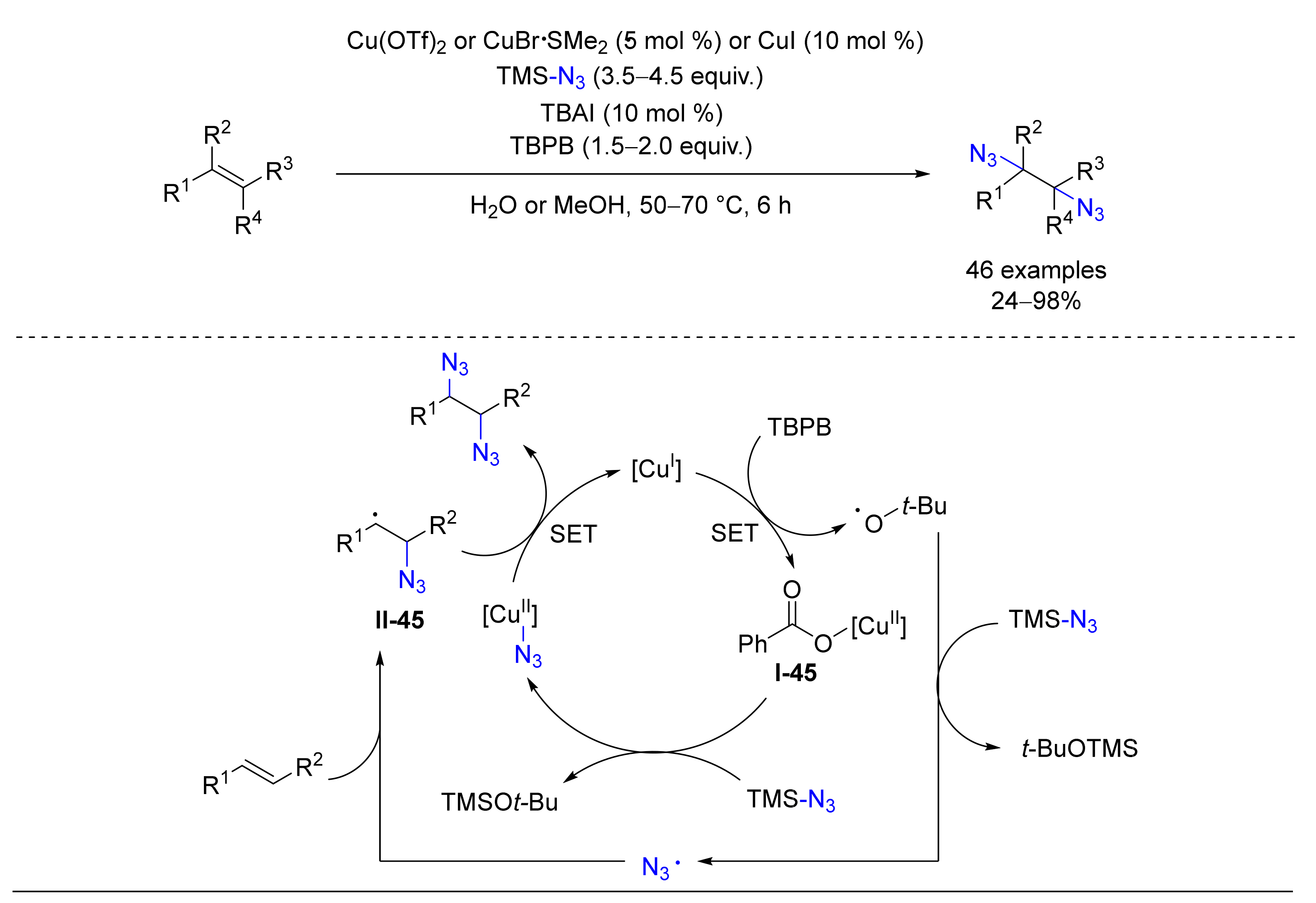
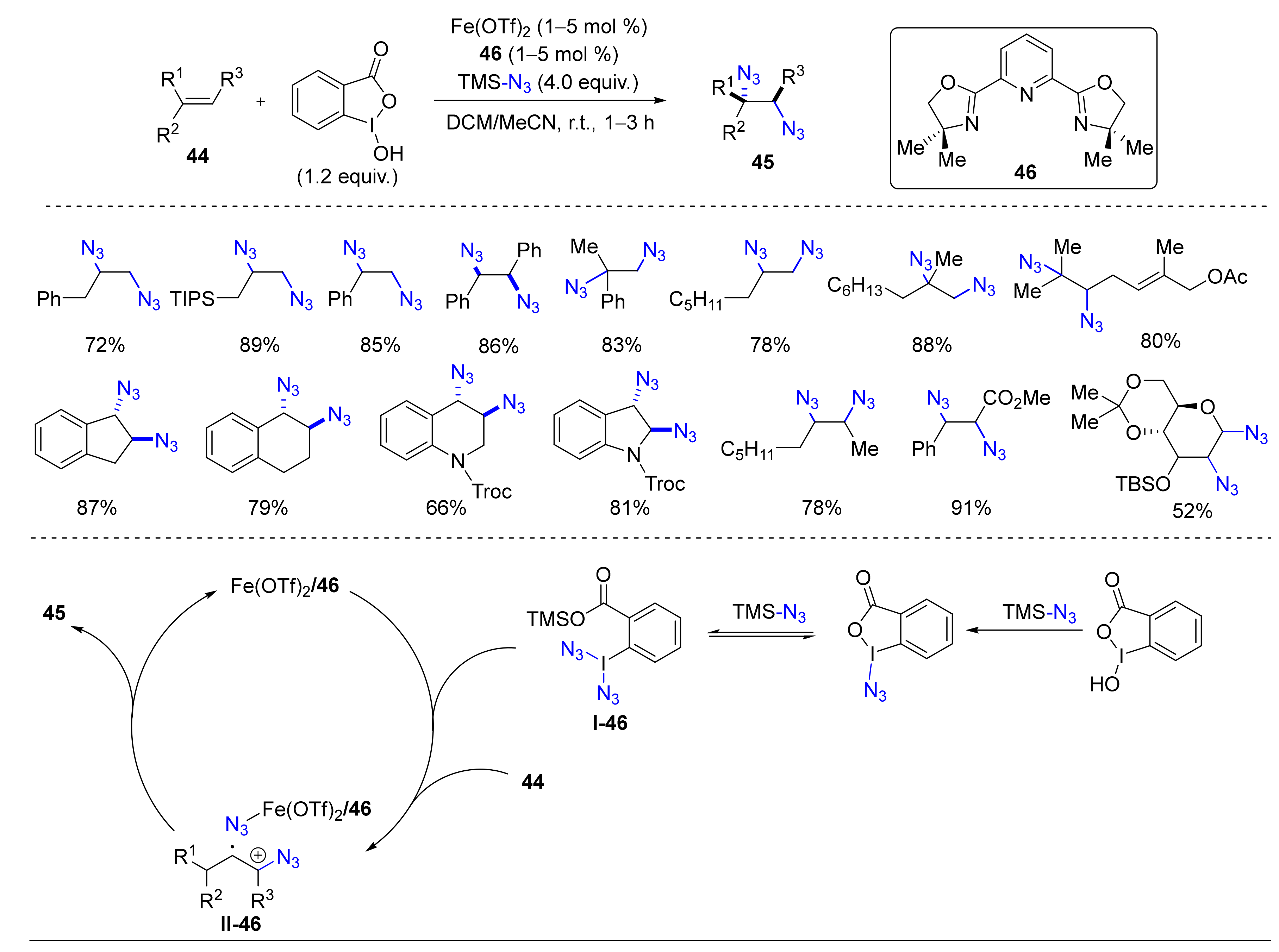
4.2. Diazidation
4.3. Hydroxylation/Azidation
4.4. Alkoxylation/Azidation
4.5. Haloalkylation/Azidation
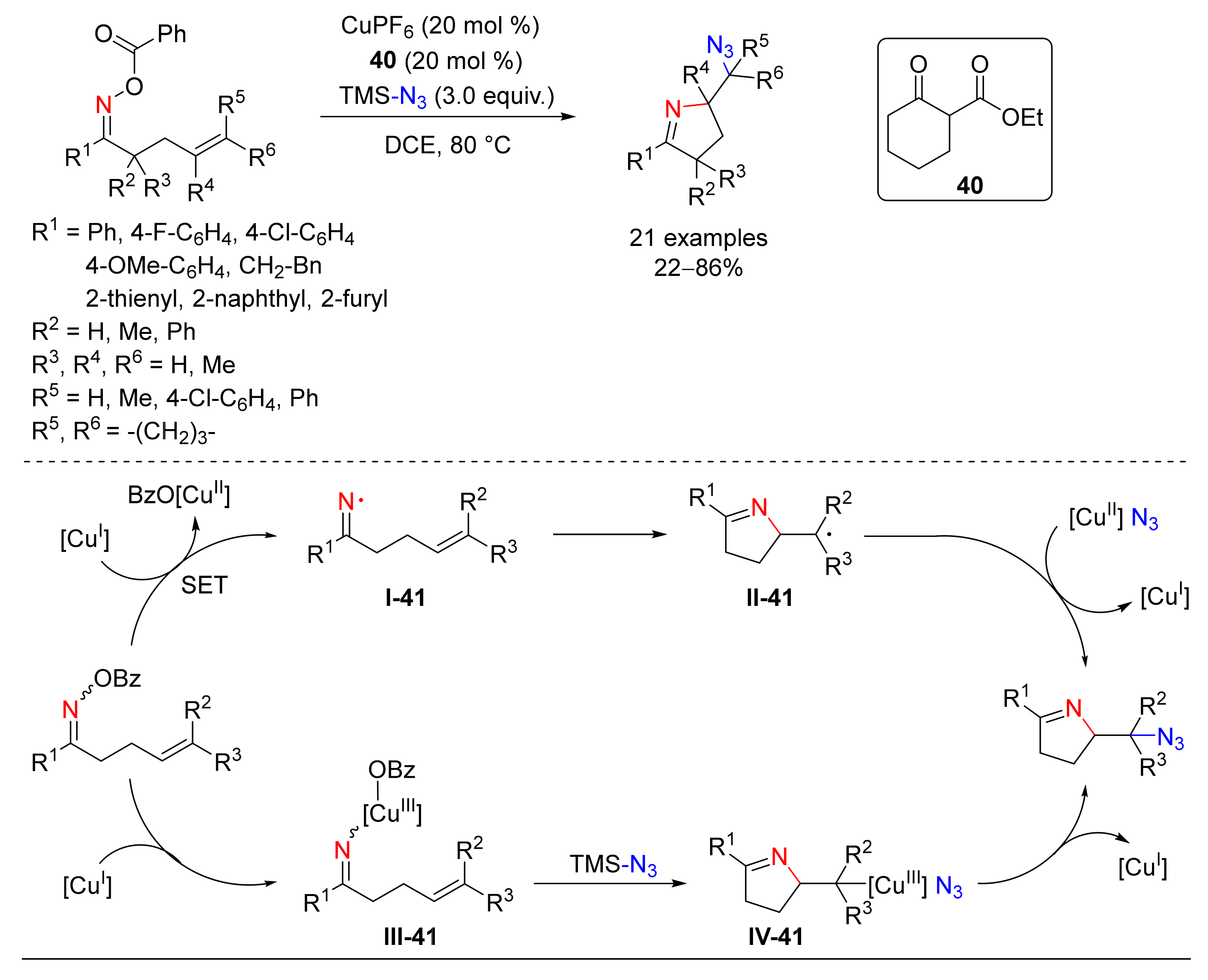
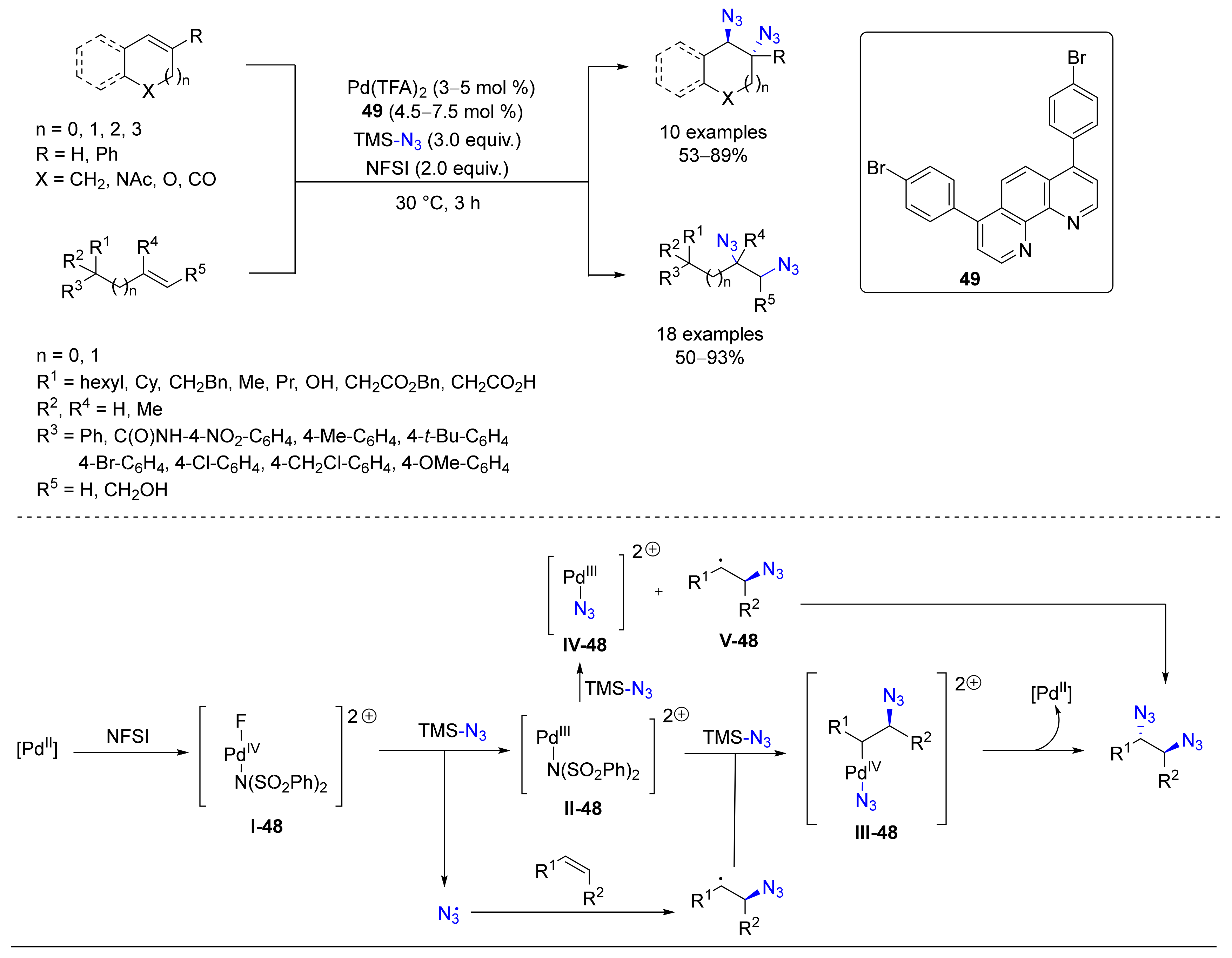
4.6. Alkylation or Arylation/Azidation
4.7. Cyanation/Azidation
5. Other Preparation of Allylic Azides
6. Hydroazidation Reactions
7. Azidation Involving Ring-Opening
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abramovitch, R.A.; Banthorpe, D.V.; Biffin, M.E.C.; Gurst, J.E.; Kyba, E.P.; Lwowski, W.; Miller, J.; Paul, D.B.; Pryde, C.A.; Reiser, A.; et al. The Chemistry of the Azido Group; Patai, S., Ed.; Wiley: New York, NY, USA, 1971. [Google Scholar]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Gololobov, Y.G.; Kasukhin, L.K. Recent advances in the staudinger reaction. Tetrahedron 1992, 48, 1353–1406. [Google Scholar] [CrossRef]
- Chen, B.; Mapp, A.K. A Phosphorimidate Rearrangement for the Facile and Selective Preparation of Allylic Amines. J. Am. Chem. Soc. 2004, 126, 5364–5365. [Google Scholar] [CrossRef]
- Wang, H.; Yu, Y.; Hong, X.; Tan, Q.; Xu, B. Rhodium-catalyzed Direct ortho C–N Bond Formation of Aromatic Azo Compounds with Azides. J. Org. Chem. 2014, 79, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Bellow, J.A.; Yousif, M.; Cabelof, A.C.; Lord, R.L.; Groysman, S. Reactivity Modes of an Iron Bis(alkoxide) Complex with Aryl Azides: Catalytic Nitrene Coupling vs. Formation of Iron(III) Imido Dimers. Organometallics 2015, 34, 2917–2923. [Google Scholar] [CrossRef]
- Azides and Nitrenes—Reactivity and Utility; Scriven, E.F.V. (Ed.) Academic Press: New York, NY, USA, 1984. [Google Scholar]
- Müller, P.; Fruit, C. Enantioselective Catalytic Aziridinations and Asymmetric Nitrene Insertions into CH Bonds. Chem. Rev. 2003, 103, 2905–2920. [Google Scholar] [CrossRef]
- DeGennaro, L.; Trinchera, P.; Luisi, R. Recent Advances in the Stereoselective Synthesis of Aziridines. Chem. Rev. 2014, 114, 7881–7929. [Google Scholar] [CrossRef]
- Zheng, Z.-J.; Wang, D.; Xu, Z.; Xu, L.-W. Synthesis of bi- and bis-1,2,3-triazoles by copper-catalyzed Huisgen cycloaddition: A family of valuable products by click chemistry. Beilstein J. Org. Chem. 2015, 11, 2557–2576. [Google Scholar] [CrossRef] [Green Version]
- Schulze, B.; Schubert, U.S. Beyond click chemistry-supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef]
- Broggini, G.; de Marchi, I.; Martinelli, M.; Paladino, G.; Pilati, T.; Terraneo, A. Effective Synthesis of Enantiopure [1,2,3]Triazolo[1,5-a]- and Pyrazolo[1,5-a]-pyrrolo[2,1-c][1,4]benzodiazepines by Diastereoselective Intramolecular Azide and Nitrilimine Cycloadditions. Synthesis 2005, 2005, 2246–2252. [Google Scholar] [CrossRef]
- Xie, Y.-Y.; Wang, Y.-C.; He, Y.; Hu, D.-C.; Wang, H.-S.; Pan, Y.-M. Catalyst-free synthesis of fused 1,2,3-triazole and isoindoline derivatives via an intramolecular azide–alkene cascade reaction. Green Chem. 2017, 19, 656–659. [Google Scholar] [CrossRef]
- Broggini, G.; Garanti, L.; Molteni, G.; Zecchi, G. A new entry to [1,2,4]triazolo[1,5-a][1,4]benzodiazepin-6-onesvia intramolecular nitrilimine cycloaddition to the cyano group. Tetrahedron 1998, 54, 14859–14868. [Google Scholar] [CrossRef]
- Habeeb, A.G.; Rao, P.P.N.; Knaus, E.E. Design and Synthesis of Celecoxib and Rofecoxib Analogues as Selective Cyclooxygenase-2 (COX-2) Inhibitors: Replacement of Sulfonamide and Methylsulfonyl Pharmacophores by an Azido Bioisostere. J. Med. Chem. 2001, 44, 3039–3042. [Google Scholar] [CrossRef] [PubMed]
- Geurink, P.P.; van der Linden, W.A.; Mirabella, A.C.; Gallastegui, N.; de Bruin, G.; Blom, A.E.M.; Voges, M.J.; Mock, E.D.; Florea, B.I.; van der Marel, G.A.; et al. Incorporation of Non-natural Amino Acids Improves Cell Permeability and Potency of Specific Inhibitors of Proteasome Trypsin-like Sites. J. Med. Chem. 2013, 56, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Saxon, E.; Luchansky, S.J.; Hang, H.C.; Yu, C.; Lee, S.C.; Bertozzi, C.R. Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc. 2002, 124, 14893–14902. [Google Scholar] [CrossRef]
- Lin, T.-S.; Prusoff, W.H. Synthesis and biological activity of several amino analogs of thymidine. J. Med. Chem. 1978, 21, 109–112. [Google Scholar] [CrossRef]
