Gold Functionalized Supported Ionic Liquids Catalyst for CO Oxidation





Abstract
: The present study tries to give an insight to the combination of the homogeneous and heterogeneous catalytic properties in a new class of materials. Well dispersed gold nanoparticles on an ionic liquid layer supported on a mineral carrier have been prepared. This work is concentrated on the characterizations and understanding of the interactions between all the components of the catalytic system. The application of the materials in the reaction of oxidation of carbon monoxide shows rather unexpected results—a good catalytic activity completely independent of the temperature.1. Introduction
Ionic liquids (IL) have attracted a significant scientific interest in the last decade due to their unique properties: relatively wide electrochemically stable window; good electric conductivity; high ionic mobility; broad range of room temperature liquid compositions; negligible vapor pressure; and excellent chemical and thermal stability [1,2]. Many of these materials have been applied in different fields ranging from material chemistry to electrochemistry [3,4] and organic synthesis [5] to separation approaches [6–8], as well as in catalytic applications [2,5,9] and nanostructured material synthesis [10].
One of the key challenges in the application research of the ionic liquids has been the availability and the cost issues. Therefore, the concept of immobilized ionic liquids supported on the surface of various materials offers an attractive opportunity to study the performance of ionic liquids cost effectively [1], and brings many advantages, such as easier separation of the catalyst from the reaction media and a possible re-utilization of the catalyst [9]. In addition, the high relative viscosity of many room temperature ionic liquids, promises the formation of a thin layer on the surface, facilitating the diffusion and mass transfer in gas phase reactions [1].
Moreover, ionic liquids can also be used for the formation and stabilization of metal nanoparticles [11,12]. Ionic liquids are known to possess an intrinsic high charge and polarizability allowing them to create electrostatic and steric stabilization for metal nanoparticles [8]. For instance, platinum, rhodium and iridium nanoparticles are prepared in 1-butyl-3-methyl-imidazolium ILs with control of size, near-monodispersity, shape and stabilization, and applied in catalysis [13].
The astonishing discovery of Haruta et al. [14] that very small gold nanoparticles exhibit catalytic activity for the oxidation of CO at sub-ambient temperature provokes an enormous scientific interest and makes gold the most popular metal for this catalytic application. However, the use of gold colloids for producing the active supported catalyst is not as popular as the deposition precipitation method. The advantage of using the colloidal route for preparing supported gold catalysts lies in the great number of possibilities to change the conditions of preparation in order to obtain metal particles with a narrow size distribution, and/or different particle shape [15,16].
There is a general agreement on the fact that the gold nanoparticles are kinetically unstable and have higher tendencies for agglomeration even in the presence of different stabilizing agents [17–19]. When the standard 1-butyl-3-methylimidazolium ionic liquids are used, it seems to be insufficient to prevent the agglomeration of the gold nanoparticles [20]. However, by the changes in the ionic liquid structure, addition of the ether functionalities, the presence of the ionic liquid protecting layer has been observed around the gold nanoparticles [21].
The aim of this work is to study the effect of the presence of ionic liquid on the gold based systems in the reaction of CO oxidation. The expected formation of a thin supported layer on a mineral carrier will facilitate the diffusion and mass transfer in the heterogeneous catalytic reaction and will also guarantee better dispersion and stabilization of freshly prepared gold colloids.
2. Results and Discussion
2.1. Preparation
2.1.1. Ionic Liquid Preparation
The halide free ionic liquid 1-butyl-3-methylimidazolium methanesulfonate has been prepared by the method proposed by Cassol et al. [22]. The used reactants were methanesulfonyl chloride (Alfa Aesar), triethylamine (Alfa Aesar), 1-methylimidazole (Aldrich), 1-butyl-3-methylimidazolium methanesulfonate (Fluka), n-butanol, dichloromethane, acetone and distilled water. The method goes first through the synthesis of butyl methanesulfonate, as described below.
2.1.2. Preparation of Butyl Methanesulfonate
Methanesulfonyl chloride (61.1 g, 0.53 mol) was added slowly, under vigorous stirring, to a solution of n-butanol (39.4 g, 0.53 mol) and triethylamine (53.9 g, 0.53 mol) in dichloromethane (0.5 L). An external water-ice bath was used to control the reaction mixture temperature between 10–20 °C. After addition, stirring was continued for further 2 h at room temperature. Water (200 mL) was added; the aqueous layer containing the triethylammonium chloride by-product was separated; the organic layer was washed with water (200 mL) and dried with sodium carbonate. Solvent evaporation followed by reduced pressure distillation of the residue afforded the desired butyl methanesulfonate, as a colorless liquid; yield: 55.14 g (73%).
2.1.3. Preparation of 1-Butyl-3-methylimidazolium Methanesulfonate
Butyl methanesulfonate (55.14 g, 0.36 mol) was mixed with 1-methylimidazole (29.74 g, 0.36 mol) and the reaction mixture was kept at room temperature by means of an external water bath. After 24 h, one crystal of 1-butyl-3-methyl imidazolium methanesulfonate was added and the resulting crystalline reaction mass was kept at room temperature for a further 120 h. Recrystallization was performed twice using acetone as a solvent (120 mL; from reflux temperature to freezer temperature overnight). After vacuum drying, colorless and very hygroscopic crystals of 1-butyl-3-methylimidazolium methanesulfonate were obtained; yield: 77.4 g (95%).
1H-NMR was performed of the synthesized ionic liquid by dissolving it in CDCl3 and compared to the 1H-NMR spectra of the commercially available 1-butyl-3-methylimidazolium methanesulfonate (Fluka, Louis, MO, USA). The obtained spectra were identical (not shown).
2.1.4. Supports
A commercial gamma aluminium oxide (Spheralite 505) was used after milling and sieving with the fraction 125–250 μm retained.
In order to facilitate the ionic liquid impregnation on the support surface, the number of the anchorage sites should be increased. For this purpose the phosphoric acid treatment was chosen for modification.
Five grams of γ-Al2O3 were dispersed in 100 mL of 1 mol L−1 orthophosphoric acid solution at 75 °C during 4 h [23]. Then, the solution was filtered and the resulting solid was dried at 100 °C for 3 h and subsequently calcined at 400 °C for 4 h. The resulting solid will be named γ-Al2O3-MOD.
2.1.5. Ionic Liquid Impregnation
Impregnation of 20 wt% ionic liquid onto support surface was performed in ethanol solution in an ultrasonic bath for 10 min. followed by ethanol evaporation under reduced pressure at room temperature.
2.1.6. Gold Deposition
In all preparations: HAuCl4 (Alfa Aesar, 45.92% Au) was used as gold precursor.
The gold colloids were produced by two different methods: (i) the standard Turkevich method [24]; and (ii) reduction of the gold precursor by sodium borohydride [25].
(i) In the Turkevich method, an aqueous 2 × 10−4 M gold solution was heated to boiling and then, the support was added followed by 1 mL 34 mM solution of sodium citrate. The mixture was vigorously stirred for 5 min and 1 mL of 34 mM oxalic acid solution was added to assure the complete formation of gold colloids. Then the suspension was filtered and dried at 80 °C overnight. The sample will be named as Au/support (citrate).
As the application of this method is possible only in hot aqueous solutions in order to prevent the risk of the ionic liquid removal during the preparation, a room temperature method of synthesis has been applied. (ii) In this method, a gold aqueous solution with 2 × 10−4 M concentration was mixed with the support and, 2 min later, 0.01 g of sodium borohydride was added. In these conditions, the gold reduction occurs immediately and the solution was kept at room temperature one night. The solution was then filtered and dried at 80 °C overnight. The sample will be named as Au/support (NaBH4).
As the room temperature reduction by sodium borohydride permits the utilization of other solvents, an additional preparation has been carried out by modifying the solvent, water being replaced by acetone. This allows us to diminish the immediate reduction by the addition of NaBH4 and to keep the ionic liquid on the support, as it is not soluble in this solvent. The sample will be named as Au/support (NaBH4-acetone).
The gold loading was fixed to 0.8% for all obtained samples.
2.2. Results and Discussion
The specific surface area of the supports before and after modification and its point of zero charge are presented in Table 1.
