A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate

 ,
,  and
and 
Abstract
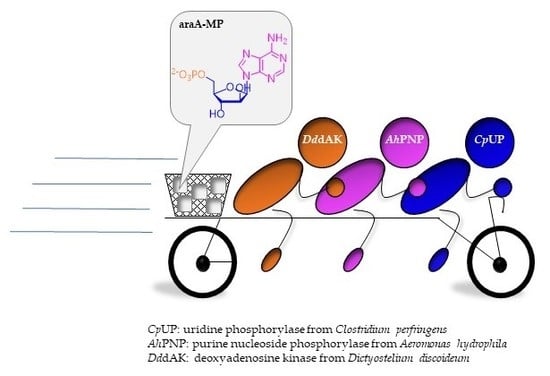
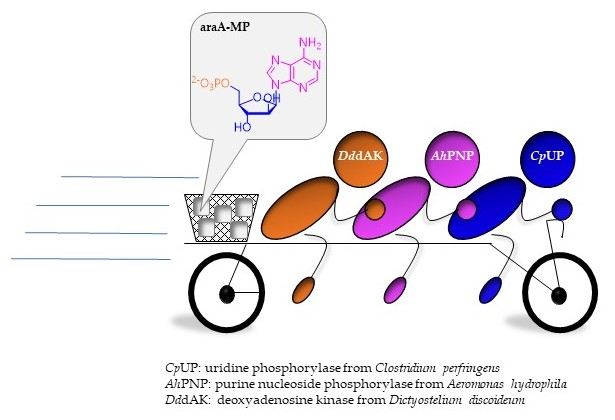
1. Introduction
2. Results and Discussion
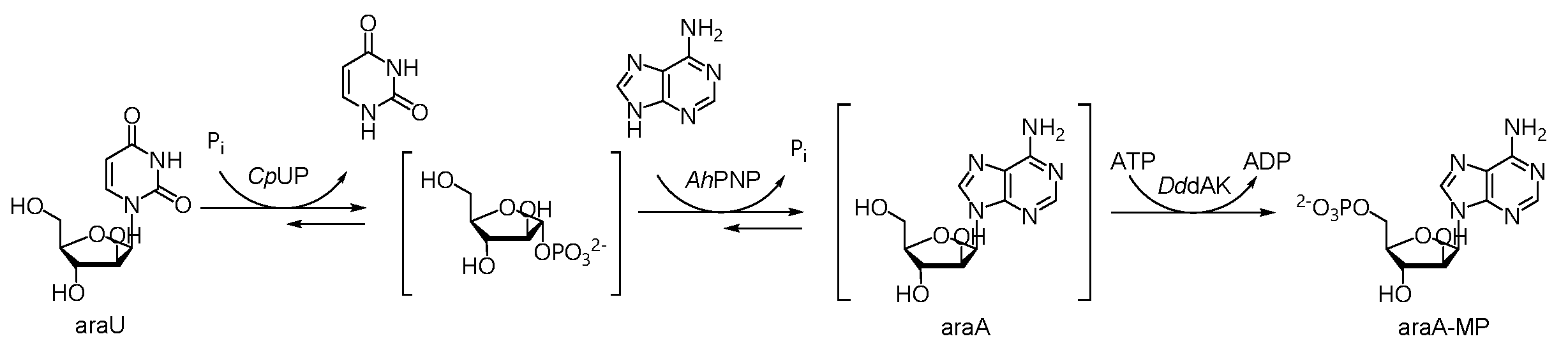
2.1. A Three-Enzyme Cascade for the Synthesis of araA-MP
2.1.1. Reaction Temperature and pH
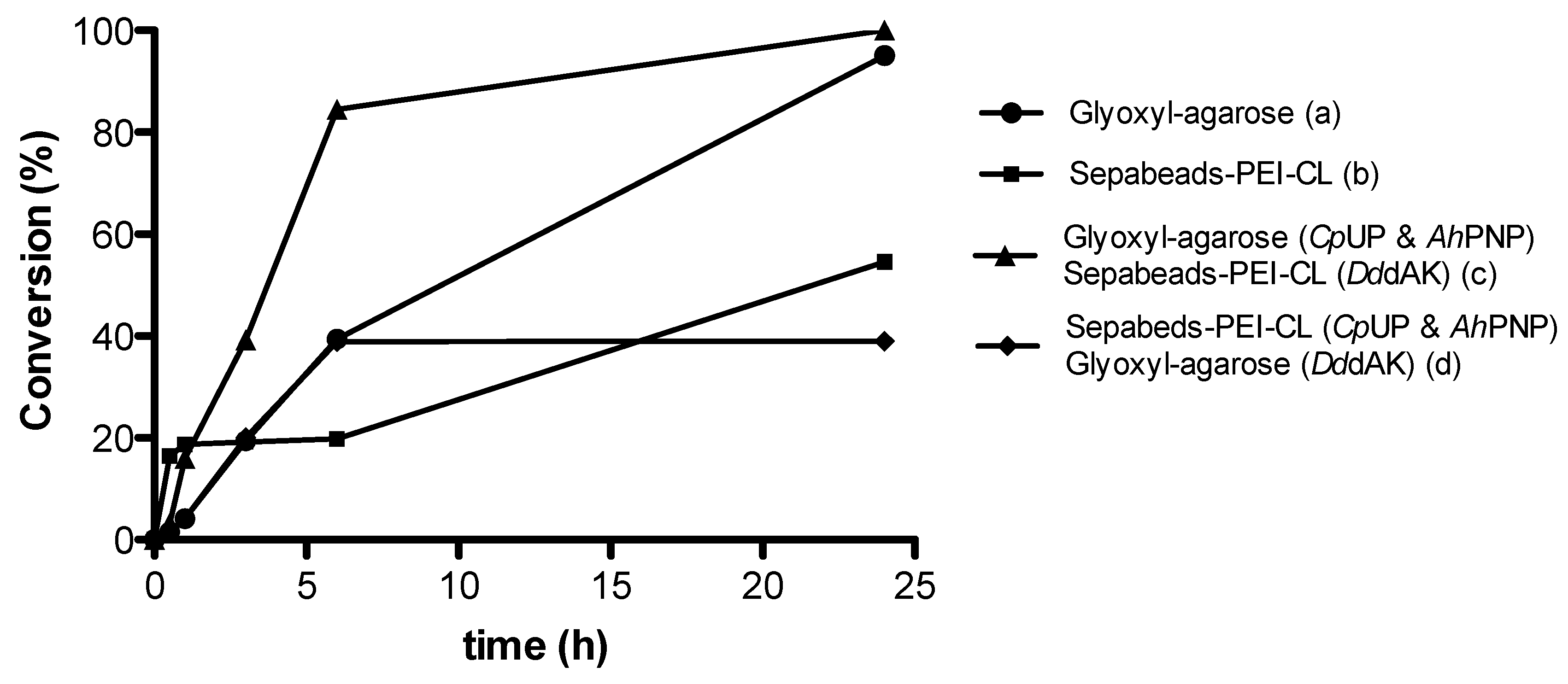
2.1.2. Immobilization Carrier
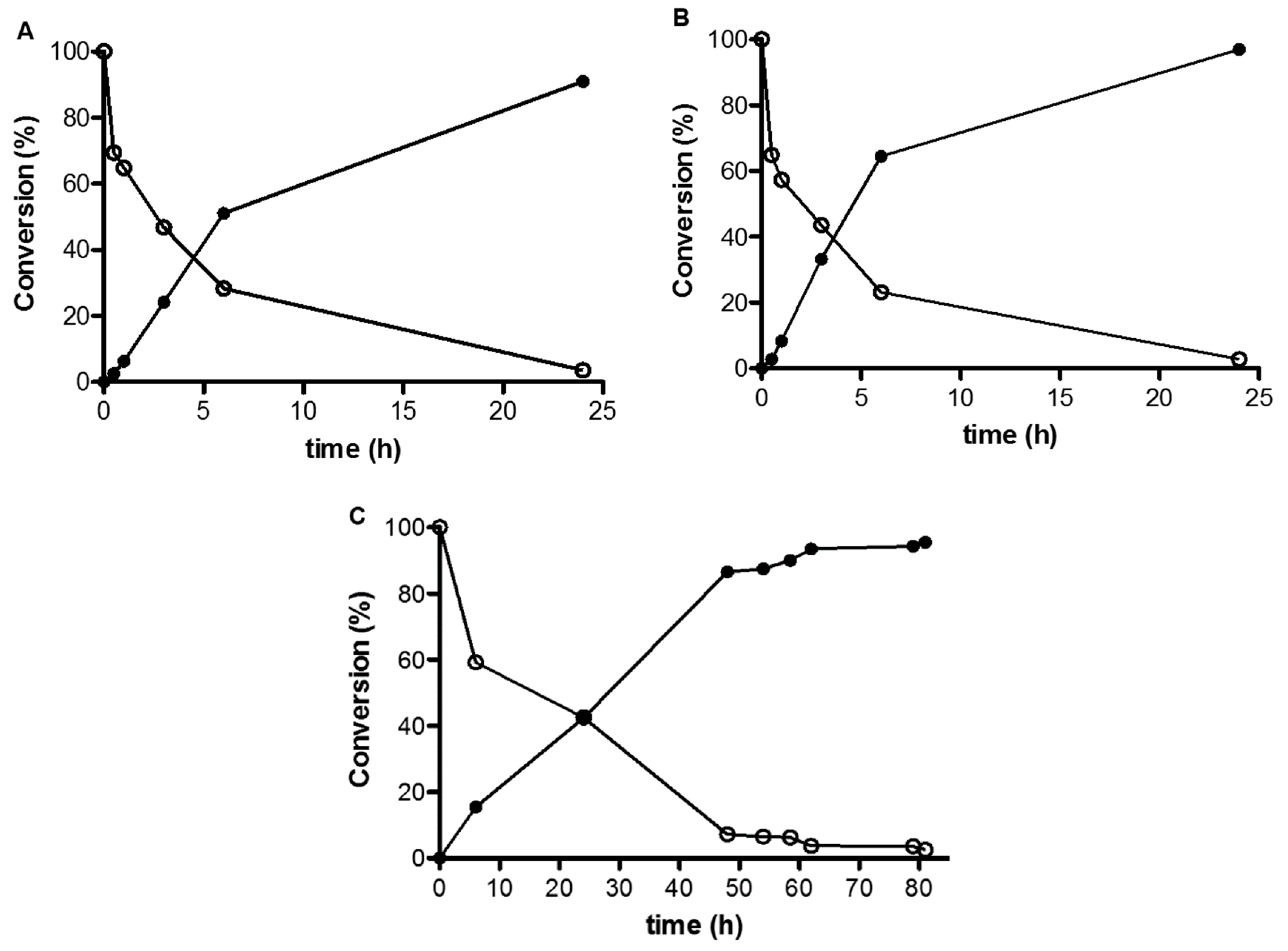
2.2. Reaction Scale-Up and Product Downstream
3. Materials and Methods
3.1. Preparation of Sepabeads®-PEI and Aldehyde Dextran
3.2. Immobilization on Sepabeads®-PEI and Cross-Linking with Aldehyde Dextran. General Procedure
3.3. Preparation of Aldehyde-Agarose (Glyoxyl-Agarose)
3.4. Immobilization on Aldehyde-Activated Agarose (Glyoxyl-Agarose). General Procedure.
3.5. Enzymatic Activity Assays
3.5.1. NP Activity Assay (Phosphorolysis Reaction)
3.5.2. DddAK Activity Assay (Phosphorylation Reaction)
3.6. Synthesis of araA-MP (Analytical Scale)
3.7. Synthesis of araA-MP (Scale-Up)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antiviral Res. 2018, 155, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Tamamyan, G.; Kadia, T.; Ravandi, F.; Borthakur, G.; Cortes, J.; Jabbour, E.; Daver, N.; Ohanian, M.; Kantarjian, H.; Konopleva, M. Frontline treatment of acute myeloid leukemia in adults. Crit. Rev. Oncol. Hematol. 2017, 110, 20–34. [Google Scholar] [CrossRef]
- Glaudemans, G.P.J.; Fletcher, H.G. Syntheses with partially benzylated sugars. III. A simple pathway to a cis-nucleoside, 9-β-d-arabinofuranosyladenine (spongoadenosine). J. Org. Chem. 1963, 28, 3004–3006. [Google Scholar] [CrossRef]
- Moreau, C.; Ashamu, G.A.; Bailey, V.C.; Galione, A.; Guse, A.H.; Potter, B.V.L. Synthesis of cyclic adenosine 5′-diphosphate ribose analogues: A C2′ endo/syn “southern” ribose conformation underlies activity at the sea urchin cADPR receptor. Org. Biomol. Chem. 2011, 9, 278–290. [Google Scholar] [CrossRef]