- Pathak, T. Azidonucleosides: Synthesis, Reactions, and Biological Properties. Chem. Rev. 2002, 102, 1623–1668. [Google Scholar] [CrossRef]
- Fletcher, M.D.; Campbell, M.M. Partially Modified Retro-Inverso Peptides: Development, Synthesis, and Conformational Behavior. Chem. Rev. 1998, 98, 763–796. [Google Scholar] [CrossRef]
- Patai, S.; Rappoport, Z. Chemistry of Halides, Pseudo-Halides and Azides, Part 1; Patai, S., Ed.; Wiley: Chichester, UK, 1995. [Google Scholar]
- Patai, S.; Rappoport, Z. Chemistry of Halides, Pseudo-Halides and Azides, Part 2; Patai, S., Ed.; Wiley: Chichester, UK, 1995. [Google Scholar]
- Biffin, J.M.E.C.; Miller, D.B. Paul in. In The Chemistry of the Azido Group; Patai, S., Ed.; Wiley: New York, NY, USA, 1971; pp. 147–176. [Google Scholar]
- Noelting, E.; Grandmougin, E.; Michel, O. Ueber die Bildung von Stickstoffwasserstoffsäure (Azoimid) aus aromatischen Azoimiden. Ber. Dtsch. Chem. Ges. 1892, 25, 3328–3342. [Google Scholar] [CrossRef] [Green Version]
- Noelting, E.; Michel, O. Direkte Ueberführung von Aminen in Diazoimide mittels Stickstoffwasserstoffsäure. Ber. Dtsch. Chem. Ges. 1893, 26, 86–87. [Google Scholar] [CrossRef] [Green Version]
- Noelting, E.; Michel, O. Ueber die Einwirkung von Diazoverbindungen auf Hydrazine. Ber. Dtsch. Chem. Ges. 1893, 26, 88–92. [Google Scholar] [CrossRef]
- Kauer, J.C.; Carboni, R.A. Aromatic azapentalenes. III. 1,3a,6,6a-Tetraazapentalenes. J. Am. Chem. Soc. 1967, 89, 2633–2637. [Google Scholar] [CrossRef]
- Chehade, K.A.H.; Spielmann, H.P. Facile and Efficient Synthesis of 4-Azidotetrafluoroaniline: A New Photoaffinity Reagent. J. Org. Chem. 2000, 65, 4949–4953. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Swenson, D.C.; Gillan, E.G. Synthesis and Structure of 2,5,8-Triazido-s-Heptazine: An Energetic and Luminescent Precursor to Nitrogen-Rich Carbon Nitrides. J. Am. Chem. Soc. 2004, 126, 5372–5373. [Google Scholar] [CrossRef]
- Lowe-Ma, C.K.; Nissan, R.A.; Wilson, W.S. Tetrazolo[1,5-a]pyridines and furazano[4,5-b]pyridine 1-oxides. J. Org. Chem. 1990, 55, 3755–3761. [Google Scholar] [CrossRef]
- Righi, G.; D’Achille, C.; Pescatore, G.; Bonini, C. New stereoselective synthesis of the peptidic aminopeptidase inhibitors bestatin, phebestin and probestin. Tetrahedron Lett. 2003, 44, 6999–7002. [Google Scholar] [CrossRef]
- Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K.A. Direct Organocatalytic Asymmetric α-Chlorination of Aldehydes. J. Am. Chem. Soc. 2004, 126, 4790–4791. [Google Scholar] [CrossRef]
- Tashiro, M.; Tsuge, A.; Sawada, T.; Makishima, T.; Horie, S.; Arimura, T.; Mataka, S.; Yamato, T. Metacyclophanes and related compounds. 26. Tetrahydroxy[2.n.2.n]metacyclophanes. Preparation, reactions, and spectra. J. Org. Chem. 1990, 55, 2404–2409. [Google Scholar] [CrossRef]
- Goswami, M.G.; de Bruin, B. Metal-Catalysed Azidation of Organic Molecules. Eur. J. Org. Chem. 2017, 2017, 1152–1176. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ebadi, A.G.; Youseftabar-Miri, L.; Hassanpour, A.; Vessally, E. Methods for direct C(sp2)–H bonds azidation. RSC Adv. 2019, 9, 25199–25215. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Groves, J.T. Taming Azide Radicals for Catalytic C–H Azidation. ACS Catal. 2015, 6, 751–759. [Google Scholar] [CrossRef]
- Chen, Z.; Rong, M.-Y.; Nie, J.; Zhu, X.-F.; Shi, B.-F.; Ma, J.-A. Catalytic alkylation of unactivated C(sp3)-H bonds for C(sp3)-C(sp3) bond formation. Chem. Soc. Rev. 2019, 48, 4921–4942. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Ferlin, F.; Ackermann, L.; Vaccaro, L. C-H functionalization reactions under flow conditions. Chem. Soc. Rev. 2019, 48, 2767–2782. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2018, 119, 2192–2452. [Google Scholar] [CrossRef]
- Borpatra, P.J.; Deka, B.; Deb, M.L.; Baruah, P.K. Recent advances in intramolecular C-O/C-N/C-S bond formation via C-H functionalization. Org. Chem. Front. 2019, 6, 3445–3489. [Google Scholar] [CrossRef]
- Broggini, G.; Borelli, T.; Giofré, S.; Mazza, A. Intramolecular Oxidative Palladium-Catalyzed Amination Involving Double C-H Functionalization of Unactivated Olefins. Synthesis 2017, 49, 2803–2818. [Google Scholar] [CrossRef]