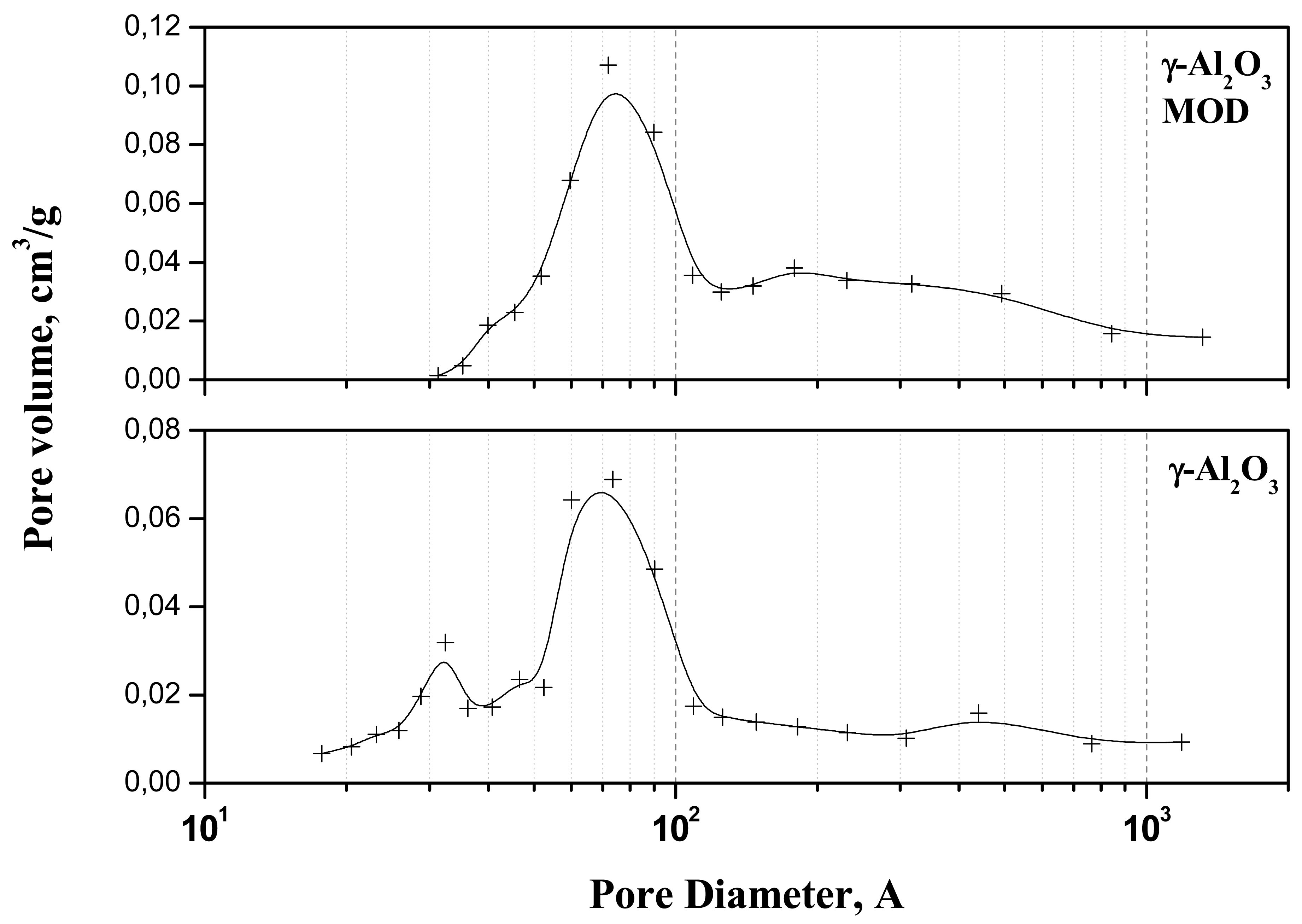
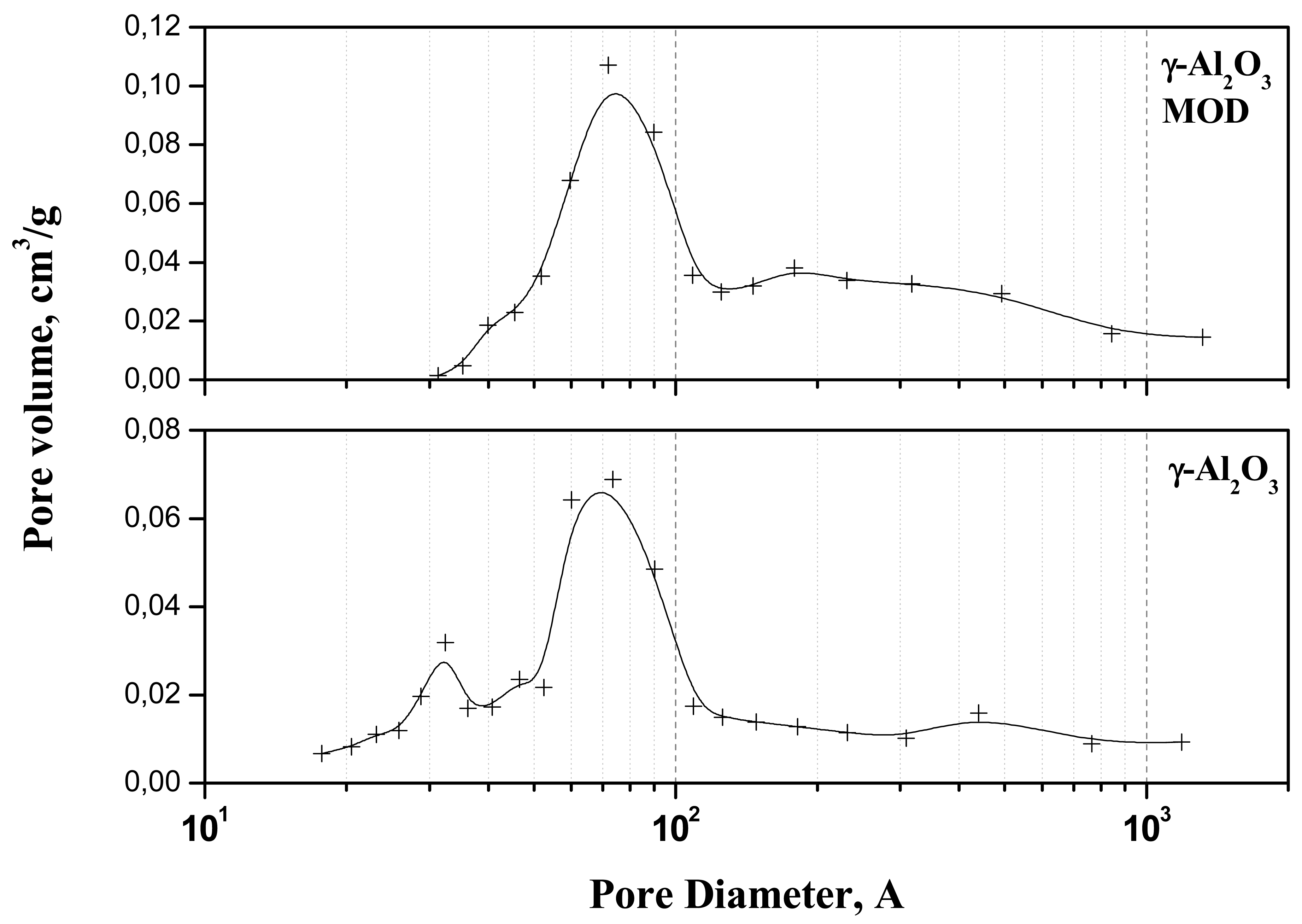
Figure 1 presents the pore volume distribution as a function of pore diameter for the bare and modified support. For the bare support, a bimodal distribution was observed with an average pore size of 67 Å. The modification of the surface by phosphoric acid provokes a general increasing of the average size of the pores to 110 Å which results in higher pore volume but in lower surface caused by the blocking of some mesopores.
The strength of the interaction of a Lewis base (PO43−) with the hard Lewis acid sites characteristic for alumina depends on the electron density and the polarizability of the base [26]. Lower polarizability and higher electron density lead to stronger interaction. The strength of interactions of the bases is as follows:
The biggest effect on the surface and, which is more important, the biggest possibility to enhance interaction modifier/alumina, should be observed when the phosphoric acid is applied and this was confirmed by the observation of significant decrease in the point of zero charge (Table 1).