- Rocchietti, S.; Ubiali, D.; Terreni, M.; Albertini, A.M.; Fernández-Lafuente, R.; Guisán, J.M.; Pregnolato, M. Immobilization and stabilization of recombinant multimeric uridine and purine nucleoside phosphorylases from Bacillus subtilis. Biomacromolecules 2004, 5, 2195–2200. [Google Scholar] [CrossRef]
- Ubiali, D.; Serra, C.D.; Serra, I.; Morelli, C.F.; Terreni, M.; Albertini, A.M.; Manitto, P.; Speranza, G. Production, characterization and synthetic application of a purine nucleoside phosphorylase from Aeromonas hydrophila. Adv. Synth. Catal. 2012, 354, 96–104. [Google Scholar] [CrossRef]
- Serra, I.; Ubiali, D.; Piškur, J.; Christoffersen, S.; Lewkowicz, E.S.; Iribarren, A.M.; Albertini, A.M.; Terreni, M. Developing a collection of immobilized nucleoside phosphorylases for the preparation of nucleoside analogues: Enzymatic synthesis of arabinosyladenine and 2′,3′-dideoxyinosine. ChemPlusChem 2013, 78, 157–165. [Google Scholar] [CrossRef]
- Serra, I.; Conti, S.; Piškur, J.; Clausen, A.R.; Munch-Petersen, B.; Terreni, M.; Ubiali, D. Immobilized Drosophila melanogaster deoxyribonucleoside kinase (DmdNK) as a high performing biocatalyst for the synthesis of purine arabinonucleotides. Adv. Synth. Catal. 2014, 356, 563–570. [Google Scholar] [CrossRef]
- Serra, I.; Ubiali, D.; Piškur, J.; Munch-Petersen, B.; Bavaro, T.; Terreni, M. Immobilization of deoxyadenosine kinase from Dictyostelium discoideum (DddAK) and its application in the 5′-phosphorylation of arabinosyladenine and arabinosyl-2-fluoroadenine. ChemistrySelect 2017, 2, 5403–5408. [Google Scholar] [CrossRef]
- Fernández-Lucas, J.; Camarasa Rius, M.J. Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives, 1st ed.; Wiley—VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 1–325. [Google Scholar]
- Slot Christiansen, L.; Munch-Petersen, B.; Knecht, W. Non-viral deoxyribonucleoside kinases—diversity and practical use. J. Genet. Genomics 2015, 42, 235–248. [Google Scholar] [CrossRef]