- Ping, L.; Chung, D.S.; Bouffard, J.; Lee, S.-G. Transition metal-catalyzed site- and regio-divergent C-H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [Google Scholar] [CrossRef]
- Théveau, L.; Schneider, C.; Fruit, C.; Hoarau, C. Orthogonal Palladium-Catalyzed Direct C−H Bond Arylation of Heteroaromatics with Aryl Halides. ChemCatChem 2016, 8, 3183–3194. [Google Scholar] [CrossRef]
- Beccalli, E.M.; Broggini, G.; Martinelli, M.; Sottocornola, S. C-C, C-O, C-N Bond Formation on sp2 Carbon by Pd(II)-Catalyzed Reactions Involving Oxidant Agents. Chem. Rev. 2007, 107, 5318–5365. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C-C and C-N bond formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [Green Version]
- Godula, K.; Sames, D. C-H Bond Functionalization in Complex Organic Synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murahashi, S.-I.; Tanigawa, Y.; Imada, Y.; Taniguchi, Y. Palladium(0) catalyzed azidation and amination of allyl acetates. Selective synthesis of allyl azides and primary allylamines. Tetrahedron Lett. 1986, 27, 227–230. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Taniguchi, Y.; Imada, Y.; Tanigawa, Y. Palladium(0)-Catalyzed Azidation of Allyl Esters. Selective Synthesis of Allyl Azides, Primary Allylamines, and Related Compounds. J. Org. Chem. 1989, 54, 3292–3303. [Google Scholar] [CrossRef]
- Aufranc, P.; Ollivier, J.; Stolle, A.; Bremer, C.; Es-Sayed, M.; de Meijere, A.; Salaün, J. Regioselective Palladium (0) Azidation and Amination of 1-Alkenylcyclopropyl Esters: A New Route to 2,3-Methanoamino Acids. Tetrahedron Lett. 1993, 34, 4193–4196. [Google Scholar] [CrossRef]
- Safi, M.; Fahrang, R.; Sinou, D. Palladium(0)-Catalyzed Azidation of Allyl Esters with Trimethylsilyl Azide. Tetrahedron Lett. 1990, 31, 527–530. [Google Scholar] [CrossRef]
- Trost, B.M.; Pulley, S.R. Asymmetric Total Synthesis of (+)-Pancratistatin. J. Am. Chem. Soc. 1995, 117, 10143–10144. [Google Scholar] [CrossRef]
- Rueping, M.; Vila, C.; Uria, U. Direct Catalytic Azidation of Allylic Alcohols. Org. Lett. 2012, 14, 768–771. [Google Scholar] [CrossRef]
- Kim, S.; Chan, L.Y.; Kim, S.; Chung, W.T.; Long, C. Fe(OTf)3-Catalyzed Reaction of Benzylic Acetates with Organosilicon Compounds. Synlett 2011, 3, 415–419. [Google Scholar] [CrossRef]
- Salunke, S.B.; Babu, N.S.; Chen, C.-T. Iron(iii) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu(0) for the subsequent click chemistry. Chem. Commun. 2011, 47, 10440–10442. [Google Scholar] [CrossRef]
- Sawama, Y.; Nagata, S.; Yabe, Y.; Morita, K.; Monguchi, Y.; Sajiki, H. Iron-Catalyzed Chemoselective Azidation of Benzylic Silyl Ethers. Chem. Eur. J. 2012, 18, 16608–16611. [Google Scholar] [CrossRef]
- Sawama, Y.; Goto, R.; Nagata, S.; Shishido, Y.; Monguchi, Y.; Sajiki, H. Chemoselective and Direct Functionalization of Methyl Benzyl Ethers and Unsymmetrical Dibenzyl Ethers by Using Iron Trichloride. Chem. Eur. J. 2014, 20, 2631–2636. [Google Scholar] [CrossRef]
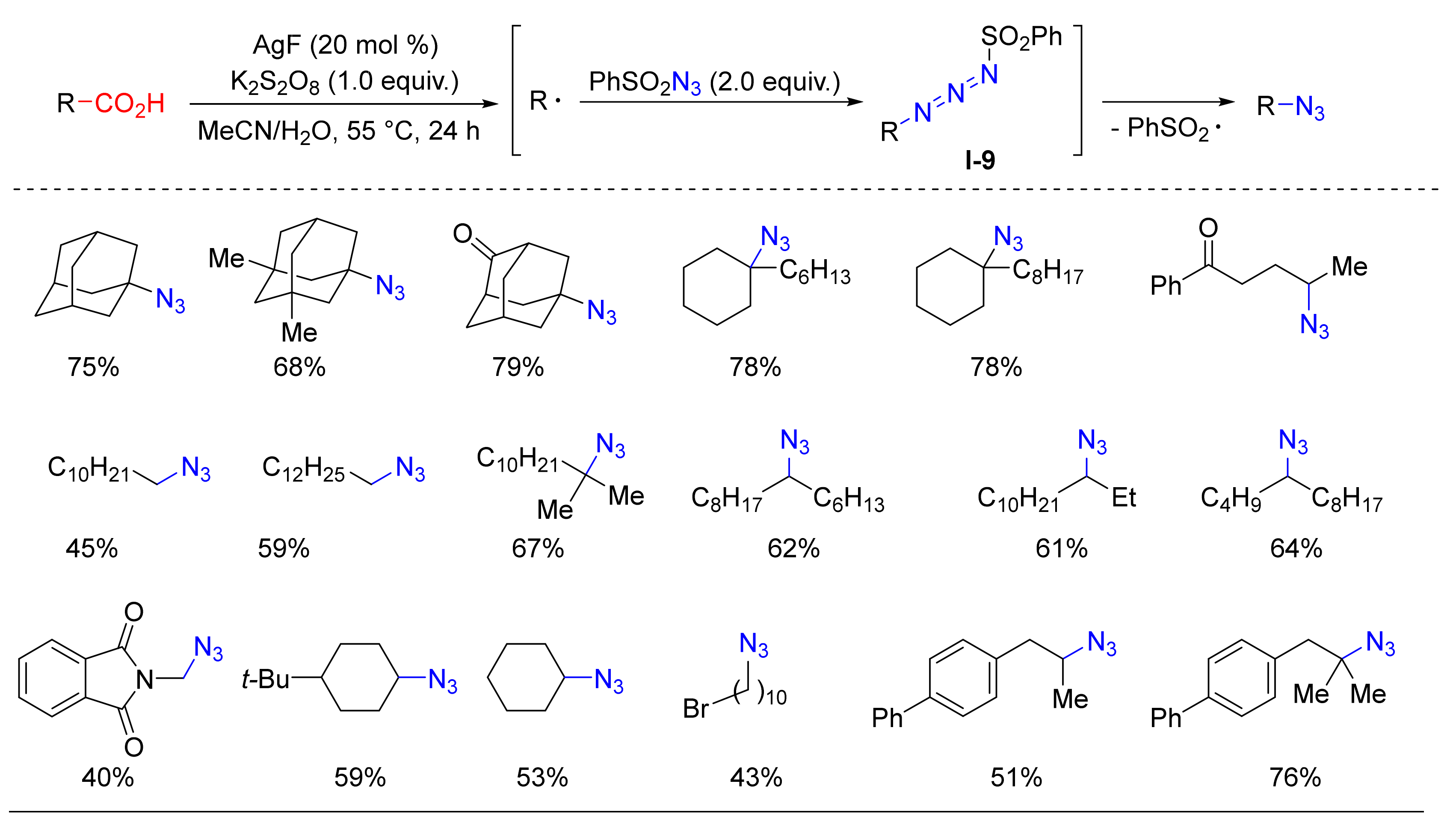
- Liu, C.; Wang, X.; Li, Z.; Cui, L.; Li, C. Silver-Catalyzed Decarboxylative Radical Azidation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2015, 137, 9820–9823. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, X.; Huang, X.; Shen, T.; Zhang, Y.; Sun, X.; Zou, M.; Song, S.; Jiao, N. Silver-Catalyzed Decarboxylative Azidation of Aliphatic Carboxylic Acids. Org. Lett. 2015, 17, 4702–4705. [Google Scholar] [CrossRef] [PubMed]
- Khedar, P.; Pericherla, K.; Kumar, A. Copper Triflate: An Efficient Catalyst for Direct Conversion of Secondary Alcohols into Azides. Synlett 2014, 25, 515–518. [Google Scholar] [CrossRef]
- Zhu, W.; Ma, D. Synthesis of aryl azides and vinyl azides via proline-promoted CuI-catalyzed coupling reaction. Chem. Commun. 2004, 7, 888–889. [Google Scholar] [CrossRef]
- Tao, C.-Z.; Cui, X.; Li, J.; Liu, A.-X.; Liu, L.; Guo, Q.-X. Copper-catalyzed synthesis of aryl azides and 1-aryl-1,2,3-triazoles from boronic acids. Tetrahedron Lett. 2007, 48, 3525–3529. [Google Scholar] [CrossRef]
- Li, Y.; Gao, L.-X.; Han, F.-S. Reliable and Diverse Synthesis of Aryl Azides through Copper-Catalyzed Coupling of Boronic Acids or Esters with TMSN3. Chem. Eur. J. 2010, 16, 7969–7972. [Google Scholar] [CrossRef]