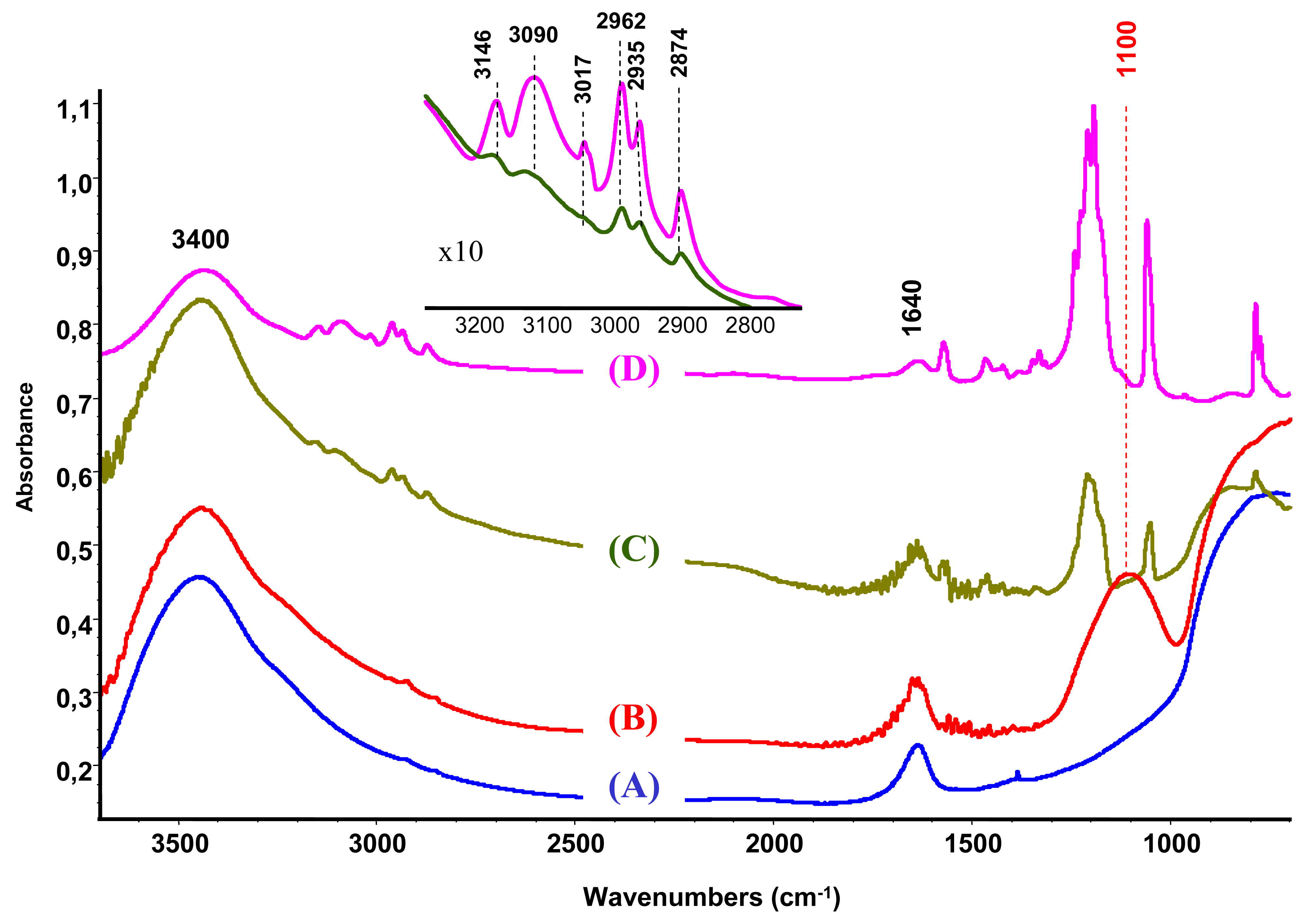
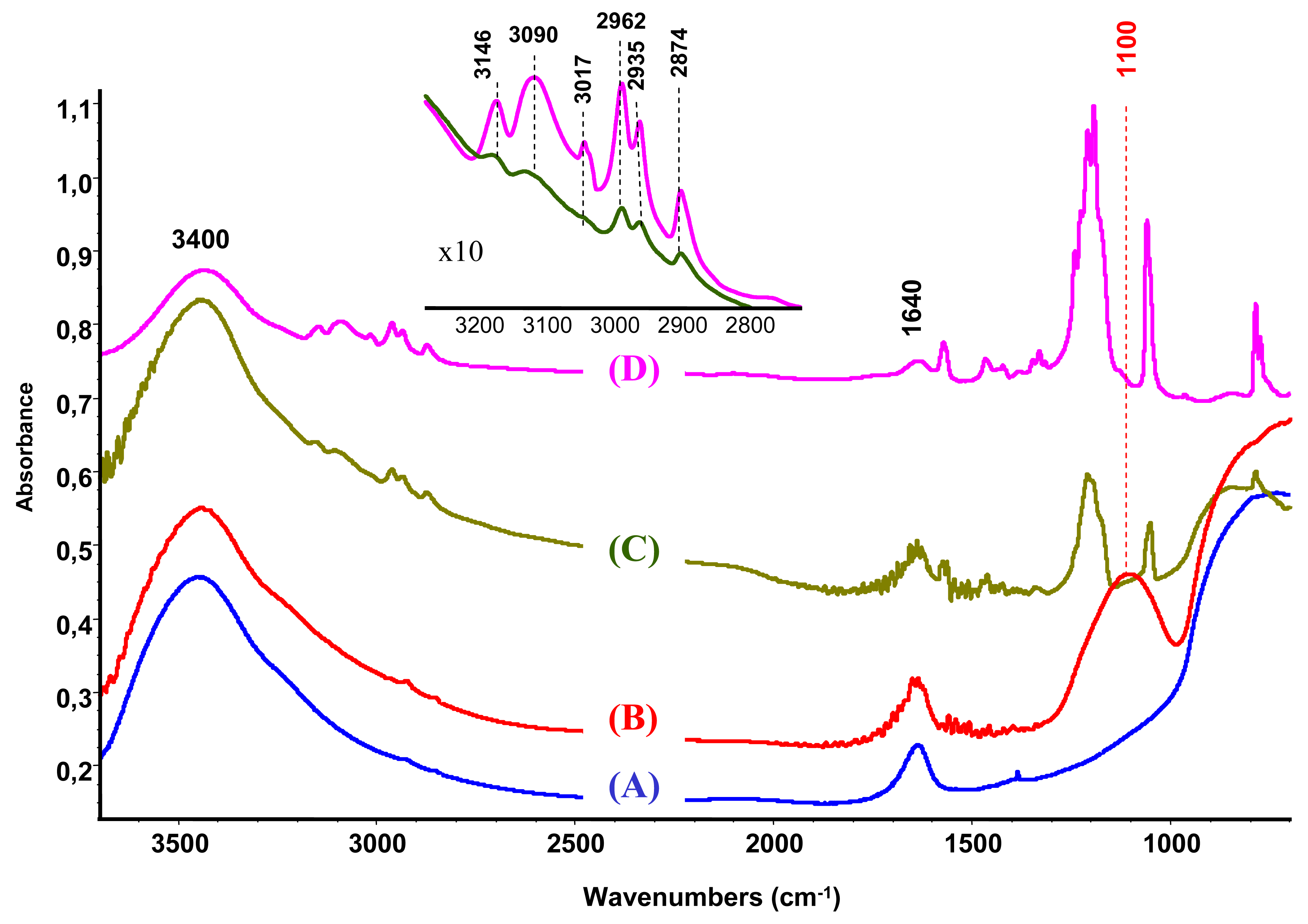
The characterization by IR spectroscopy of the modified support compared to bare unmodified alumina (Figure 2(A,B)) reveals a new band at 1100 cm−1 attributed to the P–O groups [27,28].