- Zhou, X.; Szeker, K.; Jiao, L.-Y.; Oestreich, M.; Mikhailopulo, I.A.; Neubauer, P. Synthesis of 2,6-dihalogenated purine nucleosides by thermostable nucleoside phosphorylases. Adv. Synth. Catal. 2015, 357, 1237–1244. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, W.; Zhang, C.; Yang, Z.; Neubauer, P.; Mikhailopulo, I.A.; Huang, Z. Biocatalytic synthesis of seleno-, thio- and chloro-nucleobase modified nucleosides by thermostable nucleoside phosphorylases. Catal. Commun. 2019, 121, 32–37. [Google Scholar] [CrossRef]
- Hellendahl, K.F.; Kamel, S.; Wetterwald, A.; Neubauer, P.; Wagner, A. Human deoxycytidine kinase is a valuable biocatalyst for the synthesis of nucleotide analogues. Catalysts 2019, 9, 997. [Google Scholar] [CrossRef]
- Serra, I.; Daly, S.; Alcantara, A.R.; Bianchi, D.; Terreni, M.; Ubiali, D. Redesigning the synthesis of vidarabine via a multienzymatic reaction catalyzed by immobilized nucleoside phosphorylases. RSC Adv. 2015, 5, 23569–23577. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial biocatalytic linear cascades for preparation of organic molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef]
- Sperl, J.M.; Sieber, V. Multienzyme cascade reactions-status and recent advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Claaßen, C.; Gerlach, T.; Rother, D. Stimulus-responsive regulation of enzyme activity for one-step and multi—step syntheses. Adv. Synth. Catal. 2019, 361, 2387–2401. [Google Scholar] [CrossRef]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing biocatalytic cascades: In vitro and in vivo approaches to de novo multi-enzyme pathways. ACS Catal. 2017, 7, 710–724. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, B.; Tan, J.; Zhu, L.; Li, L. Immobilized multienzymatic systems for catalysis of cascade reactions. Process Biochem. 2016, 51, 1193–1203. [Google Scholar] [CrossRef]