- Clerc, A.; Beneteau, V.; Pale, P.; Chassaing, S. Chan-Lam-type Azidation and One-Pot CuAAC under Cui-Zeolite Catalysis. ChemCatChem 2020, 12, 2060–2065. [Google Scholar] [CrossRef]
- Raushel, J.; Pitram, S.M.; Fokin, V.V. Efficient Synthesis of Sulfonyl Azides from Sulfonamides. Org. Lett. 2008, 10, 3385–3388. [Google Scholar] [CrossRef]
- Tang, C.; Jiao, N. Copper-Catalyzed C-H Azidation of Anilines under Mild Conditions. J. Am. Chem. Soc. 2012, 134, 18924–18927. [Google Scholar] [CrossRef]
- Fang, H.; Dou, Y.; Ge, J.; Chhabra, M.; Sun, H.; Zhang, P.; Zheng, Y.; Zhu, Q. Regioselective and Direct Azidation of Anilines via Cu(II)-Catalyzed C-H Functionalization in Water. J. Org. Chem. 2017, 82, 11212–11217. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wan, W.; Ma, G.; Gao, W.; Jiang, H.; Zhu, S.; Hao, J. Room-temperature Cu(II)-catalyzed aromatic C–H azidation for the synthesis of ortho-azido anilines with excellent regioselectivity. Chem. Commun. 2014, 50, 5733–5736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, C.S.; Narula, A.K. Copper-catalysed regioselective azidation of arenes by C-H activation directed by pyridine. RSC Adv. 2015, 5, 100223–100227. [Google Scholar] [CrossRef]
- Dou, Y.; Xie, Z.; Sun, Z.; Fang, H.; Shen, C.; Zhang, P.; Zhu, Q. Copper(II)-Catalyzed Direct Azidation of N-Acylated 8-Aminoquinolines by Remote C-H Activation. ChemCatChem 2016, 8, 3570–3574. [Google Scholar] [CrossRef]
- Yao, B.; Liu, Y.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Designing a Cu(II)-ArCu(II)-ArCu(III)-Cu(I) Catalytic Cycle: Cu(II)-Catalyzed Oxidative Arene C-H Bond Azidation with Air as an Oxidant under Ambient Conditions. J. Org. Chem. 2014, 79, 11139–11145. [Google Scholar] [CrossRef]
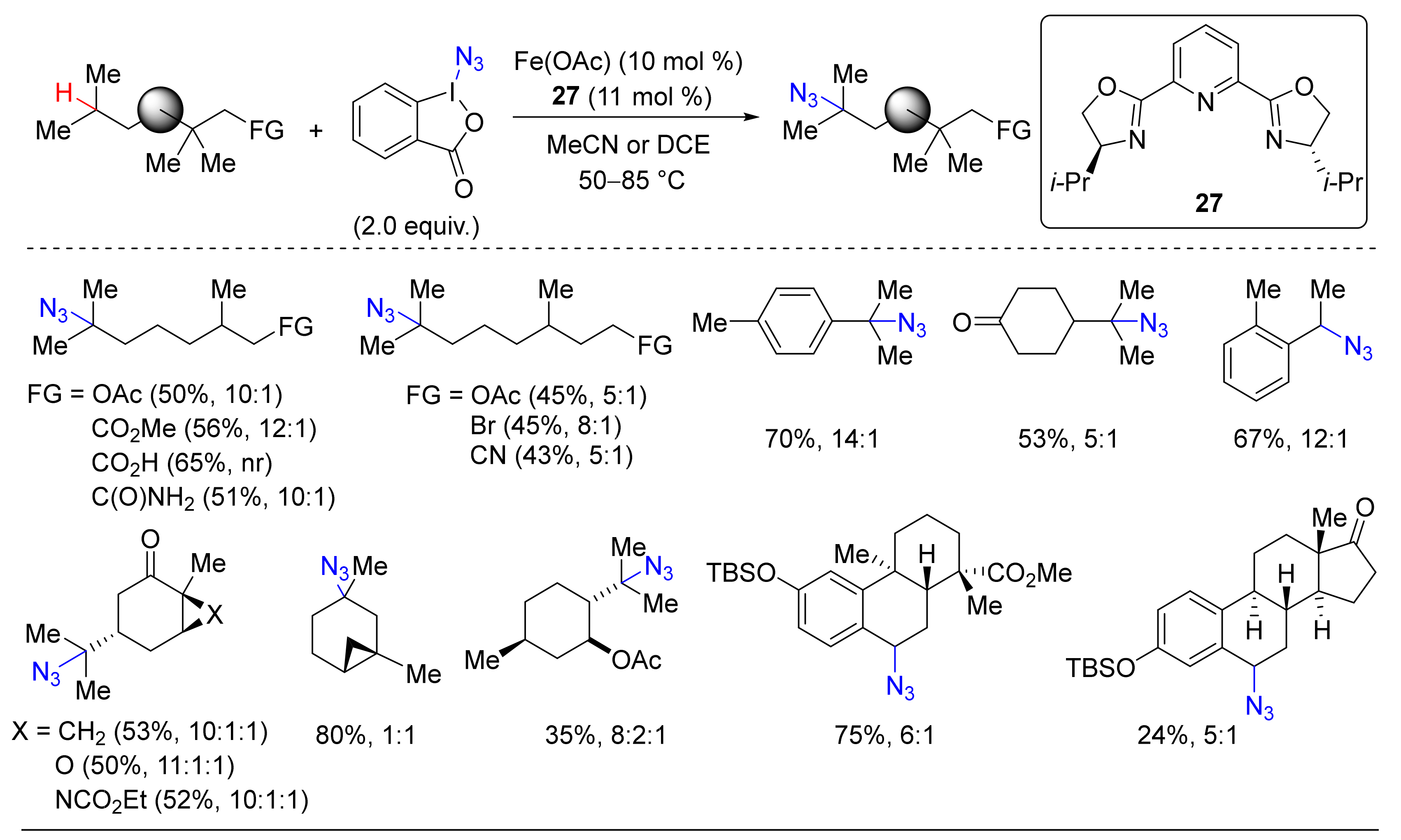
- Suh, S.-E.; Chen, S.-J.; Mandal, M.; Guzei, I.A.; Cramer, C.J.; Stahl, S.S. Site-Selective Copper-Catalyzed Azidation of Benzylic C-H Bonds. J. Am. Chem. Soc. 2020, 142, 11388–11393. [Google Scholar] [CrossRef]
- Hendrick, C.E.; Bitting, K.J.; Cho, S.; Wang, Q. Site-Selective Copper-Catalyzed Amination and Azidation of Arenes and Heteroarenes via Deprotonative Zincation. J. Am. Chem. Soc. 2017, 139, 11622–11628. [Google Scholar] [CrossRef]
- Xie, F.; Qi, Z.; Li, X. Rhodium(III)-Catalyzed Azidation and Nitration of Arenes by C-H Activation. Angew. Chem. Int. Ed. 2013, 52, 11862–11866. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fang, Z.; Chen, X.; Wang, Y. Mild Dirhodium(II)-Catalyzed Chemo- and Regioselective Azidation of Arenes. Org. Lett. 2018, 20, 5732–5736. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Z.; Chen, X.; Wang, Y. Dirhodium(II)-Catalyzed C(sp2)-H Azidation of Benzaldegydes. Chem. Eur. J. 2020, 26, 6805–6811. [Google Scholar] [CrossRef]
- Huang, X.; Bergsten, T.M.; Groves, J.T. Manganese-Catalyzed Late-Stage Aliphatic C-H Azidation. J. Am. Chem. 2015, 137, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-H.; Bleith, T.; Wadepohl, H.; Gade, L.H. Enantioselective Iron-Catalyzed Azidation of β-Keto Esters and Oxindoles. J. Am. Chem. Soc. 2013, 135, 5356–5359. [Google Scholar] [CrossRef]
- Sharma, A.; Hartwig, J.F. Metal-catalysed azidation of tertiary C–H bonds suitable for late-stage functionalization. Nature 2015, 517, 600–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimov, R.R.; Sharma, A.; Hartwig, J.F. Late Stage Azidation of Complex Molecules. ACS Cent. Sci. 2016, 2, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.-J.; Wang, C.Y.; Huang, Y.-L.; Xu, Y.L.; Wang, X.-S. Remote azidation of C(sp3)-H bonds to synthesize δ-azido sulphonamides via iron-catalyzed radical relay. Org. Biomol. Chem. 2020, 18, 5354–5357. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.-X.; Yang, G.; He, G.; Chen, G. A visible-light-promoted radical reaction system for azidation and halogenation of tertiary aliphatic C–H bonds. Chem. Sci. 2016, 7, 2679–2683. [Google Scholar] [CrossRef] [Green Version]
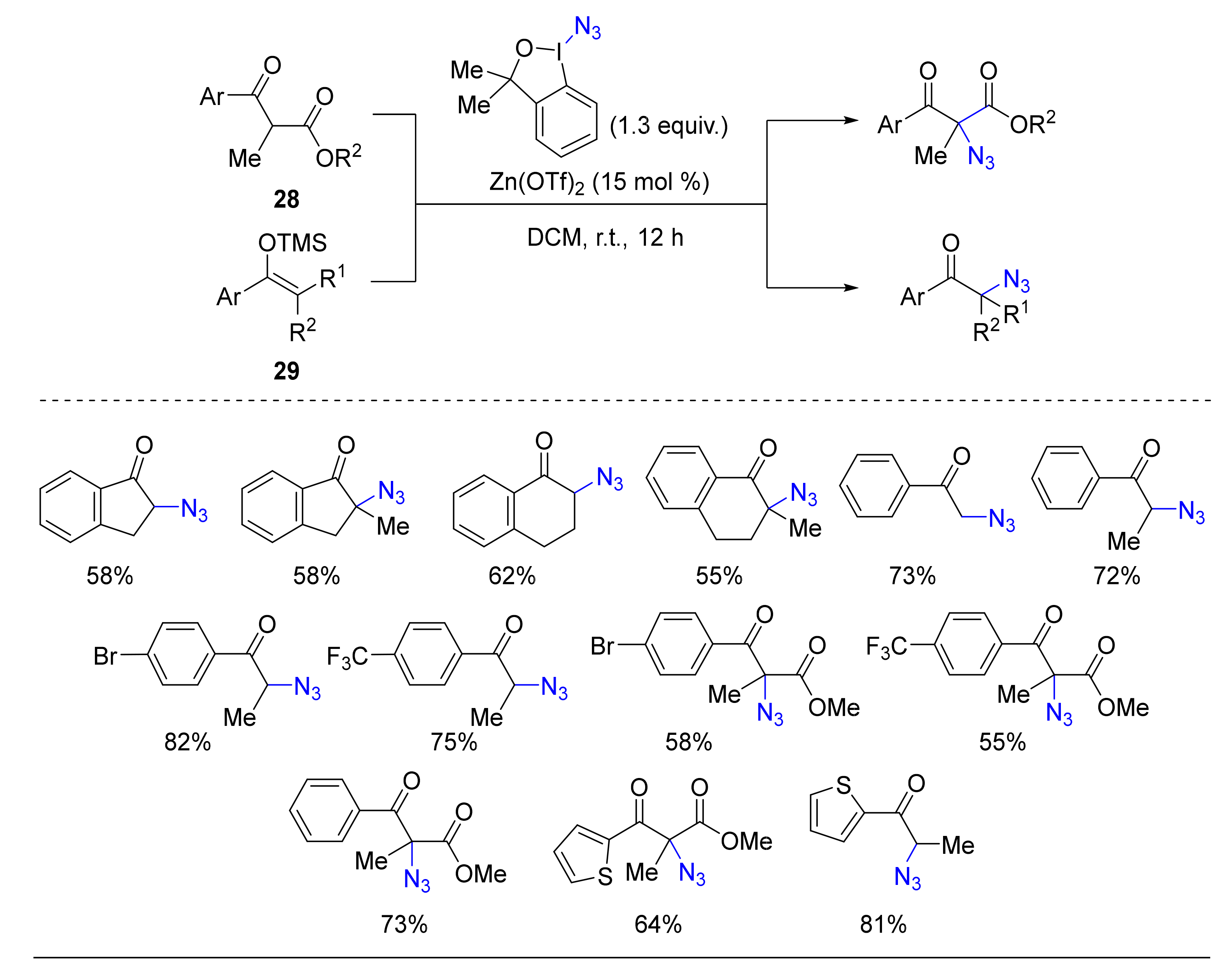
- Vita, M.V.; Waser, J. Azidation of β-Keto Esters and Silyl Enol Ethers with a Benziodoxole Reagent. Org. Lett. 2013, 15, 3246–3249. [Google Scholar] [CrossRef] [Green Version]
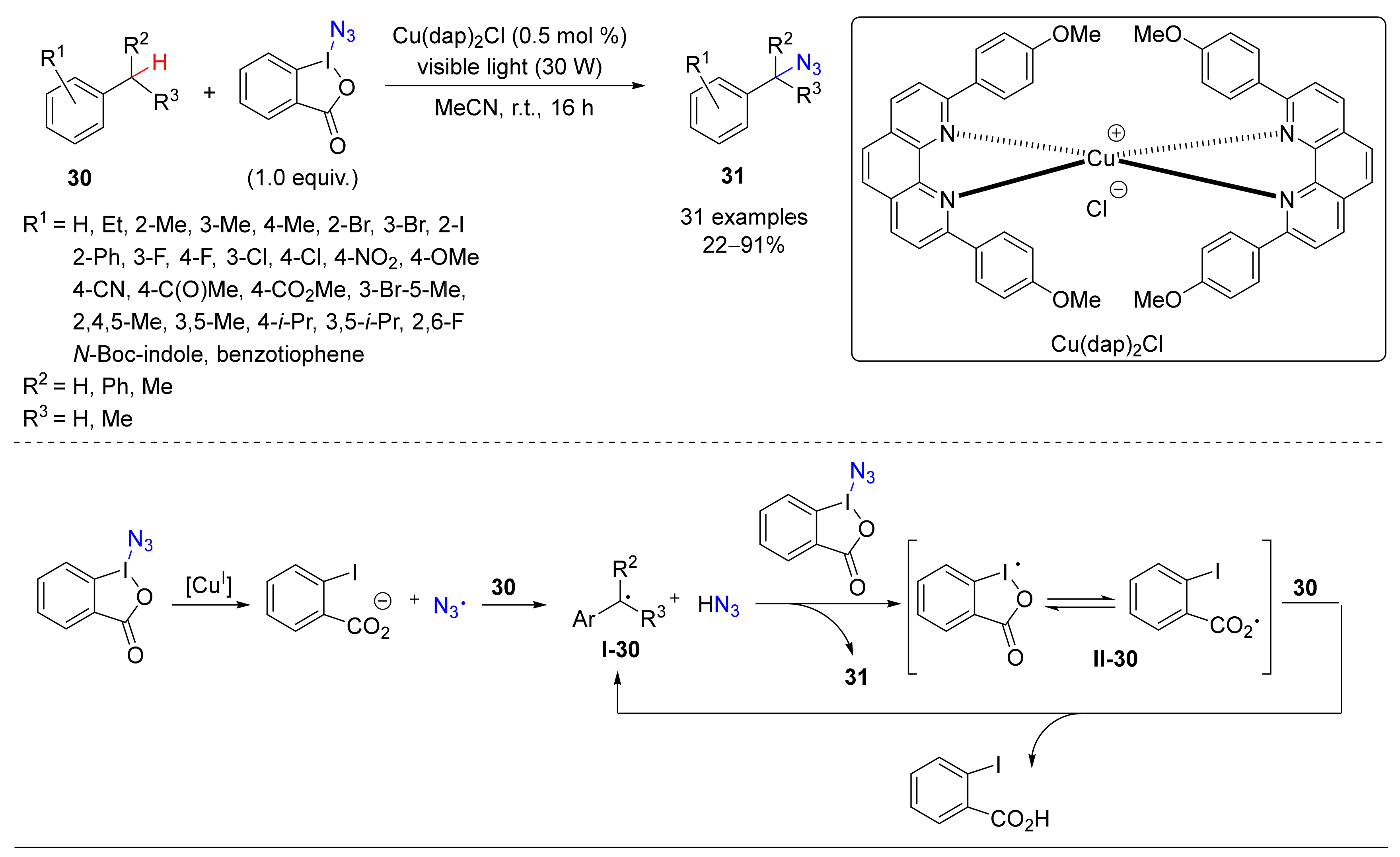