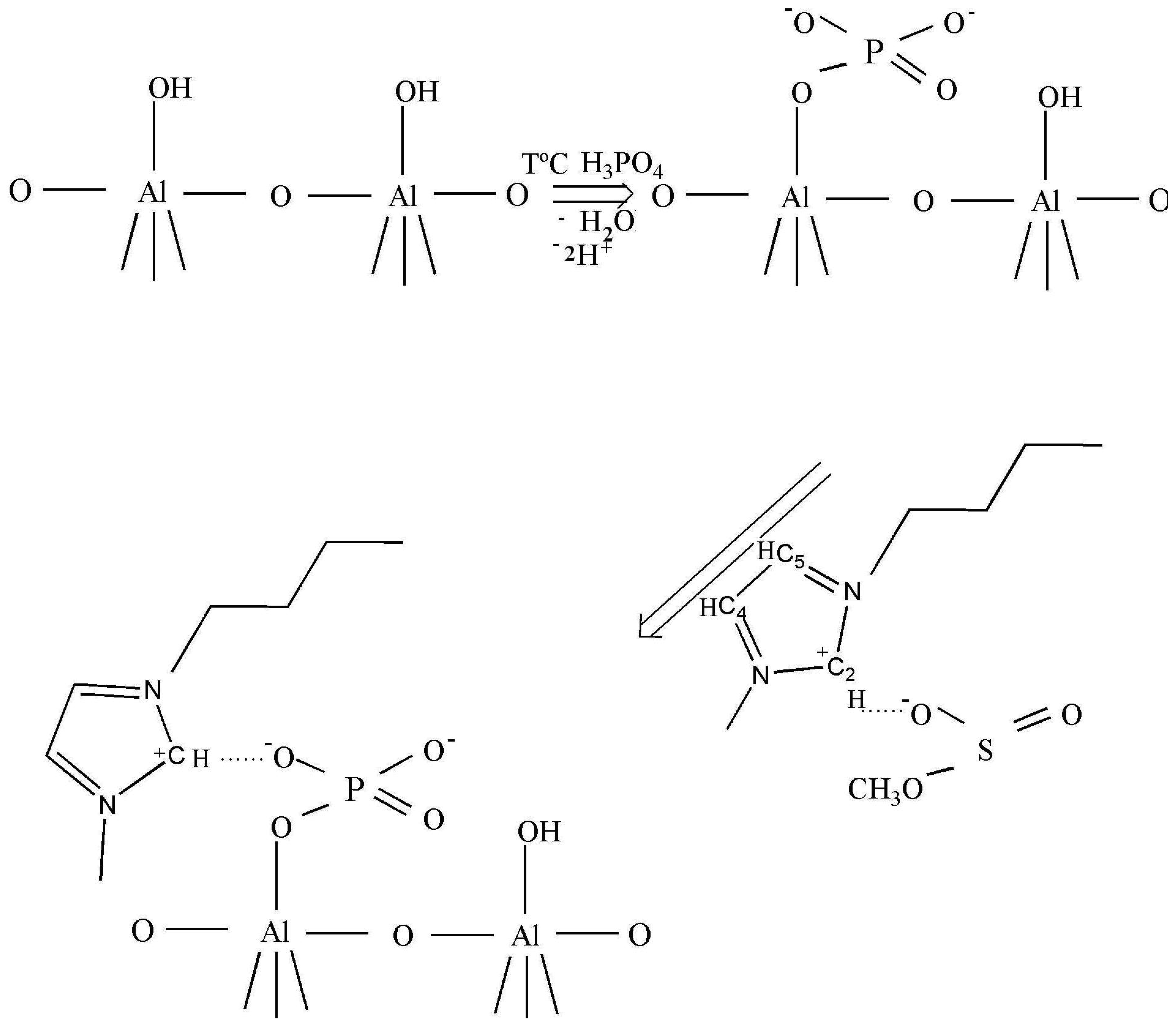
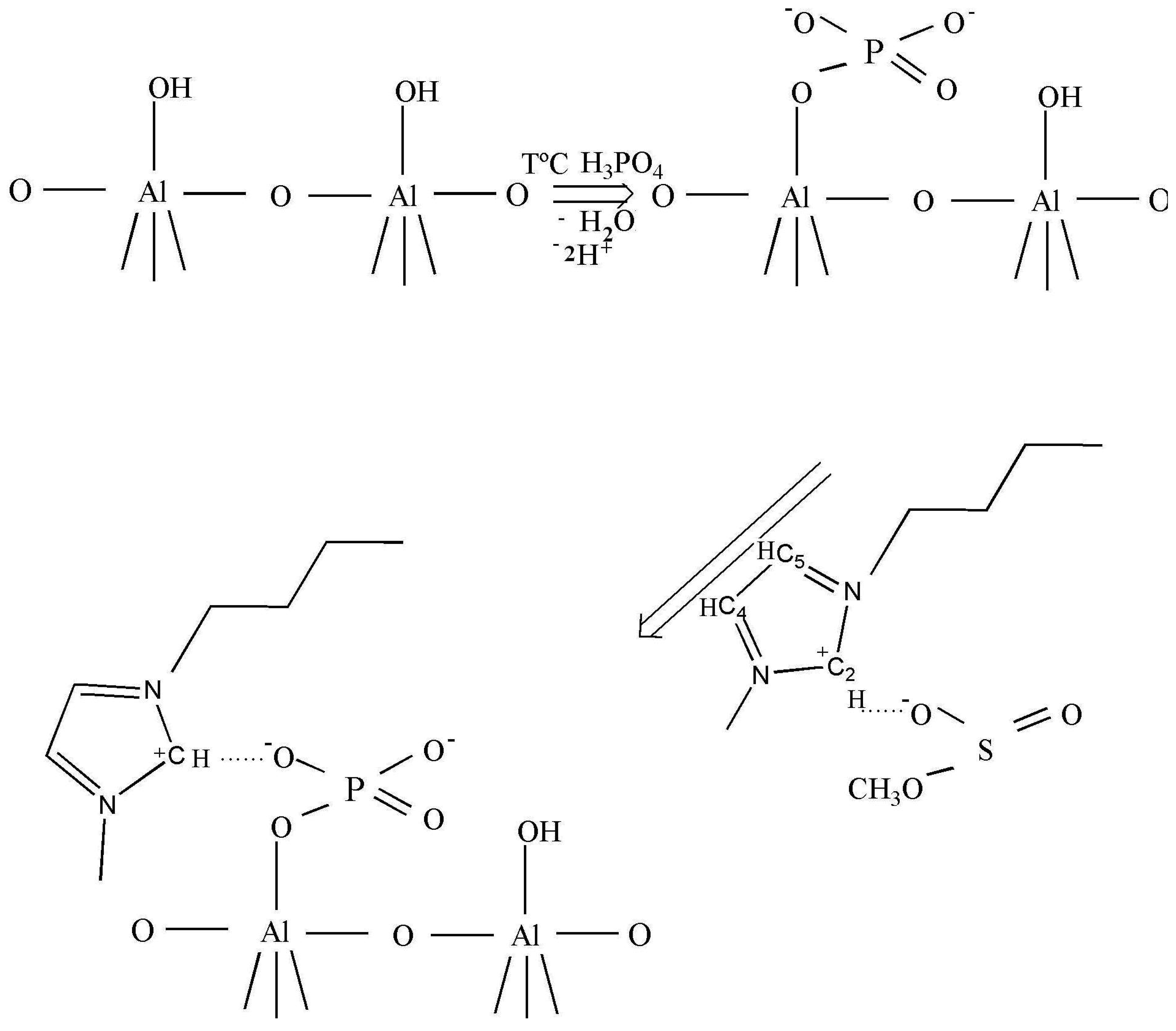
As claimed in the literature [29], the anions can be adsorbed in an oxide–water interface in an inner- or outer-sphere mode. Inner-sphere surface complexes are created as a result of direct chemical bond formation between adsorbing anion (Lewis base) and the metal ion at the surface (Lewis acid). In the case of anions with more than one donor atom (e.g., phosphate), several coordination geometries are possible: monodentate, chelating bidentate, or bridging bidentate. Other -sphere surface complexes are formed as a result of electrostatic attraction of opposite charges. Busca et al. [30] reported a selective interaction of the phosphoric acid on the basic sites which excludes the formation of H-bonded phosphate species, preventing the formation of the two types of bidentates. The formation of only ionically bonded species can be envisaged (Scheme 1, row 1).