- Bonomi, P.; Bavaro, T.; Serra, I.; Tagliani, A.; Terreni, M.; Ubiali, D. Modulation of the microenvironment surrounding the active site of penicillin G acylase immobilized on acrylic carriers improves the enzymatic synthesis of cephalosporins. Molecules 2013, 18, 14349–14365. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Lozano, S.; Benítez-Mateos, A.I.; López-Gallego, F. Co-immobilized phosphorylated cofactors and enzymes as self-sufficient heterogeneous biocatalysts for chemical processes. Angew. Chem. Int. Ed. 2017, 56, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Mateos, A.I.; Contente, M.L.; Velasco-Lozano, S.; Paradisi, F.; López-Gallego, F. Self-sufficient flow-biocatalysis by coimmobilization of pyridoxal 5′-phosphate and ω-transaminases onto porous carriers. ACS Sustain. Chem. Eng. 2018, 6, 13151–13159. [Google Scholar] [CrossRef]
- Tacias-Pascacio, V.G.; Ortiz, C.; Rueda, N.; Acosta, N.; Aranaz, I. Dextran aldehyde in biocatalysis: More than a mere immobilization system. Catalysts 2019, 9, 622. [Google Scholar] [CrossRef]
- Ren, S.; Li, C.; Jiao, X.; Jia, S.; Jiang, Y.; Bilal, M.; Cui, J. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade catalysis in membranes with enzyme immobilization for multi-enzymatic conversion of CO2 to methanol. New Biotechnol. 2015, 32, 319–3279. [Google Scholar] [CrossRef]
- Garny, S.; Beeton-Kempen, N.; Gerber, I.; Verschoor, J.; Jordaan, J. The co-immobilization of P450-type nitric oxide reductase and glucose dehydrogenase for the continuous reduction of nitric oxide via cofactor recycling. Enzyme Microb. Technol. 2016, 85, 71–81. [Google Scholar] [CrossRef]
- Li, H.; Xiao, W.; Xie, P.; Zheng, L. Co-immobilization of enoate reductase with a cofactor-recycling partner enzyme. Enzyme Microb. Technol. 2018, 109, 66–73. [Google Scholar] [CrossRef]
- Solé, J.; Caminal, G.; Schürmann, M.; Álvaro, G.; Guillén, M. Co-immobilization of P450 BM3 and glucose dehydrogenase on different supports for application as a self-sufficient oxidative biocatalyst. J. Chem. Technol. Biotechnol. 2018, 94, 244–255. [Google Scholar] [CrossRef]