- Rabet, P.T.G.; Fumagalli, G.; Boyd, S.; Greaney, M.F. Benzylic C-H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett. 2016, 18, 1646–1649. [Google Scholar] [CrossRef] [Green Version]
- Dhineshkumar, J.; Samaddar, P.; Prabhu, K.R. A copper catalyzed azidation and peroxidation of β-naphthols via an oxidative dearomatization strategy. Chem. Commun. 2016, 52, 11084–11087. [Google Scholar] [CrossRef]
- Dhineshkumar, J.; Gadde, K.; Prabhu, K.R. Substituent-Directed Regioselective Azidation: Copper-Catalyzed C–H Azidation and Iodine-Catalyzed Dearomatizative Azidation of Indole. J. Org. Chem. 2018, 83, 228–235. [Google Scholar] [CrossRef]
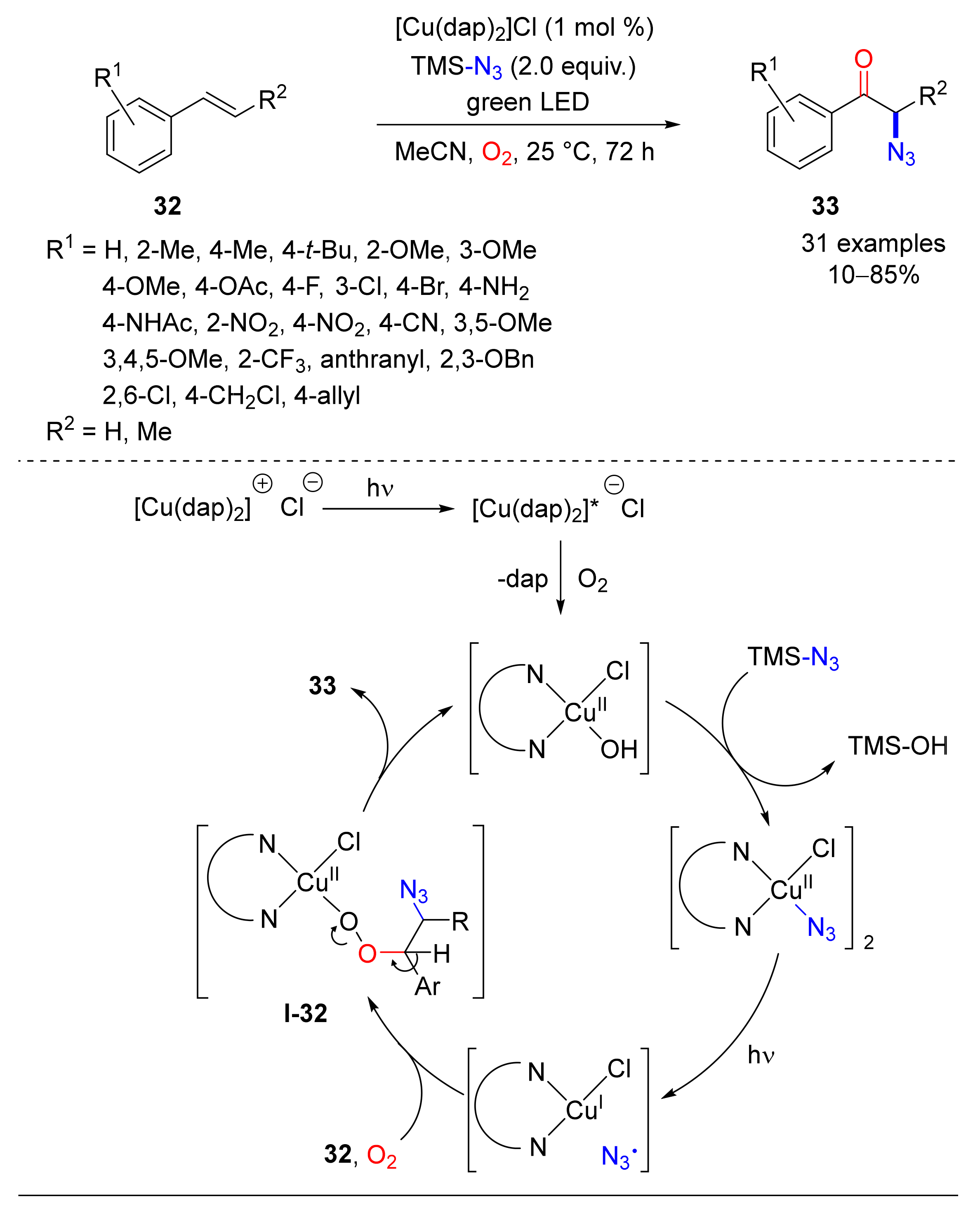
- Hossain, A.; Vidyasagar, A.; Eichinger, C.; Lankes, C.; Phan, J.; Rehbein, J.; Reiser, O. Visible-Light-Accelerated Copper(II)-Catalyzed Regio- and Chemoselective Oxo-Azidation of Vinyl Arenes. Angew. Chem. Int. Ed. 2018, 57, 8288–8292. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, F.C.; Turnpenny, B.W.; Chemler, S.R. Copper-promoted and copper-catalyzed intermolecular alkene diamination. Angew. Chem. Int. Ed. 2010, 49, 6365–6368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liang, Y.; Dong, J.; Deng, Y.; Zhao, C.; Su, Z.; Guan, W.; Bi, X.; Liu, Q.; Fu, J. Directed Copper-Catalyzed Intermolecular Aminative Difunctionalization of Unactivated Alkenes. J. Am. Chem. Soc. 2019, 141, 18475–18485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Studer, A. Copper-Catalyzed Intermolecular Aminoazidation of Alkenes. Org. Lett. 2014, 16, 1790–1793. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q. N-Fluorobenzenesulfonimide: An Efficient Nitrogen Source for C-N Bond Formation. Synthesis 2015, 47, 159–174. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Zhang, G.; Yang, S.; Li, Y.; Zhang, Q. Copper-catalyzed 1,3-aminoazidation of arylcyclopropanes: A facile access to 1,3-diamine derivatives. Org. Chem. Front. 2019, 6, 2934–2938. [Google Scholar] [CrossRef]
- Lei, B.; Wang, X.; Ma, L.; Li, Y.; Li, Z. NFSI-participated intermolecular aminoazidation of alkene through iron catalysis. Org. Biomol. Chem. 2018, 16, 3109–3113. [Google Scholar] [CrossRef]
- Shen, K.; Wang, Q. Copper-Catalyzed Alkene Aminoazidation as a Rapid Entry to 1,2-Diamines and Installation of an Azide Reporter onto Azahetereocycles. J. Am. Chem. Soc. 2017, 139, 13110–13116. [Google Scholar] [CrossRef]
- Olding, A.; Ho, C.C. Recent Applications of the Zhdankin Reagent in Organic Synthesis. Aust. J. Chem. 2019, 72, 646–648. [Google Scholar] [CrossRef] [Green Version]
- Abi-Fayssal, S.; Giungi, A.; Berhal, F.; Prestat, G. Iron-Catalyzed Intra-intermolecular Aminoazidation of Alkenes. Org. Process. Res. Dev. 2019, 24, 695–703. [Google Scholar] [CrossRef]
- Foschi, F.; Loro, C.; Sala, R.; Oble, J.; Presti, L.L.; Beccalli, E.M.; Poli, G.; Broggini, G. Intramolecular Aminoazidation of Unactivated Terminal Alkenes by Palladium-Catalyzed Reactions with Hydrogen Peroxide as the Oxidant. Org. Lett. 2020, 22, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qi, X.; Hou, C.; Chen, P.; Liu, G. Palladium(II)-Catalyzed Enantioselective Azidation of Unactivated Alkenes. Angew. Chem. Int. Ed. 2020, 59, 17239–17244. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Li, W.; Xuan, Z.; Yu, W. Copper-Catalyzed Cyclization and Azidation of γ,δ-Unsaturated Ketone O-Benzoyl Oximes. Adv. Synth. Catal. 2014, 357, 64–70. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Y.; Wang, H.; Li, X.; Wan, B. Transition-Metal Controlled Diastereodivergent Radical Cyclization/Azidation Cascade of 1,7-Enynes. J. Org. Chem. 2016, 81, 4412–4420. [Google Scholar] [CrossRef]
- Ouyang, X.-H.; Song, R.-J.; Liu, Y.; Hu, M.; Li, J.-H. Copper-Catalyzed Radical [2 + 2 + 1] Annulation of Benzene-Linked 1,n-Enynes with Azide: Fused Pyrroline Compounds. Org. Lett. 2015, 17, 6038–6041. [Google Scholar] [CrossRef]
- Lu, M.-Z.; Wang, C.-Q.; Loh, T.-P. Copper-Catalyzed Vicinal Oxyazidation and Diazidation of Styrenes under Mild Conditions: Access to Alkyl Azides. Org. Lett. 2015, 17, 6110–6113. [Google Scholar] [CrossRef]
- Zhou, H.; Jian, W.; Qian, B.; Ye, C.; Li, D.; Zhou, J.; Bao, H. Copper-Catalyzed Ligand-Free Diazidation of Olefins with TMSN3 in CH3CN or in H2O. Org. Lett. 2017, 19, 6120–6123. [Google Scholar] [CrossRef]