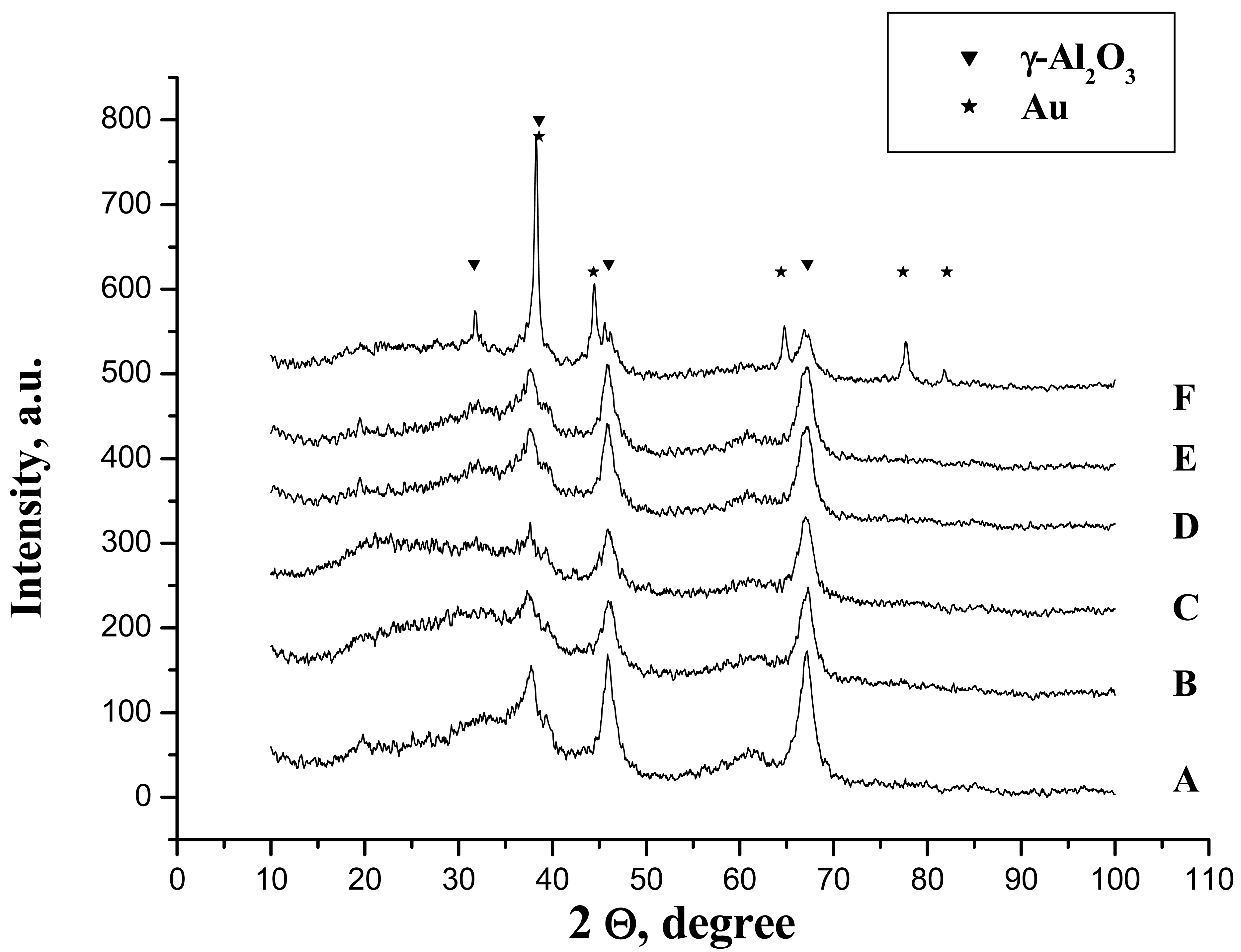
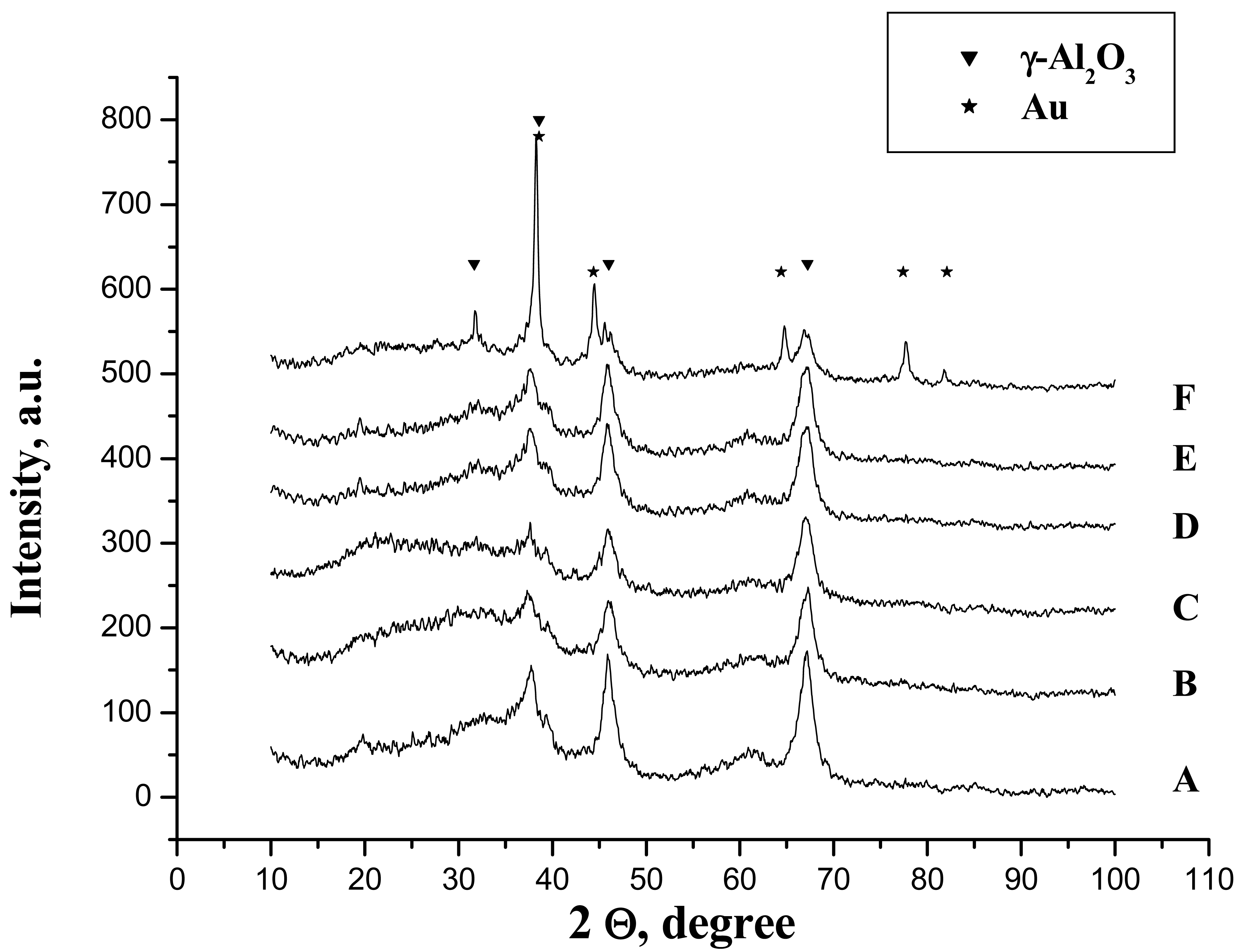
One can suppose that in this case, the biggest part of the phosphates is chemically bonded but the formation of the hydrogen bonded phosphates cannot be excluded. However, the phosphoric acid treatment does not provoke any chemical changes and the only detectable phase by XRD was γ-Al2O3 and no formation of aluminium phosphate was observed (Figure 3(A,B)).
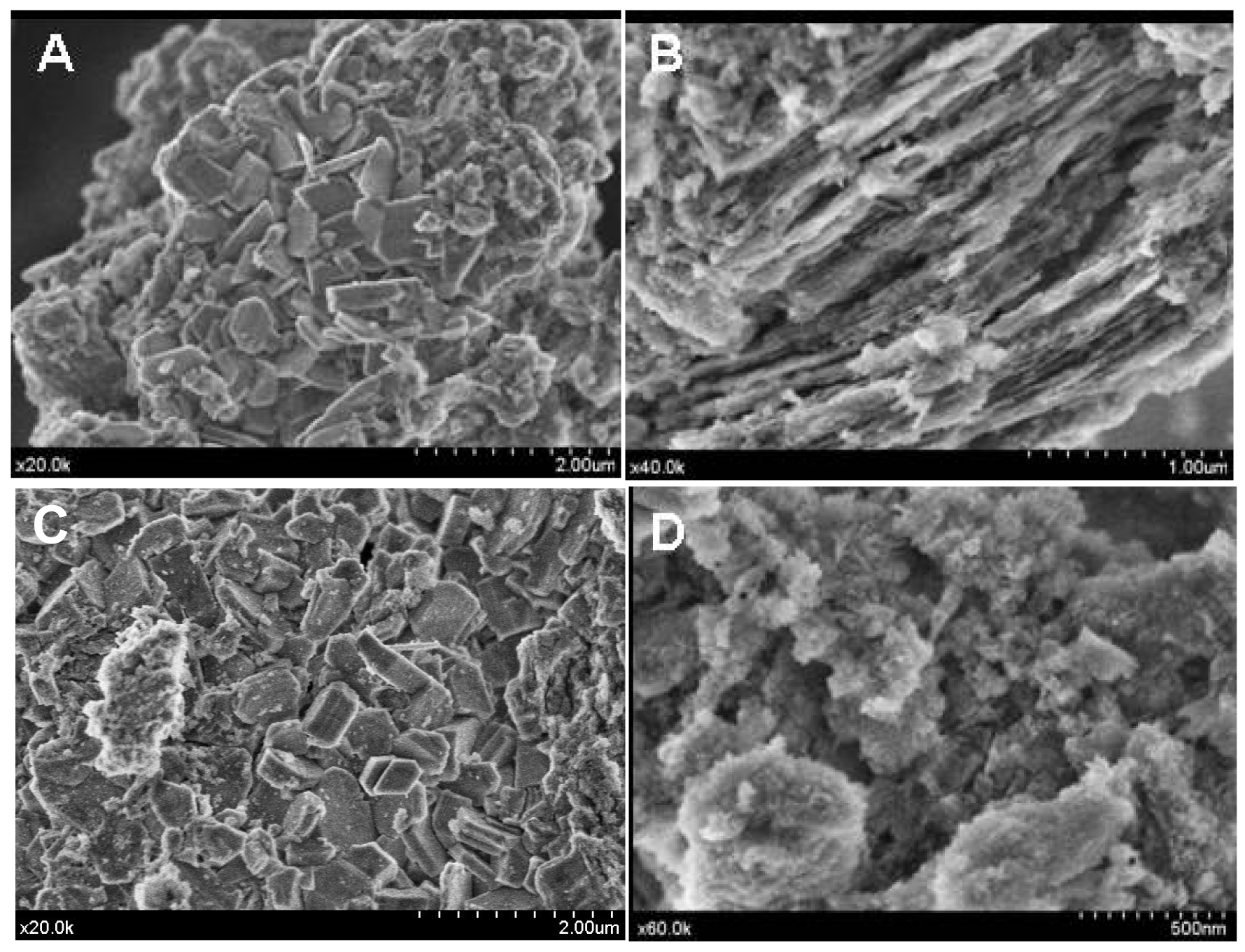
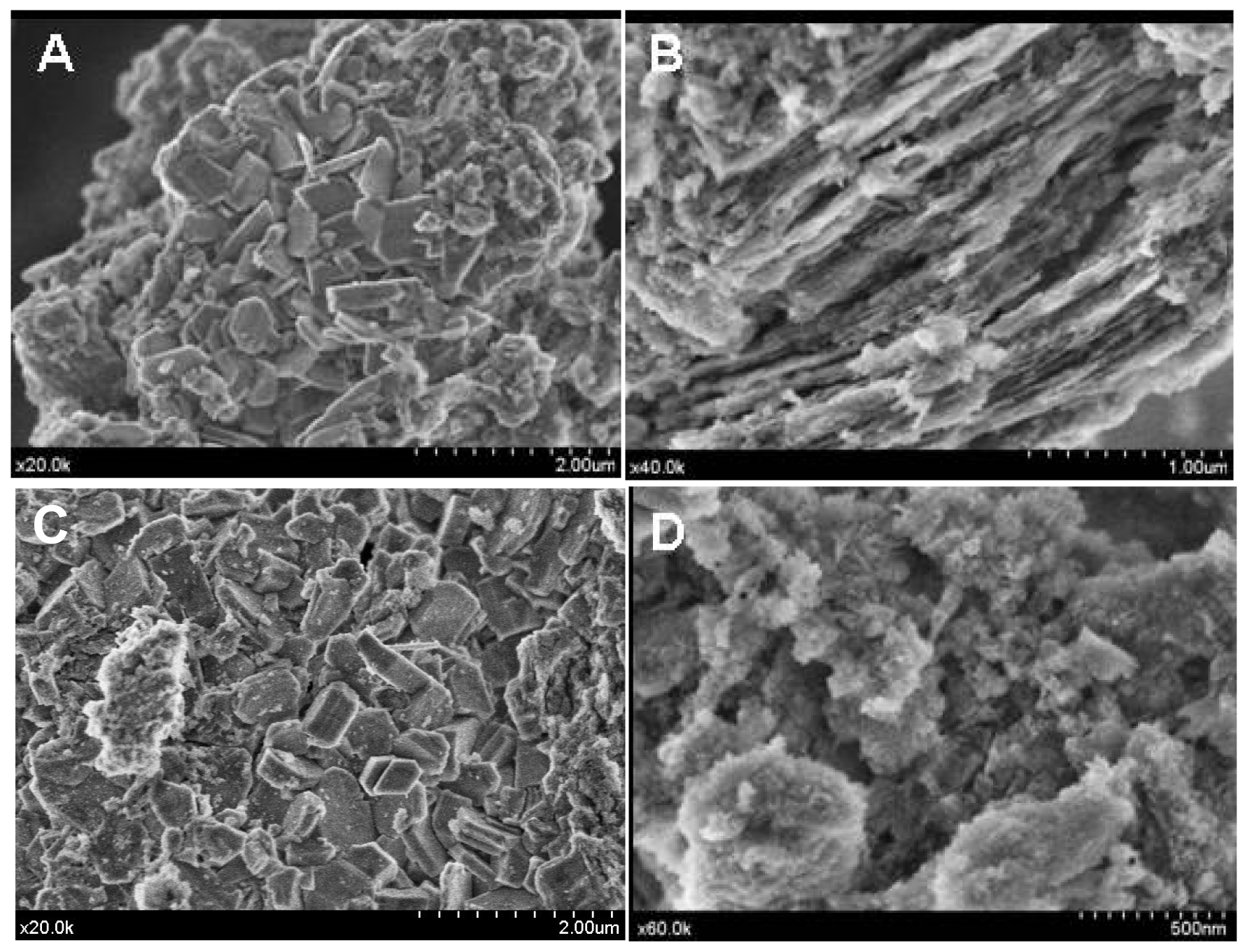
However, the acid treatment of the support provokes changes in its morphology as could be seen from the SEM images (Figure 4(A,B)).
The bare alumina presents a heterogeneous morphology, composed of a mixture of grains and plates, which became rather laminar and ordered after acidic treatment but seemed to lose a part of its crystallinity.
2.3. Ionic Liquid Deposition
The deposition of the ionic liquids depends on the specific surface of the support [9,31]. A high specific surface area provides a large number of adsorption sites and will increase the quantity of the adsorbed ionic liquid. De Castro et al. [9] have found a 20 weight percent maximal value for the quantity of deposited ionic liquids adsorbed on the gamma alumina surface. However, in this study the modification of alumina provides additional adsorption sites resulted in 50 wt% maximal ionic liquid loading. Nevertheless, this sample, was not be used since the possibility of the formation of multiple layers of ionic liquid on the support are higher with the increase of the deposited amount. The lower the quantity of ionic liquid adsorbed, the higher the possibility of a good anchoring and subsequent fine dispersion of the gold nanoparticles. That is why a sample with 20 wt% deposited ionic liquid was chosen for the study.
The specific surface area changes dramatically when the ionic liquid is supported, a value of one tenth being obtained (Table 1). However, the outgas treatment provokes ionic liquid melting and the pores filling, which results in a lower surface area and a quasi inexistent pore volume.