- Petrovicová, T.; Markošová, K.; Hegyi, Z.; Smonou, I.; Rosenberg, M.; Rebroš, M. Co-immobilization of ketoreductase and glucose dehydrogenase. Catalysts 2018, 8, 168. [Google Scholar] [CrossRef]
- Faber, K. Biotransformations in Organic Chemistry: A Textbook, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–423. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sandrini, M.P.B.; Söderbom, F.; Mikkelsen, N.E.; Piškur, J. Dictyostelium discoideum salvages purine deoxyribonucleosides by highly specific bacterial-like deoxyribonucleoside kinases. J. Mol. Biol. 2007, 369, 653–664. [Google Scholar] [CrossRef]
- Serra, I.; Serra, C.D.; Rocchietti, S.; Ubiali, D.; Terreni, M. Stabilization of thymidine phosphorylase from Escherichia coli by immobilization and post immobilization techniques. Enzyme Microb. Technol. 2011, 49, 52–58. [Google Scholar] [CrossRef]
- Guisán, J.M. Aldehyde-agarose gels as activated supports for immobilization-stabilization of enzymes. Enzyme Microb. Technol. 1988, 10, 375–382. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzyme Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Immobilization Carrier | Conversion |
|---|---|---|
| a | Glyoxyl-agarose | 97% |
| b | Sepabeads®-PEI-CL | 56% |
| c | Glyoxyl-agarose (CpUP & AhPNP) Sepabeads®-PEI-CL (DddAK) | >99% |
| d | Sepabeads®-PEI-CL (CpUP & AhPNP) Glyoxyl-agarose (DddAK) | 39% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts 2020, 10, 60. https://doi.org/10.3390/catal10010060
Robescu MS, Serra I, Terreni M, Ubiali D, Bavaro T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts. 2020; 10(1):60. https://doi.org/10.3390/catal10010060
Chicago/Turabian StyleRobescu, Marina Simona, Immacolata Serra, Marco Terreni, Daniela Ubiali, and Teodora Bavaro. 2020. "A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate" Catalysts 10, no. 1: 60. https://doi.org/10.3390/catal10010060
APA StyleRobescu, M. S., Serra, I., Terreni, M., Ubiali, D., & Bavaro, T. (2020). A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts, 10(1), 60. https://doi.org/10.3390/catal10010060