- Minisci, F.; Galli, R. Influence of the electrophilic character on the reactivity of free radicals in solution reactivity of alkoxy, hydroxy, alkyl and azido radicals in presence of olefins. Tetrahedron Lett. 1962, 3, 533–538. [Google Scholar] [CrossRef]
- Minisci, F.; Galli, R. Reactivity of alkoxy and hydroxy radicals in presence of olefins and oxidation-reduction systems. Introduction of azido, chloro and acyloxy groups in allylic position and azido-chlorination of olefins. Tetrahedron Lett. 1963, 6, 357–360. [Google Scholar] [CrossRef]
- Yuan, Y.-A.; Lu, D.-F.; Chen, Y.R.; Zu, H. Iron-Catalyzed Direct Diazidation for a Broad Range of Olefins. Angew. Chem. Int. Ed. 2015, 55, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.-J.; Zhu, C.-L.; Lu, D.-F.; Xu, H. Iron-Catalyzed Direct Olefin Diazidation Via Peroxyester Activation Promoted by Nitrogen-Based Ligands. ACS Catal. 2018, 8, 4473–4482. [Google Scholar] [CrossRef]
- Peng, H.; Yuan, Z.; Chen, P.; Liu, G. Palladium-Catalyzed Intermolecular Oxidative Diazidation of Alkenes. Chin. J. Chem. 2017, 35, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Fu, N.; Sauer, G.S.; Saha, A.; Loo, A.; Lin, S. Metal-catalyzed electrochemical diazidation of alkenes. Science 2017, 357, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, X.; Song, S.; Zhu, Y.; Liang, Y.-F.; Jiao, N. Mn-Catalyzed Highly Efficient Aerobic Oxidative Hydroxyazidation of Olefins: A Direct Approach to β-Azido Alcohols. J. Am. Chem. Soc. 2015, 137, 6059–6066. [Google Scholar] [CrossRef]
- Fumagalli, G.; Rabet, P.T.G.; Boyd, S.; Greaney, M.F. Three-Component Azidation of Styrene-Type Double Bonds: Light-Switchable Behavior of a Copper Photoredox Catalyst. Angew. Chem. Int. Ed. 2015, 54, 11481–11484. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Wang, T.; Jiao, N. Copper-Catalyzed Oxoazidation and Alkoxyazidation of Indoles. Org. Lett. 2014, 16, 2302–2305. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, W.; Li, G.; Hong, L.; Wang, R. Copper-catalyzed cascade azidation–cyclization of tryptophols and tryptamines. Chem. Commun. 2015, 51, 12293–12296. [Google Scholar] [CrossRef]
- Zhu, R.; Buchwald, S.L. Versatile Enantioselective Synthesis of Functionalized Lactones via Copper-Catalyzed Radical Oxyfunctionalization of Alkenes. J. Am. Chem. Soc. 2015, 137, 8069–8077. [Google Scholar] [CrossRef] [Green Version]
- Alazet, S.; le Vaillant, F.; Nicolai, S.; Courant, T.; Waser, J. Divergent Access to (1,1) and (1,2)-Azidolactones from Alkenes using Hypervalent Iodine Reagents. Chem. Eur. J. 2017, 23, 9501–9504. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yu, H.; Xu, Z.; Jiang, X.; Lin, L.; Wang, R. Copper-Catalyzed Oxyazidation of Unactivated Alkenes: A Facile Synthesis of Isoxazolines Featuring an Azido Substituent. Org. Lett. 2014, 16, 1562–1565. [Google Scholar] [CrossRef]
- Qian, L.-L.; Yi, R.; Min, X.-T.; Hu, Y.-C.; Wan, B.; Chen, Q.C. Rapid assembly of 3-azidomethylfurans from 2-(1-alkynyl)-2-alken-1-ones enabled by silver catalysis. Tetrahedron 2020, 76, 131327. [Google Scholar] [CrossRef]
- Bertho, S.; Rey-Rodriguez, R.; Colas, C.; Retailleau, P.; Gillaizeau, I. Regio- and Stereoselective Iron-Catalyzed Oxyazidation of Enamides Using a Hypervalent Iodine Reagent. Chem. Eur. J. 2017, 23, 17674–17677. [Google Scholar] [CrossRef]
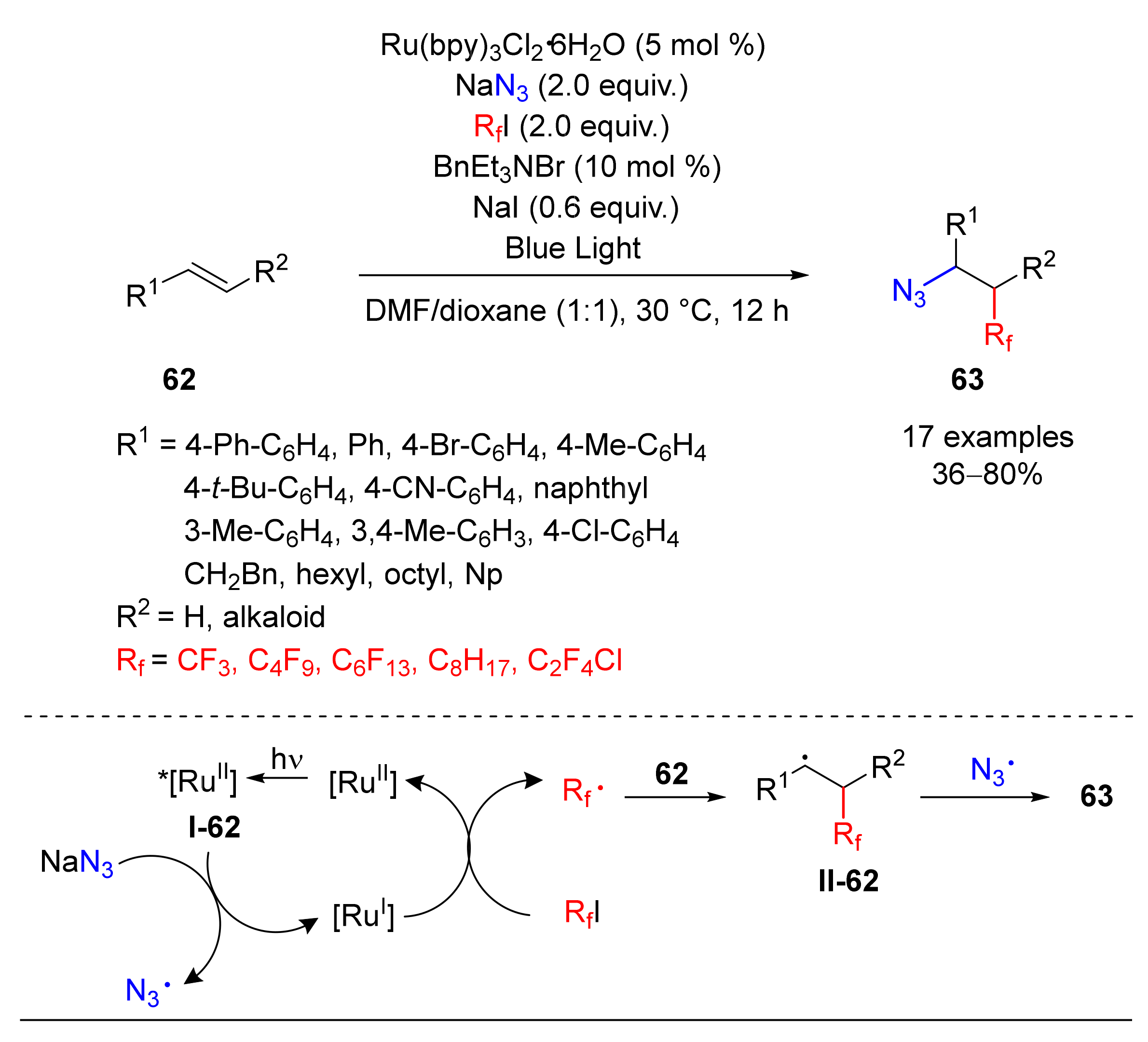
- Wang, F.; Qi, X.; Liang, Z.; Chen, P.; Liu, G. Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides. Angew. Chem. Int. Ed. 2014, 53, 1881–1886. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, N.; Chen, P.; Ye, J.; Liu, G. Copper-Catalyzed Trifluoromethylazidation of Alkynes: Efficient Access to CF3-Substituted Azirines and Aziridines. Angew. Chem. Int. Ed. 2015, 54, 9356–9360. [Google Scholar] [CrossRef]
- Yang, M.; Wang, W.; Liu, Y.; Feng, L.; Jua, X. Copper-Catalyzed Three-Component Azidotrifluoromethylation/Difunctionalization of Alkenes. Chin. J. Chem. 2014, 32, 833–837. [Google Scholar] [CrossRef]
- Huang, L.; Lin, J.-S.; Tan, B.; Liu, X.-Y. Alkene Trifluoromethylation-Initiated Remote α-Azidation of Carbonyl Compounds toward Trifluoromethyl γ-Lactam and Spirobenzofuranone-Lactam. ACS Catal. 2015, 5, 2826–2831. [Google Scholar] [CrossRef]