As the ionic liquid did not possess a crystalline structure, no structural changes have been observed (Figure 3). However, heterogeneity of the surface was observed. In some places, the morphology seems to change and to recover the initial morphology of the non-treated alumina (Figure 4(C)) while, in other parts, it keeps the morphology of the modified alumina (Figure 4(D)). This interesting observation led us to suggest that the part of the support surface is liberated from the weakly bonded phosphates through the H-bond and which are now bonded to the ionic liquids as a counter ion.
2.4. Gold Addition
The gold amount was fixed to 0.8 wt% and verified by X-ray fluorescence. The specific surface area and its pore volume of the gold prepared samples are listed in Table 1. When citrate was used for the gold reduction, the sample recovers the specific surface area of the initial alumina indicating the complete liberation of the surface by ionic liquid loss during the deposition procedure.
The use of NaBH4 as a reducing agent at room temperature in gold solutions, no matter the solvent, increases the surface and pore volume compared to those of the supported ionic liquid system demonstrating the presence of the ionic liquid in the samples. As expected, the lower surface area of the sample prepared in acetone solution indicates the higher amount of the ionic liquid present in the sample.
In aqueous media, no matter the reducing agent, the diffractions of metallic gold have not been observed (Figure 3(D,E)). This could be provoked either by the lower particle size or by the low quantity of the gold present in the sample (less than 1%).
Nevertheless, the sample prepared in acetone solution clearly presents the diffraction lines attributed to metallic gold suggesting far bigger particle size. In addition, the diffraction lines of the gamma alumina become more stretched and intensive and a new diffraction peak appears at 2Θ = 32° (Figure 3(F)). This line has been reported by De Yu et al. [32] for calculated gamma alumina XRD pattern, although, to our knowledge, no experimental XRD pattern has yet been reported. The utilization of acetone in combination with a strong reducing agent seems to influence the crystallization of gamma alumina.