- Carboni, A.; Dagousset, G.; Magnier, E.; Masson, G. Photoredox-Induced Three-Component Oxy-, Amino-, and Carbotrifluoromethylation of Enecarbamates. Org. Lett. 2014, 16, 1240–1243. [Google Scholar] [CrossRef]
- Dagousset, G.; Carboni, A.; Magnier, E.; Masson, G. Photoredox-Induced Three-Component Azido- and Aminotrifluoromethylation of Alkenes. Org. Lett. 2014, 16, 4340–4343. [Google Scholar] [CrossRef]
- Geng, X.; Lin, F.; Wang, X.; Jiao, N. Azidofluoroalkylation of Alkenes with Simple Fluoroalkyl Iodides Enabled by Photoredox Catalysis. Org. Lett. 2017, 19, 4738–4741. [Google Scholar] [CrossRef]
- Lu, L.; Huang, W.-Y. The reaction of perfluoroalkanesulfinates VII. Fenton reagent-initiated addition of sodium perfluoroalkanesulfinates to alkenes. Chin. J. Chem. 2010, 10, 365–372. [Google Scholar] [CrossRef]
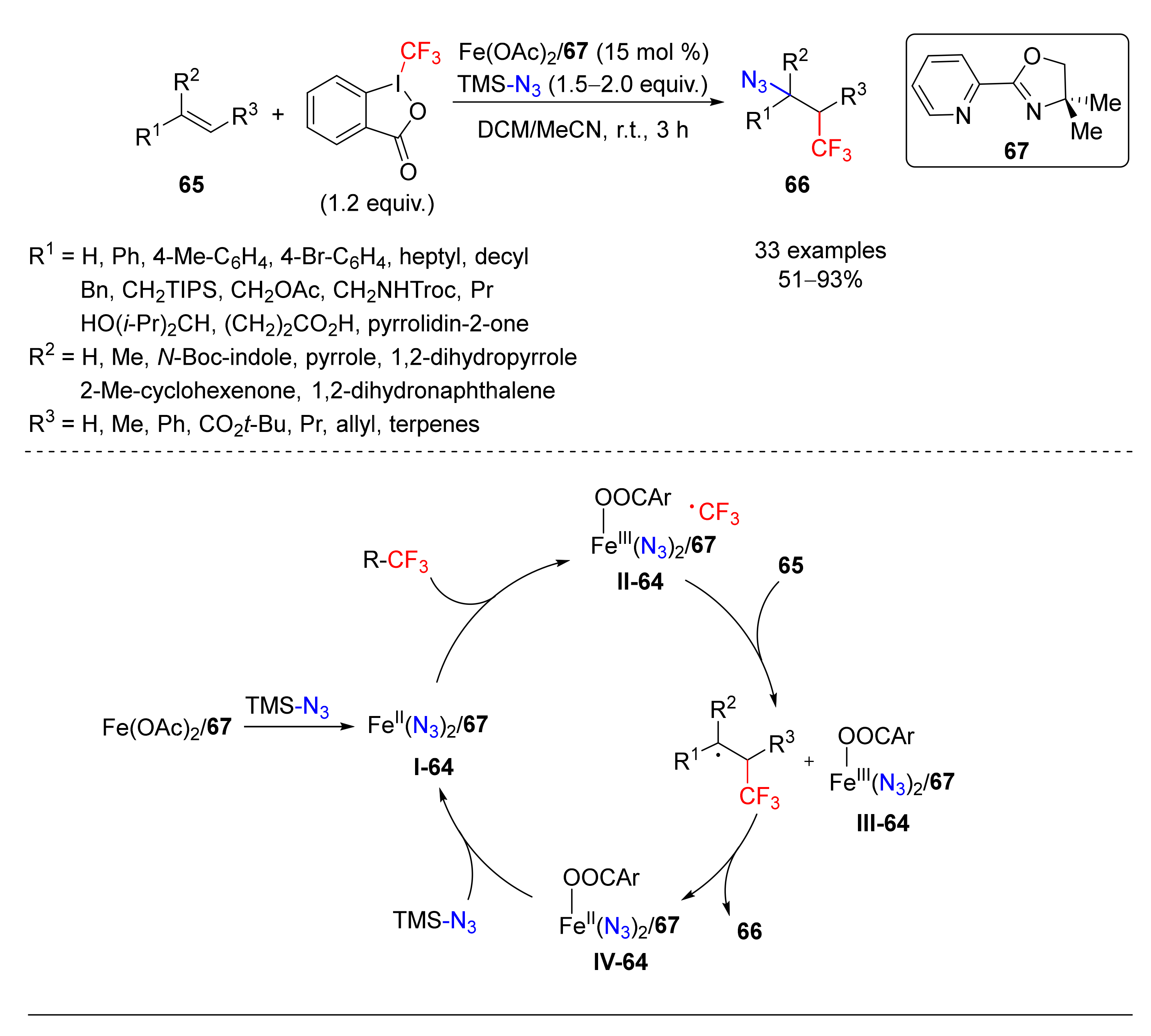
- Zhu, C.-L.; Wang, C.; Qin, Q.-X.; Yruegas, S.; Martin, C.D.; Xu, H.; Yruegas, S. Iron(II)-Catalyzed Azidotrifluoromethylation of Olefins and N-Heterocycles for Expedient Vicinal Trifluoromethyl Amine Synthesis. ACS Catal. 2018, 8, 5032–5037. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Ramkumar, N.; Chiou, M.-F.; Jian, W.; Li, Y.; Su, J.-H.; Zhang, X.; Bao, H. Iron-catalyzed carboazidation of alkenes and alkynes. Nat. Commun. 2019, 10, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, M.-F.; Xiong, H.; Li, Y.; Bao, H.; Zhang, X. Revealing the Iron-Catalyzed β-Methyl Scission of tert-Butoxyl Radicals via the Mechanistic Studies of Carboazidation of Alkenes. Molecules 2020, 25, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Chen, J.; Chu, L. Solvent-tuned Chemoselective carboazidation and diazidation of alkenes via iron catalysis. Org. Chem. Front. 2019, 6, 512–516. [Google Scholar] [CrossRef]
- Hajra, S.; Sinha, D.; Bhowmick, M. Metal triflate catalyzed highly regio- and stereoselective 1,2-bromoazidation of alkenes using NBS and TMSN3 as the bromine and azide sources. Tetrahedron Lett. 2006, 47, 7017–7019. [Google Scholar] [CrossRef]
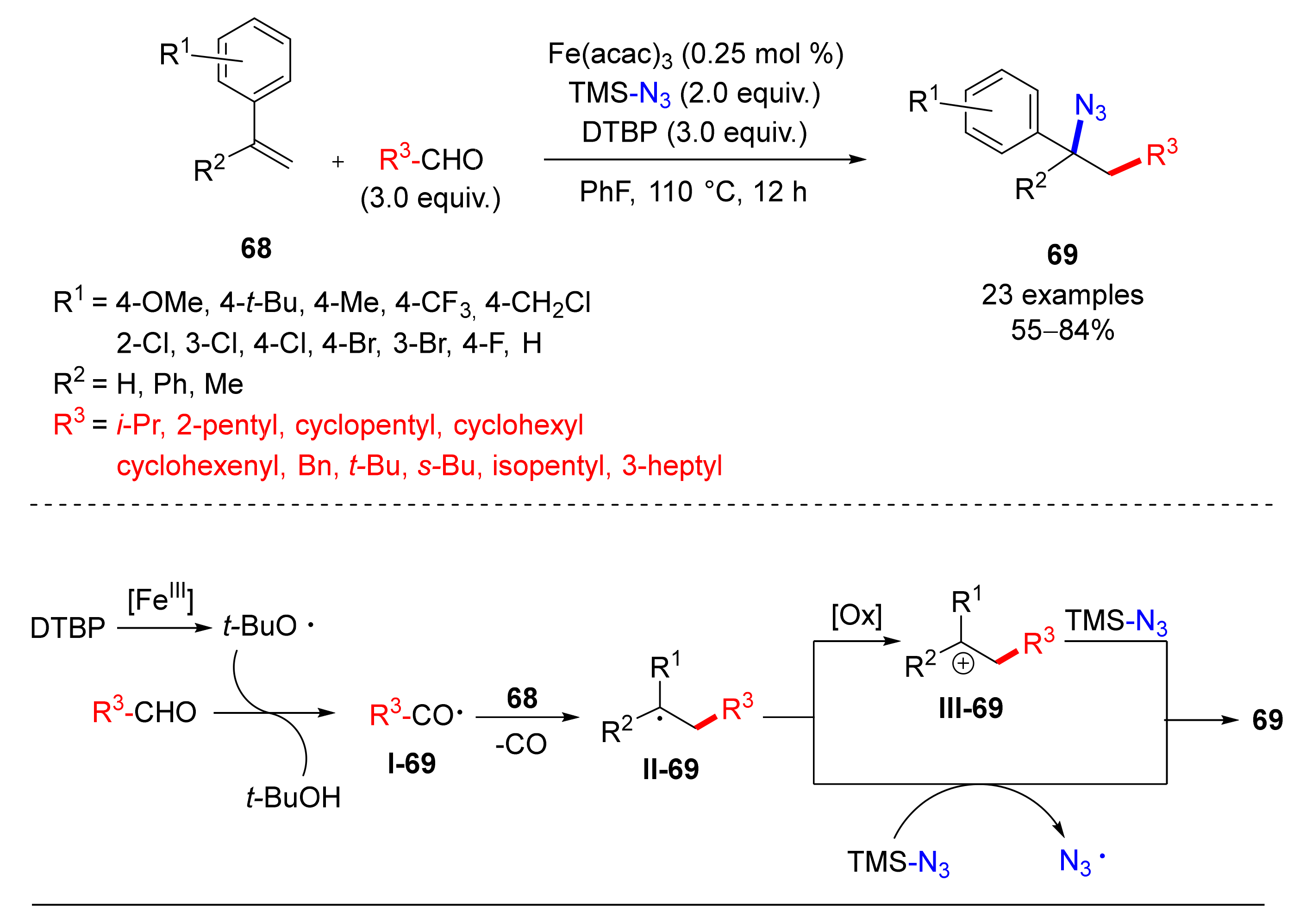
- Li, W.-Y.; Wang, Q.-Q.; Yang, L. Fe-Catalyzed radical-type difunctionalization of styrenes with aliphatic aldehydes and trimethylsisyl azide via a decarbonilative alkylation-azidation cascade. Org. Biomol. Chem. 2017, 15, 9987–9991. [Google Scholar] [CrossRef]
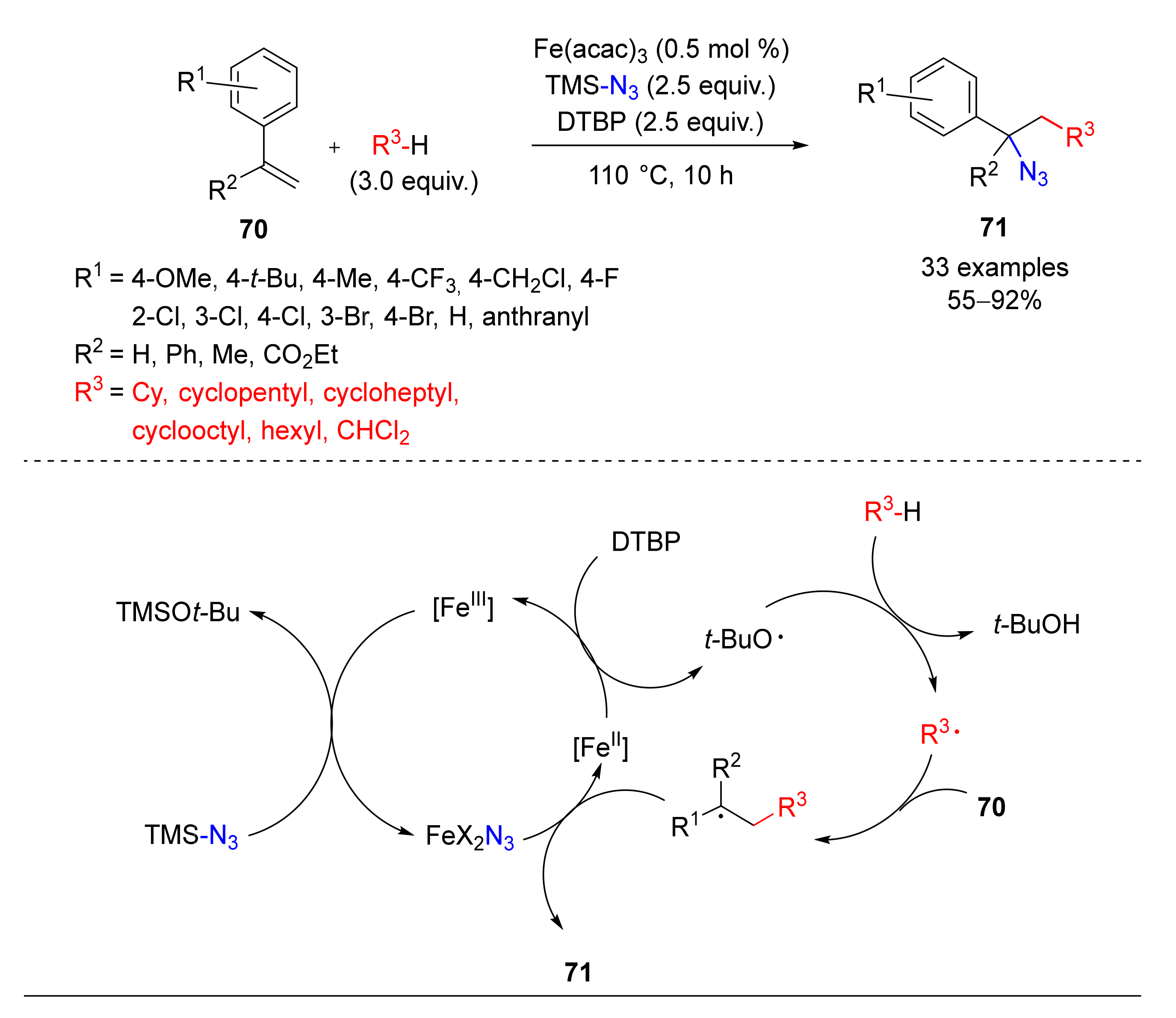
- Li, W.-Y.; Wu, C.-S.; Zhou, W.; Yang, L. Fe-catalyzed three-component carboazidation of alkenes with alkanes and trimethylsilyl azide. Chem. Commun. 2018, 54, 11013–11016. [Google Scholar] [CrossRef]
- Yang, B.; Ren, X.; Shen, X.; Li, T.; Lu, Z. Visible Light-Promoted Three-Component Carboazidation of Unactivated Alkenes with TMSN3 and Acrylonitrile. Chin. J. Chem. 2018, 36, 1017–1023. [Google Scholar] [CrossRef]
- Wei, X.-H.; Li, Y.-M.; Zhou, A.-X.; Yang, T.-T.; Yang, S.-D. Silver-Catalyzed Carboazidation of Arylacrylamides. Org. Lett. 2013, 15, 4158–4161. [Google Scholar] [CrossRef]
- Yuan, Y.; Shen, T.; Wang, K.; Jiao, N. Ag-Promoted Azido-Carbocyclization of Activated Alkenes via C-H Bond Cleavage. Chem. Asian J. 2013, 8, 2932–2935. [Google Scholar] [CrossRef]
- Xu, L.; Mou, X.-Q.; Chen, Z.-M.; Wang, S.-H. Copper-catalyzed intermolecular azidocyanation of aryl alkenes. Chem. Commun. 2014, 50, 10676–10679. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, W.; Wu, W.; Jiang, H. Palladium-catalyzed regioselective azidation of allylic C–H bonds under atmospheric pressure of dioxygen. Org. Biomol. Chem. 2014, 12, 3340. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Cheng, C.-H. Palladium-catalyzed intermolecular carboazidation of allenes with aryl iodides and trimethylsilyl azide. J. Chem. Soc. Perkin Trans. 1 2000, 22, 3799–3807. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, M.; Grigg, R.; Sridharan, V.; Vicker, N. Cascade and Sequential Palladium Catalysed Cyclisation-Azide Capture-1,3-Dipolar Cycloaddition Route to Complex Triazoles. Tetrahedron Lett. 1998, 39, 435–438. [Google Scholar] [CrossRef]
- Hurtado-Rodrigo, C.; Hoehne, S.; Muñoz, M.P. A new gold-catalysed azidation of allenes. Chem. Commun. 2014, 50, 1494–1496. [Google Scholar] [CrossRef] [Green Version]