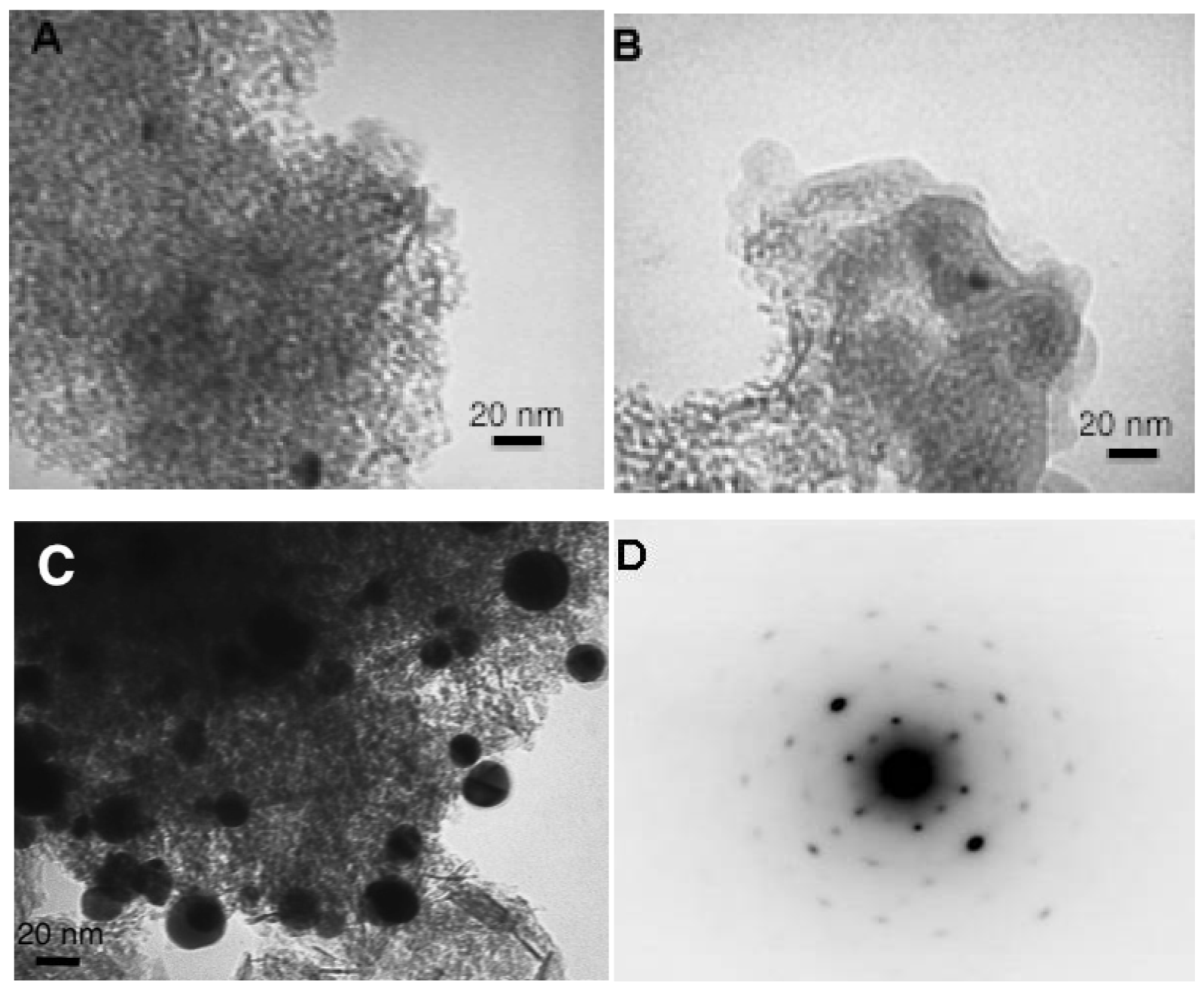
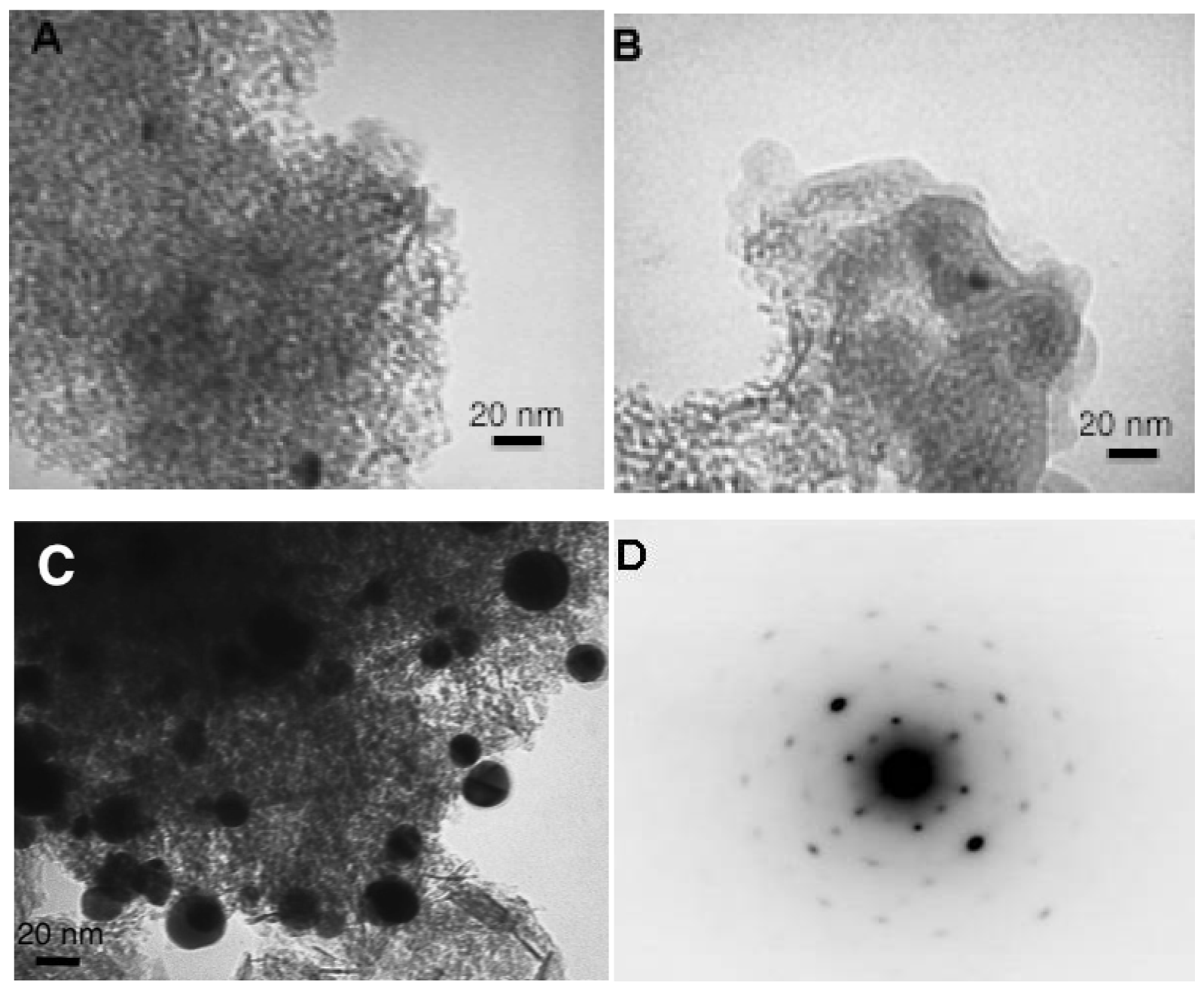
The gold particles size has been studied by Transmission Electron Microscopy (Figure 5).
In aqueous solutions the change of the reducing agent from citrate to NaBH4 did not seem to influence the gold particle size. However, it should be noted that the restrictions of the microscope did not permit observation of particles smaller than 3 nm. The use of water as a solvent resulted in lower gold particle sizes at around 6.5 nm for both samples. The change to acetone provokes an increase of the particle size to 9.5 nm in average but the agglomerates with average size higher than 40 nm have been observed and excluded from the particle size distribution. For the samples prepared in the gold solutions at room temperature (Figure 5(B,C)), the presence of a fine organic layer can also be observed, which confirms the presence of IL.