- Porta, E.O.J.; Vallejos, M.M.; Bracca, A.B.J.; Labadie, G.R. Experimental and theoretical studies of the [3,3]-sigmatropic rearrangement of prenyl azides. RSC Adv. 2017, 7, 47527–47538. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, A. Transition States and Activation Barriers for [3,3]-Sigmatropic Shift of Allyl Azides. Org. Chem. J. 2010, 1, 6–14. [Google Scholar]
- Khrakovsky, D.A.; Tao, C.; Johnson, M.W.; Thornbury, R.T.; Shavick, S.L.; Toste, F.D. Enantioselectivem Stereodivrgent Hydroazidation and Hydroamination of Allenes Catalyzed by Acyclic Diaminocarbene (ADC) Gold(I) Complexes. Angew. Chem. Int. Ed. 2016, 55, 6079–6083. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ding, N.; Tan, X.; Li, X.; Zhao, Z. Iron(II)-chloride-catalyzed regioselective azidation of allenamides with TMSN3. Chem. Commun. 2020, 56, 7507–7510. [Google Scholar] [CrossRef]
- Waser, J.; Nambu, H.; Carreira, E.M. Cobalt-Catalyzed Hydroazidation of Olefins: Convenient Access to Alkyl Azides. J. Am. Chem. Soc. 2005, 127, 8294–8295. [Google Scholar] [CrossRef]
- Waser, J.; Boris, G.; Nambu, H.; Carreira, E.M. Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J. Am. Chem. Soc. 2006, 128, 11693–11712. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-J.; Yu, W. Anti-Markovnikov Hydroazidation of Alkenes by Visible-Light Photoredox Catalysis. Chem. Eur. J. 2019, 25, 3510–3514. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Wang, T.; Qin, C.; Jiao, N. Silver-Catalyzed Nitrogenation of Alkynes: A Direct Approach to Nitriles through C≡C Bond Cleavage. Angew. Chem. Int. Ed. 2013, 52, 6677–6680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liao, P.; Bi, X. General Silver-Catalyzed Hydroazidation of Terminal Alkynes by Combining TMS-N3 and H2O: Synthesis of Vinyl Azides. Org. Lett. 2014, 16, 3668–3671. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Zhang, L.; Liao, P.; Song, J.; Bi, X. Silver(I)-Catalyzed Hydroazidation of Ethynyl Carbinols: Synthesis of 2-Azidoallyl Alcohols. Angew. Chem. Int. Ed. 2014, 53, 5305–5309. [Google Scholar] [CrossRef]
- Pal, P.; Mainkar, P.S.; Nayani, K.; Chandrasekhar, S. Mn-catalyzed radical initiated domino transformation of alkynylated cyclohexadienones with TMSN3 and O2 to bicyclic azido alcohols. Chem. Commun. 2020, 56, 3453–3456. [Google Scholar] [CrossRef]
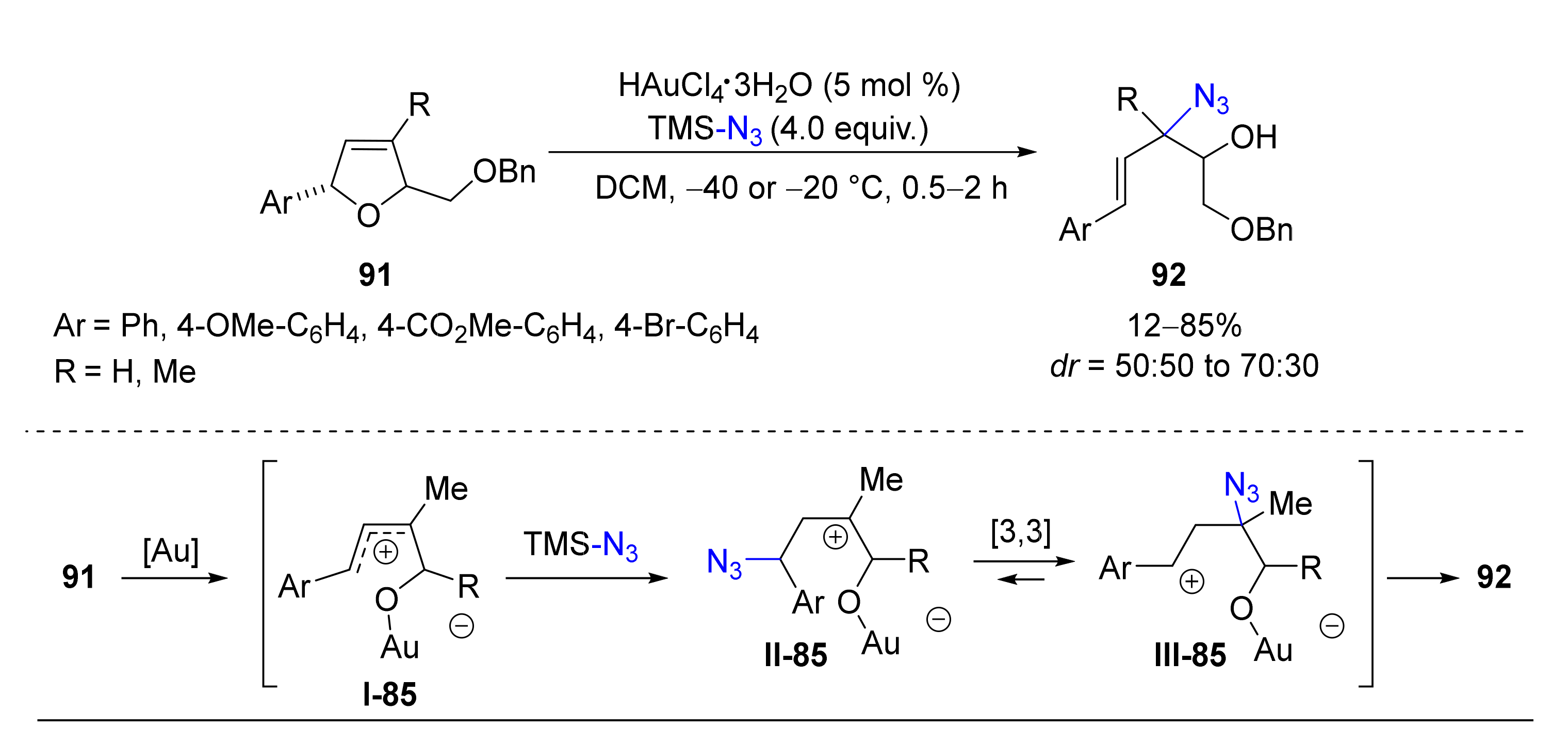
- Sawama, Y.; Sawama, Y.; Krause, N. Highly Regioselective Gold-Catalyzed Ring-Opening Allylation and Azidation of Dihydrofurans. Org. Lett. 2009, 11, 5034–5037. [Google Scholar] [CrossRef]
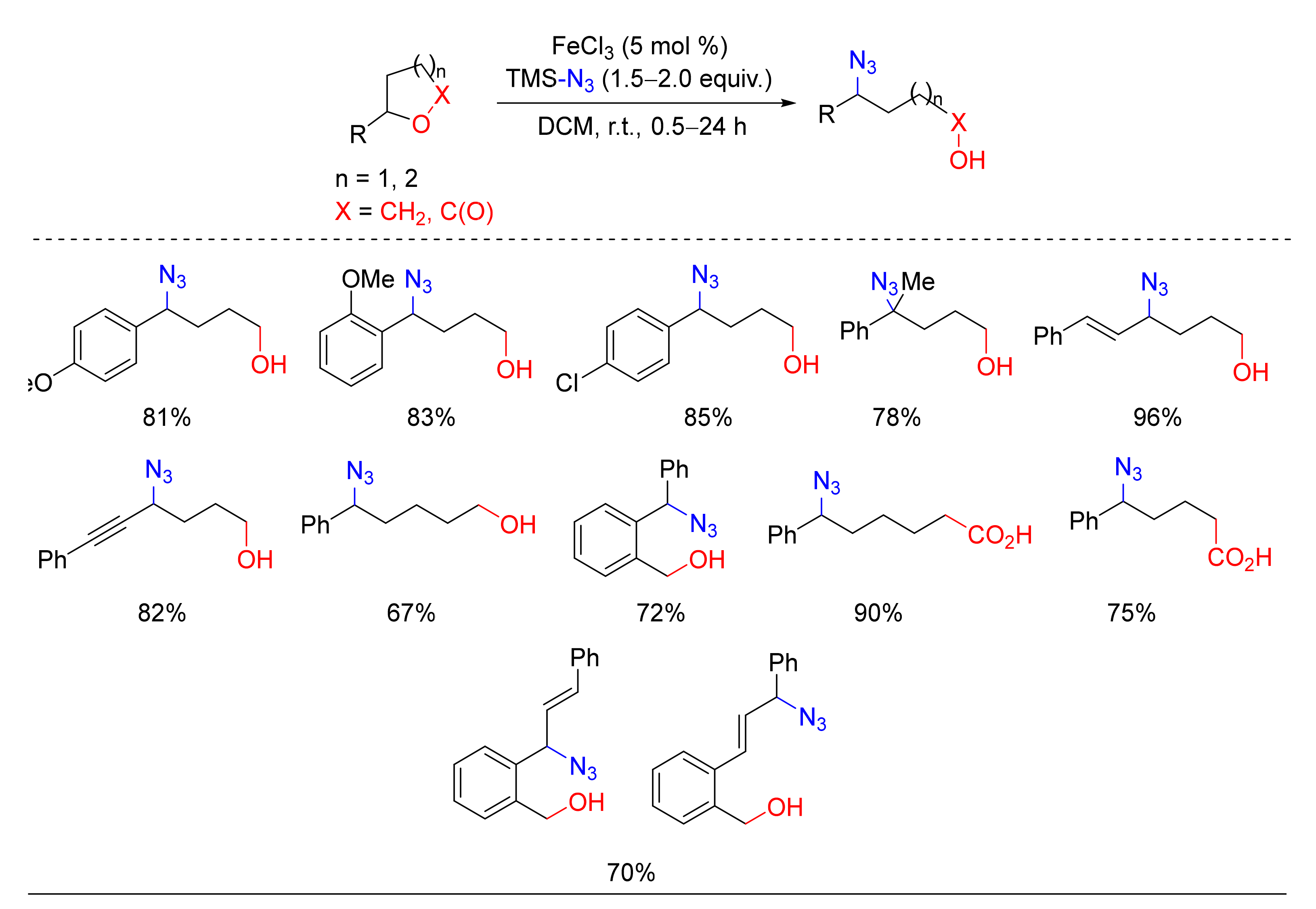
- Sawama, Y.; Shibata, K.; Sawama, Y.; Takubo, M.; Monguchi, Y.; Krause, N.; Sajiki, H. Iron-Catalyzed Ring-Opening Azidation and Allylation of O-Heterocycles. Org. Lett. 2013, 15, 5282–5285. [Google Scholar] [CrossRef]
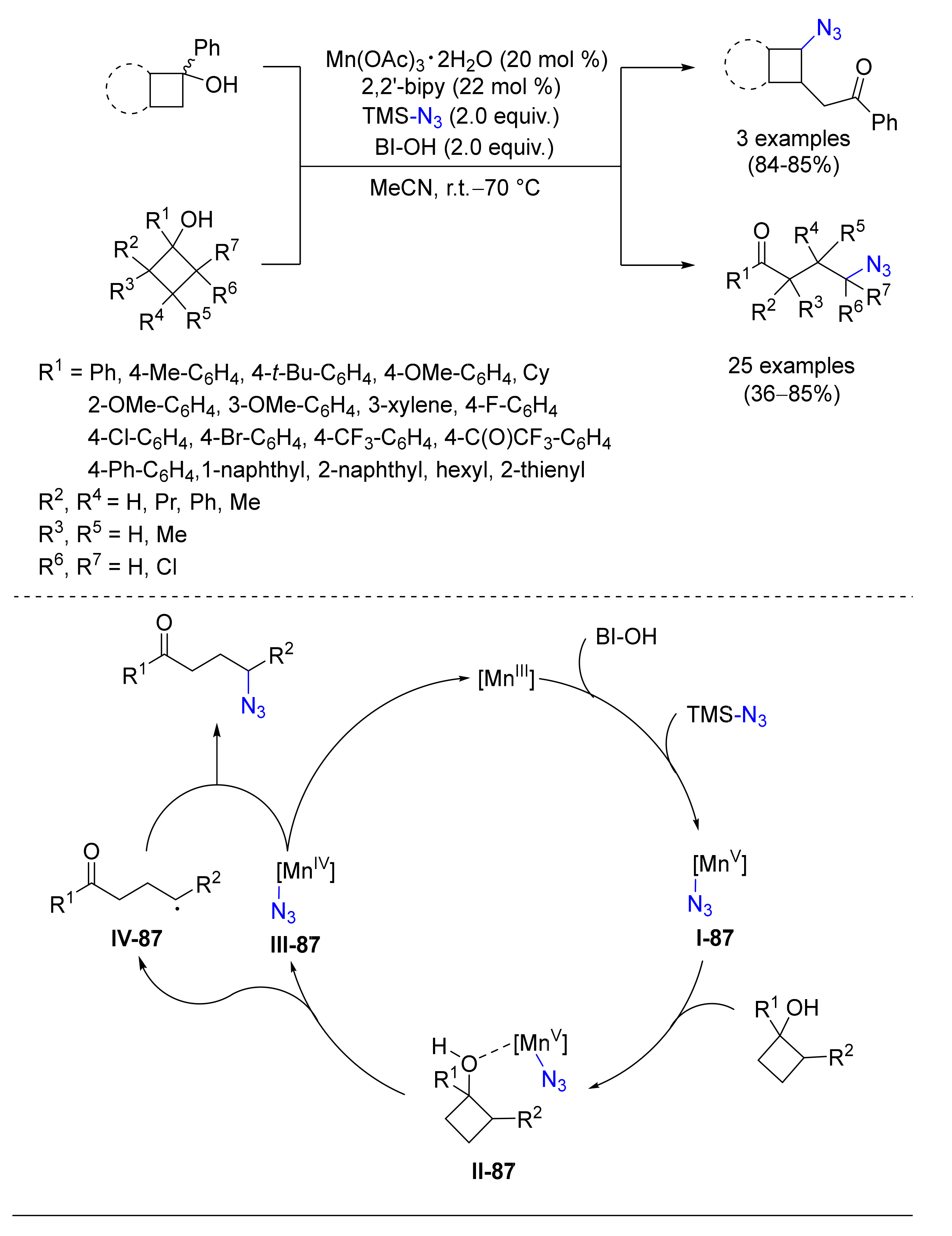
- Ren, R.; Zhao, H.; Huan, L.; Zhu, C. Manganese-Catalyzed Oxidative Azidation of Cyclobutanols: Regiospecific Synthesis of Alkyl Azides by C-C Bond Cleavage. Angew. Chem. Int. Ed. 2015, 54, 12692–12696. [Google Scholar] [CrossRef]
























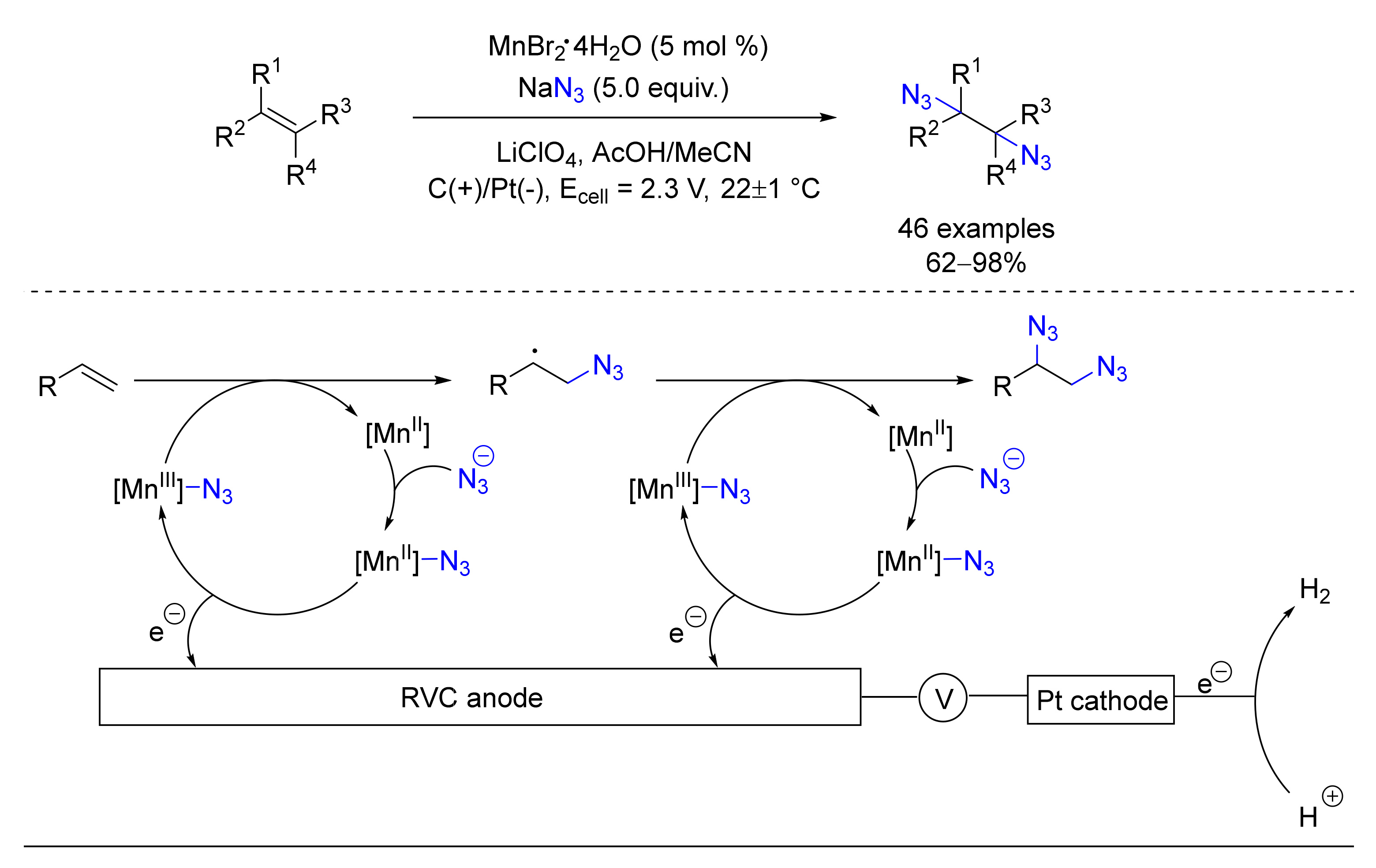
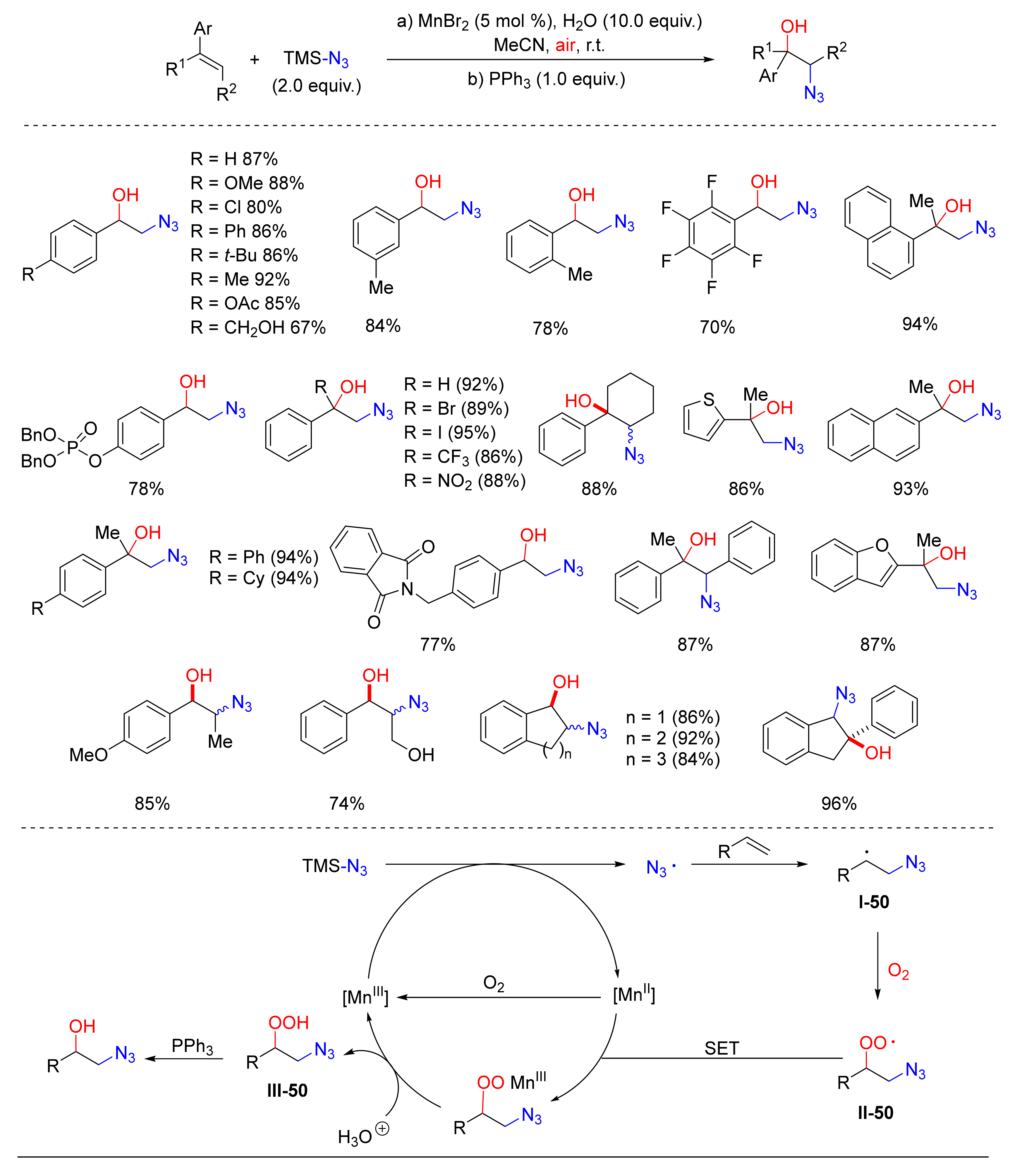
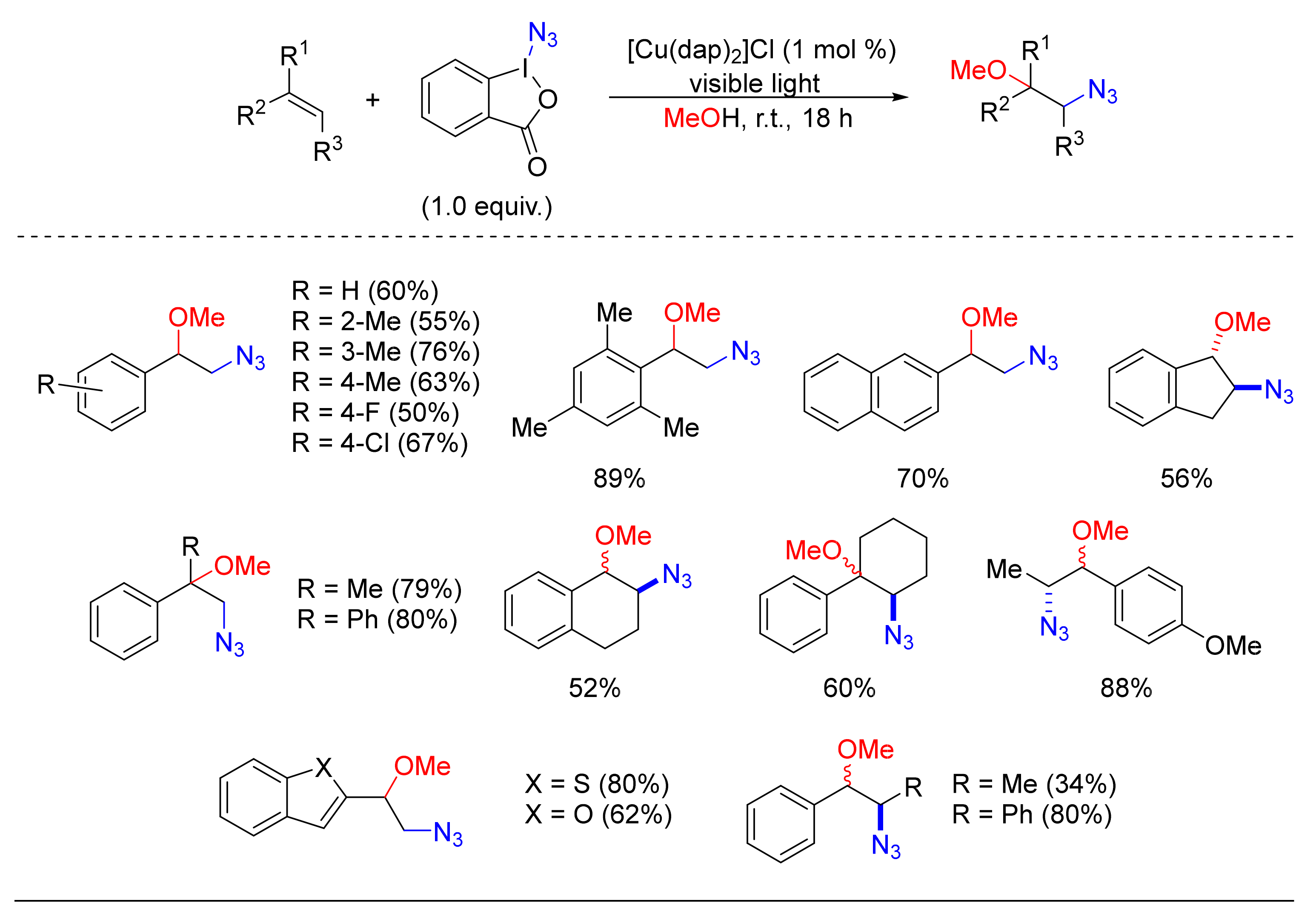
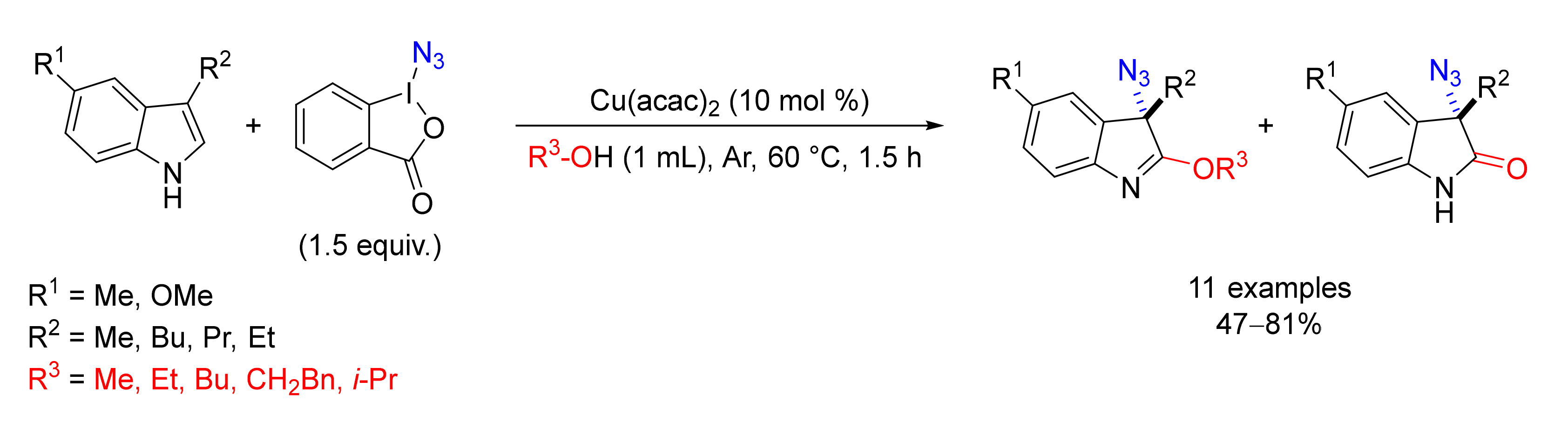











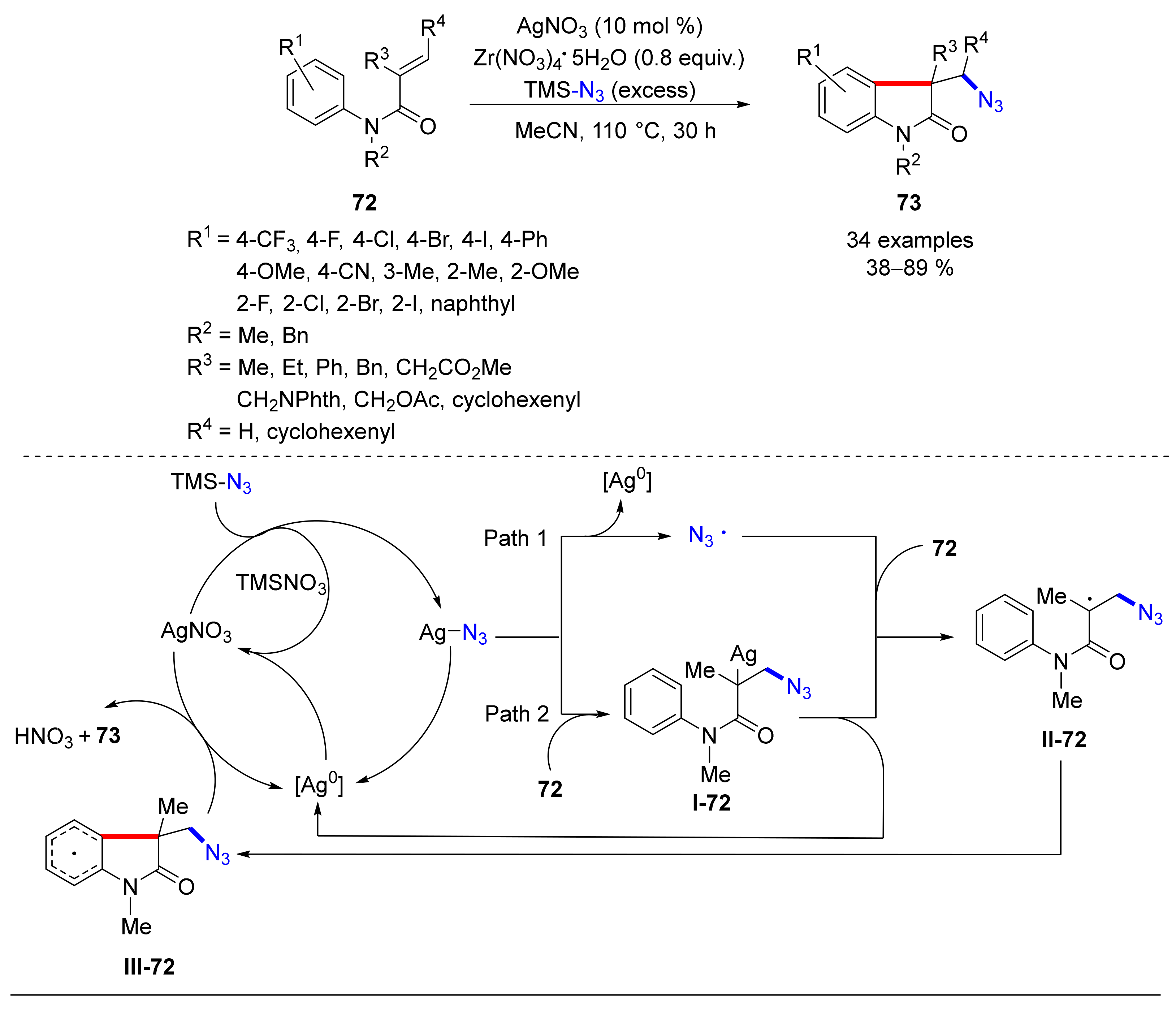


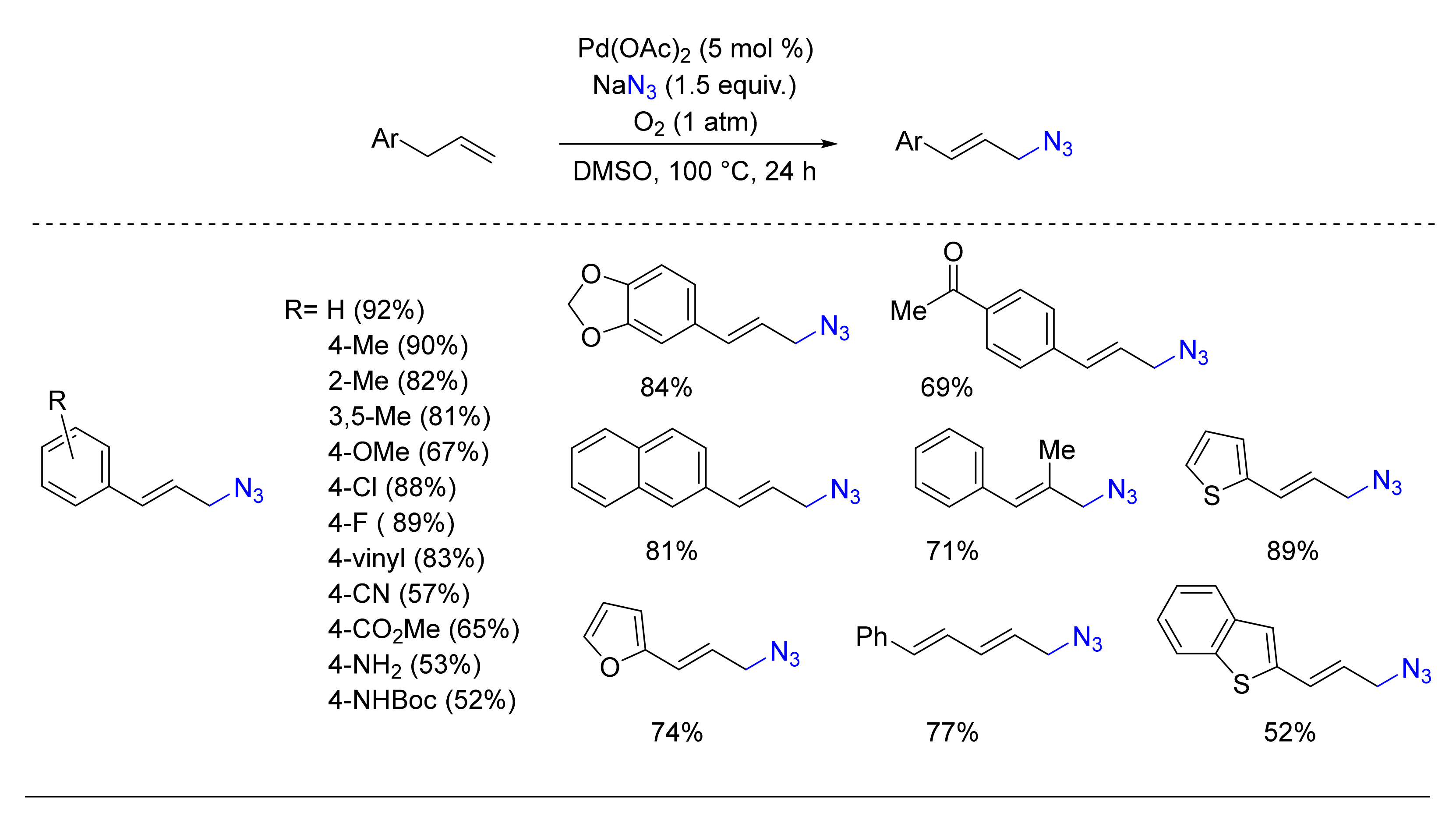
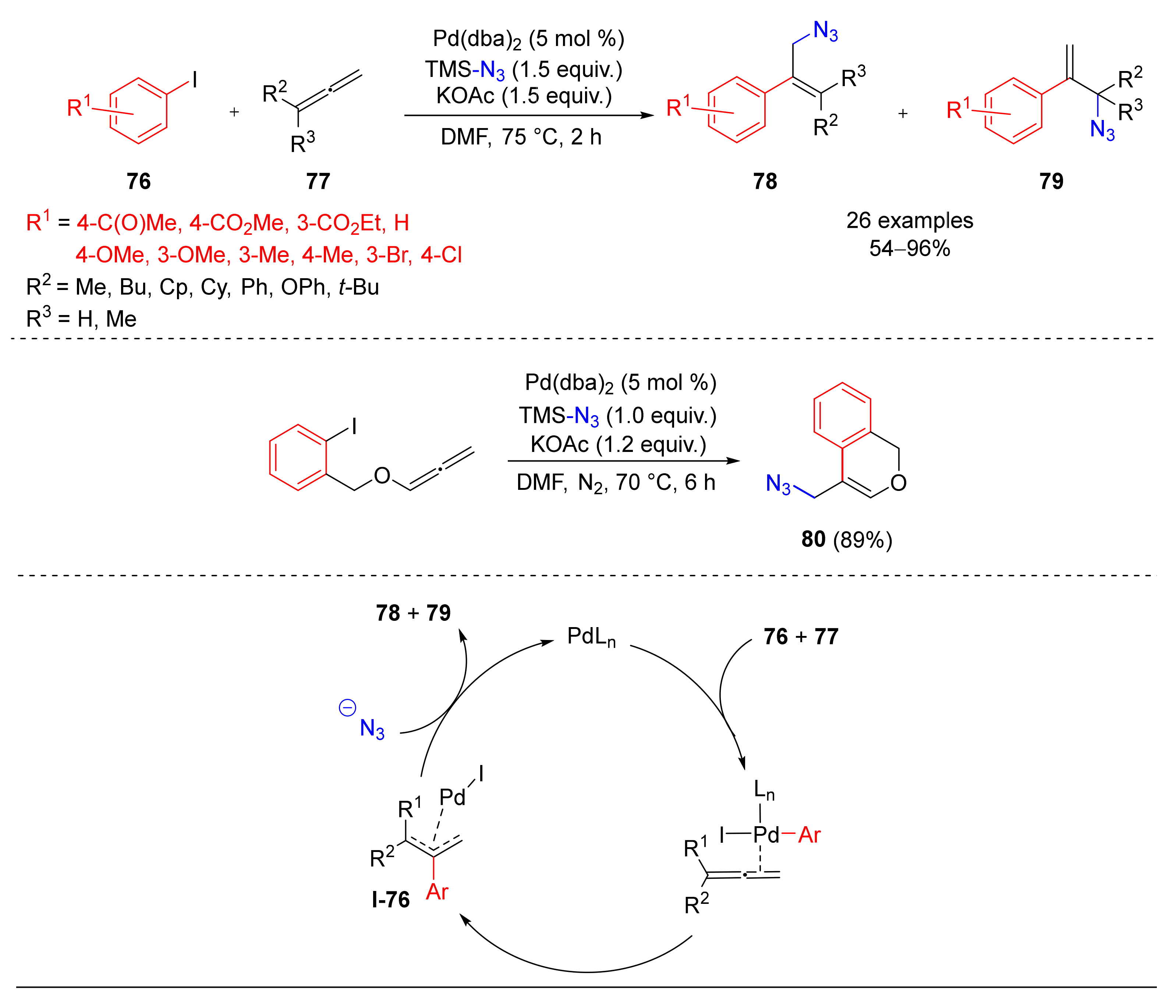
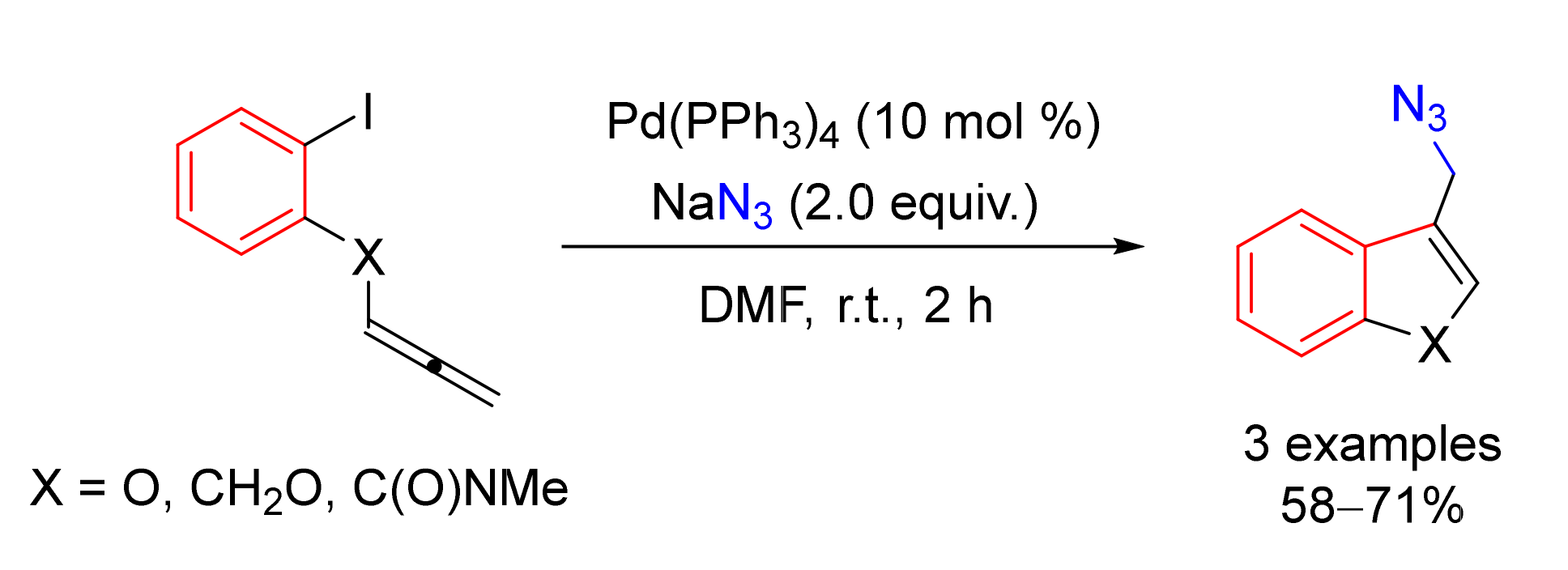

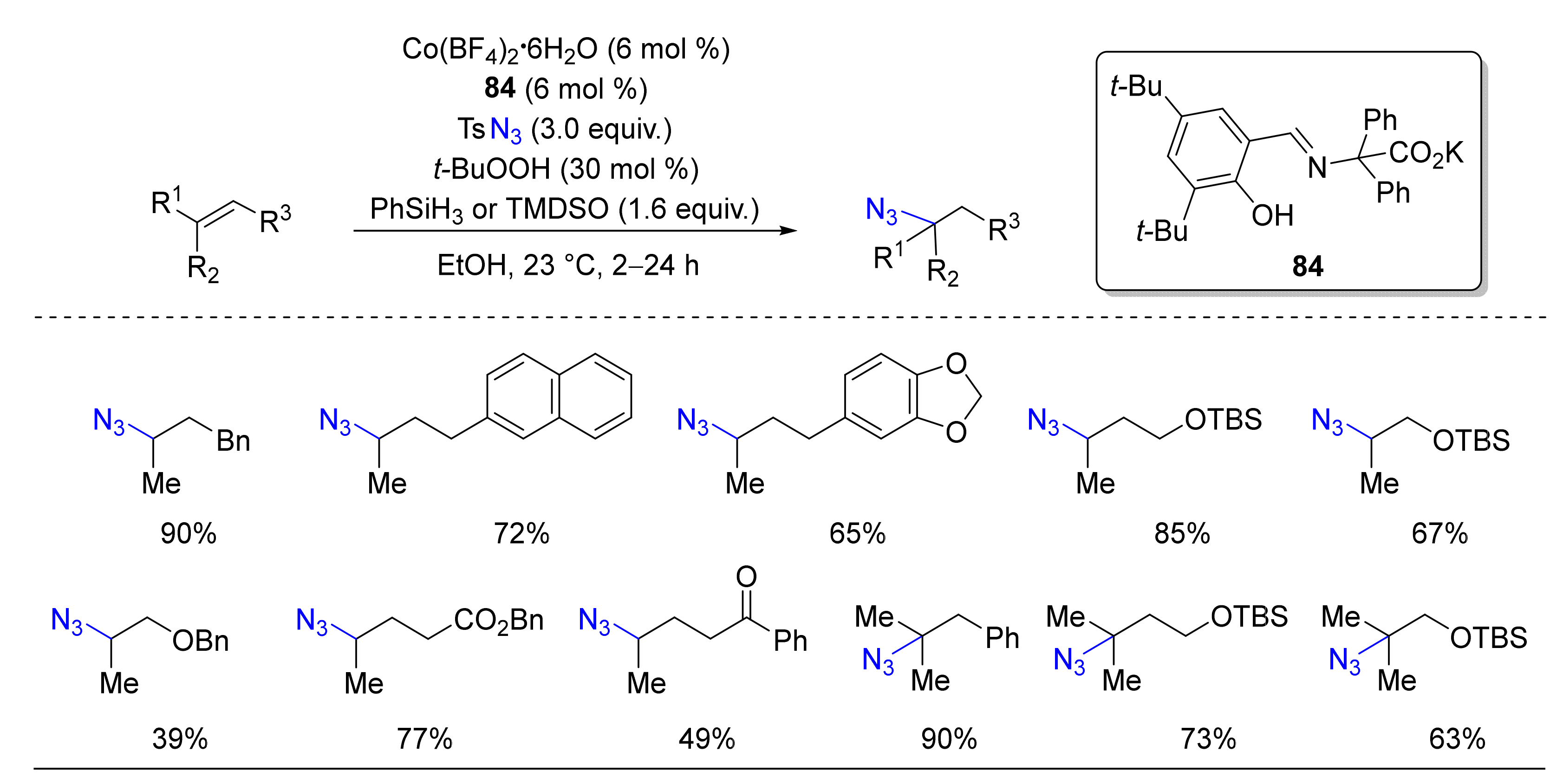
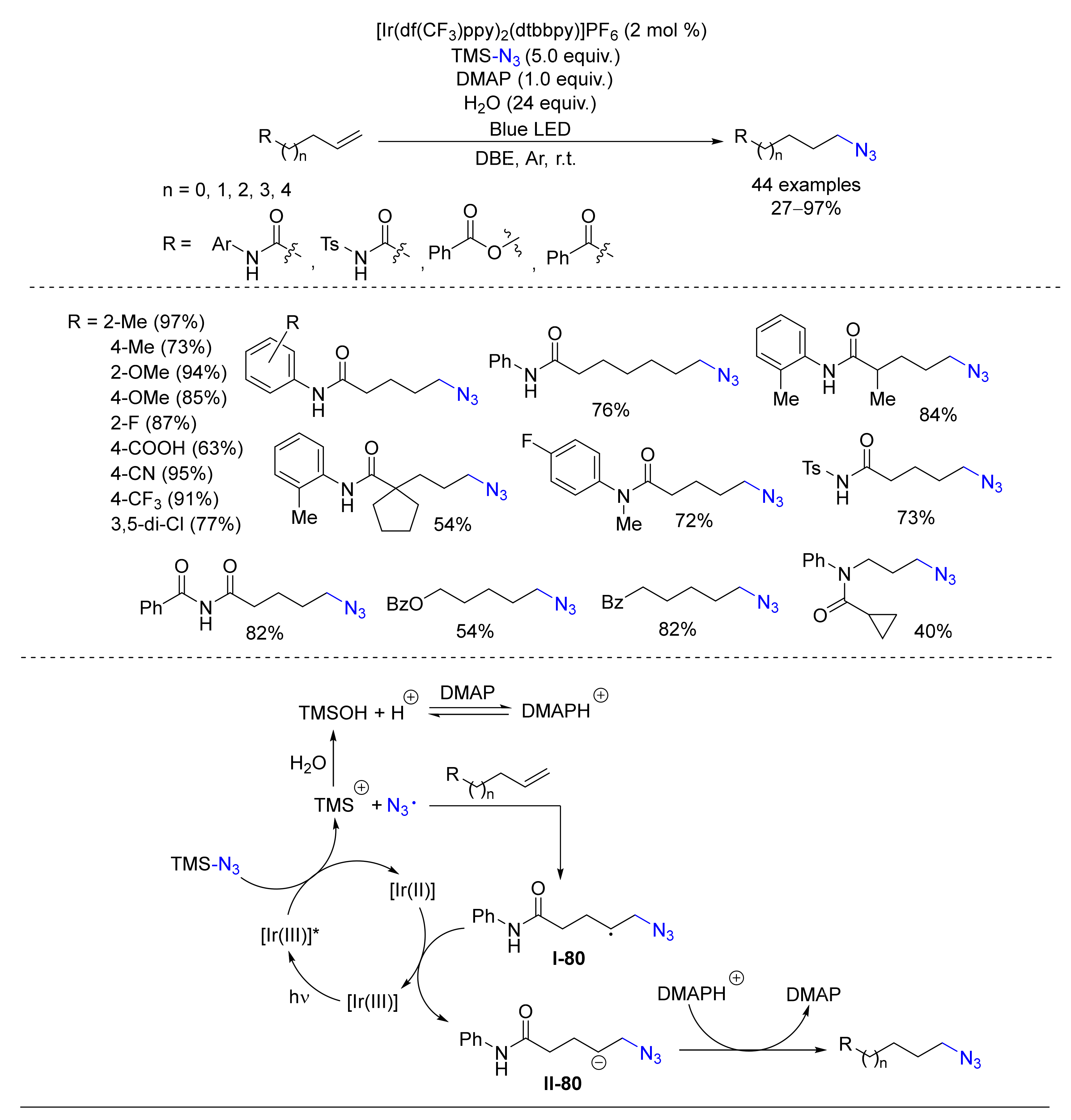

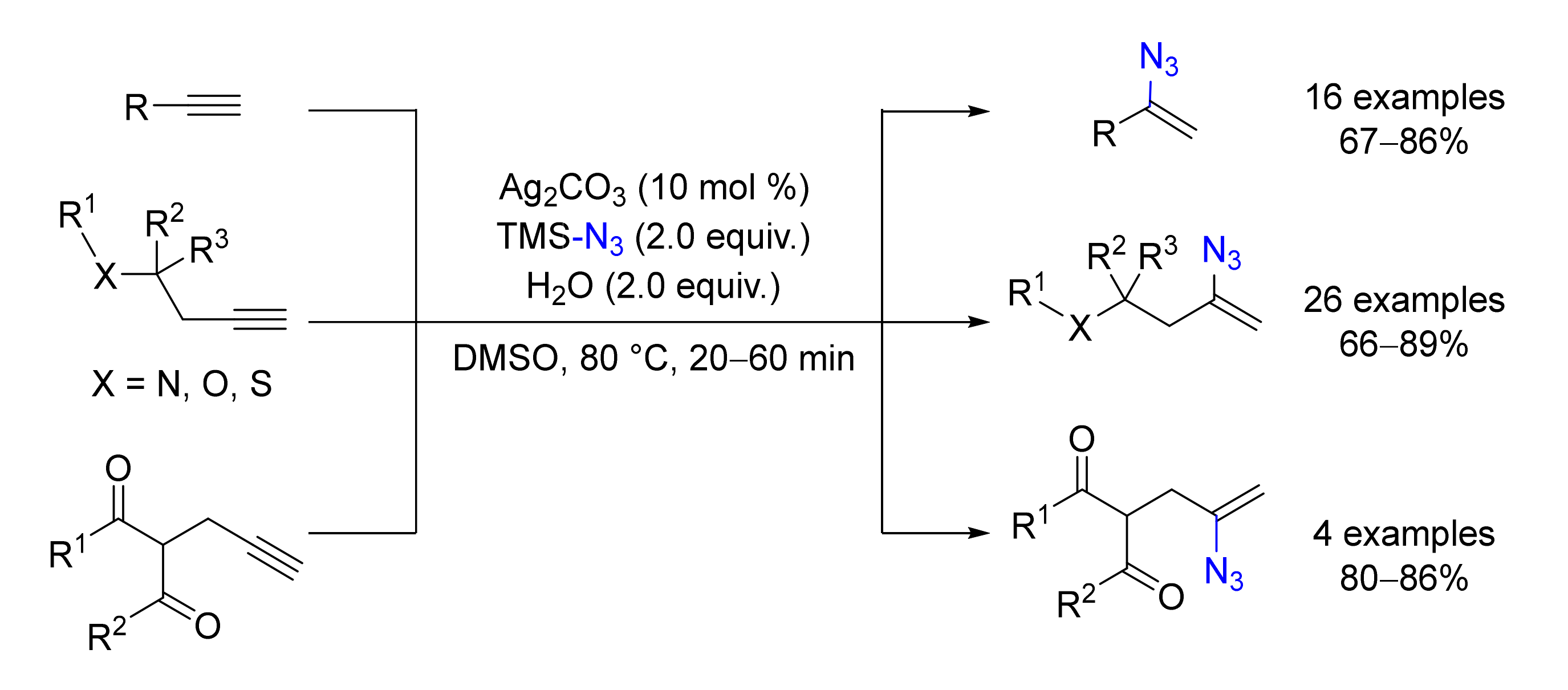

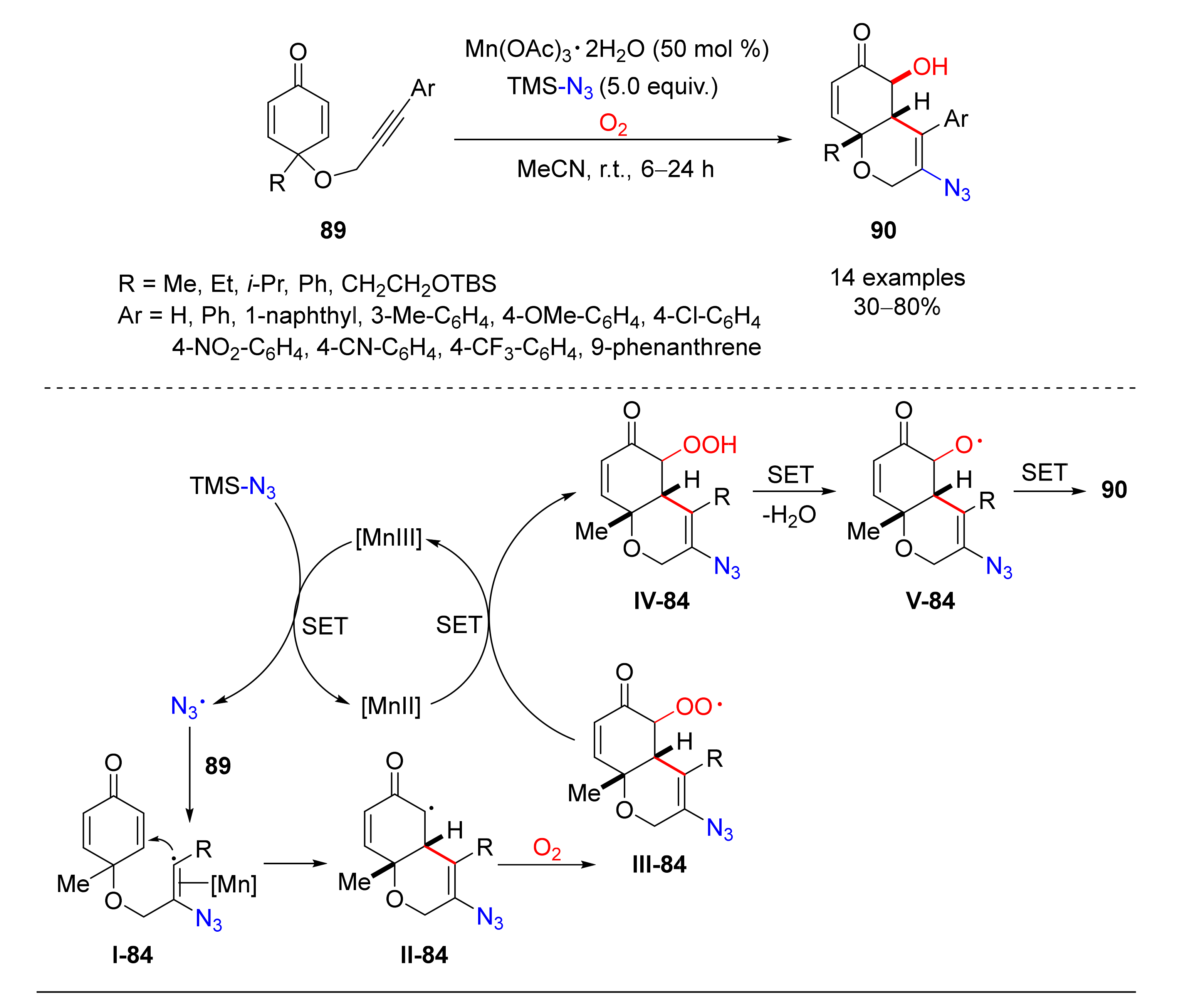
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala, R.; Loro, C.; Foschi, F.; Broggini, G. Transition Metal Catalyzed Azidation Reactions. Catalysts 2020, 10, 1173. https://doi.org/10.3390/catal10101173
Sala R, Loro C, Foschi F, Broggini G. Transition Metal Catalyzed Azidation Reactions. Catalysts. 2020; 10(10):1173. https://doi.org/10.3390/catal10101173
Chicago/Turabian StyleSala, Roberto, Camilla Loro, Francesca Foschi, and Gianluigi Broggini. 2020. "Transition Metal Catalyzed Azidation Reactions" Catalysts 10, no. 10: 1173. https://doi.org/10.3390/catal10101173
APA StyleSala, R., Loro, C., Foschi, F., & Broggini, G. (2020). Transition Metal Catalyzed Azidation Reactions. Catalysts, 10(10), 1173. https://doi.org/10.3390/catal10101173