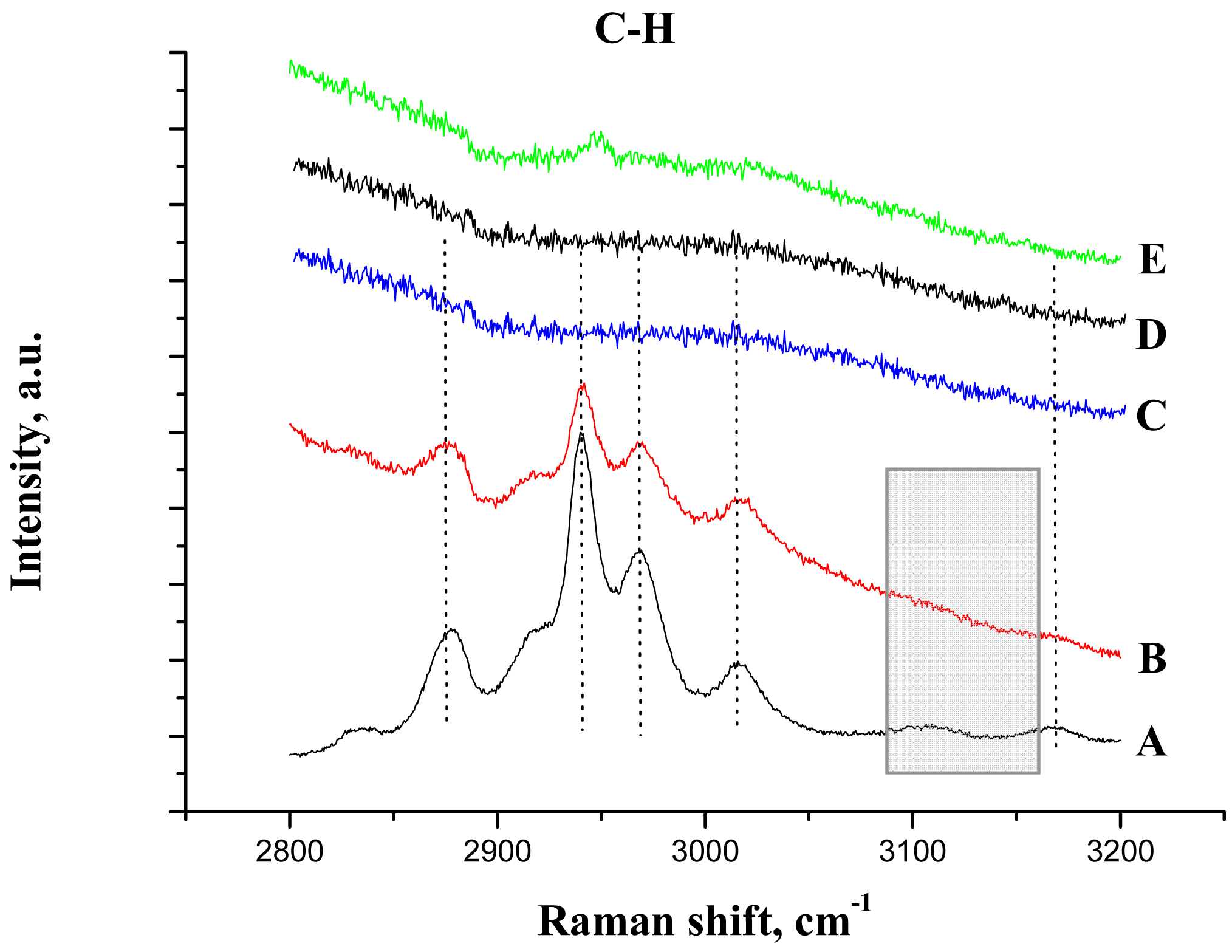
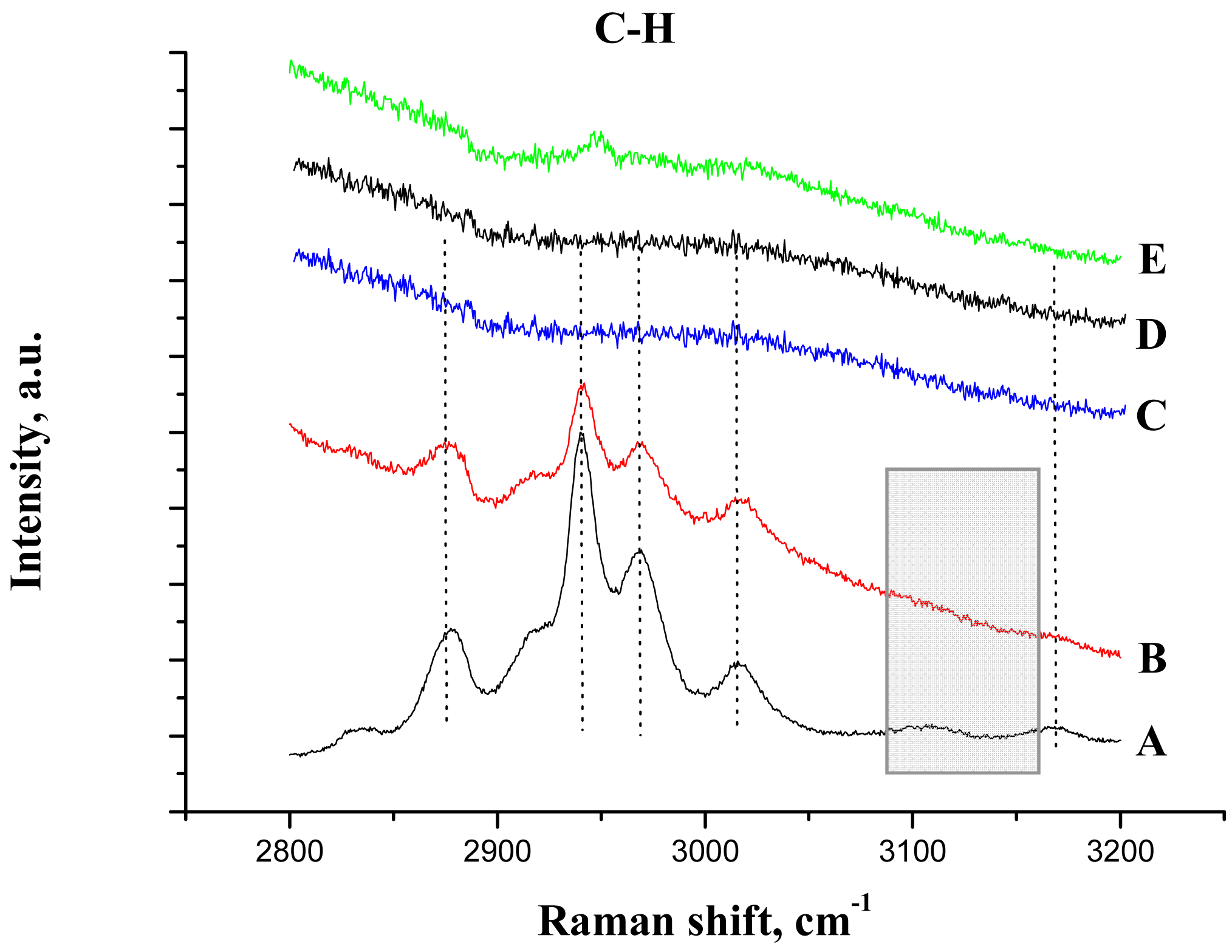
The IR study of the supported ionic liquid compared to the IR spectra of the synthesized unsupported ionic liquid is presented in Figure 2(C,D). For the IL only, the bands at 3146, 3090 and 3017 cm−1 are attributed to the ν(CH) vibrations of C4, C5 and C2 (as shown in Scheme 1, on the right) of the imidazolium ring and those at 2962, 2935 and 2874 cm−1 corresponds to ν(CH) of butyl and methyl groups bonded to the ring [33,34]. On the base of theoretical calculations of IR spectra of an isolated gas phase methyl-ethyl imidazolium ion, the ν(C-H) stretching of the C2 atom appears at 3118 cm−1 [34]. Moreover, it was evidenced that the frequency of this band strongly depends on the counter ion nature, being the charge to volume ratio, the most important factor influencing the shift of this band. For instance, a shift from 3118 cm−1 to 3049 cm−1 was detected when chloride acts as a counter ion, while this band appears at 3063 cm−1 if chloride is replaced by bromide. In our case, the counter ion is methanesulfonate, with oxygen atoms able to interact with the imidazolium. Taking into account this information and the higher charge to volume ratio, the produced shift of this band from the theoretical value (3118 cm−1) to 3017 cm−1 is not surprising.
After the ionic liquid deposition onto the support, the maximum of the bands positioned at 3146 and 3090 cm−1 ν(C4H C5H) suffer a slight shift to a higher wave number (being practically constant in relative intensity). In addition, the band at 3017 cm−1 disappears, while the bands due to the methyl and butyl groups' vibration keep the same position. This fact is also observed in the bending region (not shown because of the low intensity). These observations suggest that the interaction with the phosphate-modified support proceeds via the C2 atom of the imidazolium ring (disappearance of the 3017 cm−1 band), being the frequencies of vibration of the substituting groups the least affected (Scheme 1, second row). Therefore, we can conclude that the immobilization process of the imidazolium ring, initially equilibrated by methanesulfonate, proceed now through interaction with the phosphate groups from the support, which provoke the disappearance of the ν(C2-H) band.
When gold is attached to the support/ionic liquid system from aqueous solutions, no trace of ionic liquid vibrations could be observed and the obtained spectrum is practically identical to the IR spectra of the modified alumina. This result shows that the formed bond between the support and IL is strongly electrostatic (attraction between negatively charged alumina surface and the imidazolium ring). However, in acetone solution the vibrations due to the IL are still present, which indicates the occurrence of this phase either as bonded species or as a physical mixture.
In addition, Raman spectroscopy was used to study the IL/support interaction. Figure 6 shows the C-H bonding region for all the samples and Table 2 the assignments of the bands, as reported previously for butyl methyl imidazolium based ionic liquids [33,35,36].
After the deposition of IL on the alumina support, all Raman lines of the bare IL are preserved, except the vibration band at 3107 cm−1 due to the vibration of CH bond between the two nitrogen atoms in the imidazolium ring (the C2H bond, as shown in Scheme 1). This observation is not completely unexpected and agrees with the IR data, and confirms that the bonding of the IL goes through electrostatic attraction between the positive charge at this place in the imidazolium ring and the negatively charged alumina surface. The bands caused by the vibrations of butyl and methyl radicals bonded to the imidazolium ring, did not change after the deposition on the support, which was expected for the short chain radicals where the possibility of steric attraction or repulsion is close to zero.
After gold deposition in acetone solution (Figure 6(E)), a weak signal was observed indicating the presence of ionic liquid. However, all Raman lines were red shifted signalizing a perturbation of the imidazolium ring by the gold addition. In aqueous solution, no signals were detected either by masking of the signals by the strong fluorescence or by the ionic liquid removal from the support (Figure 6(C,D)).
2.5. CO Oxidation
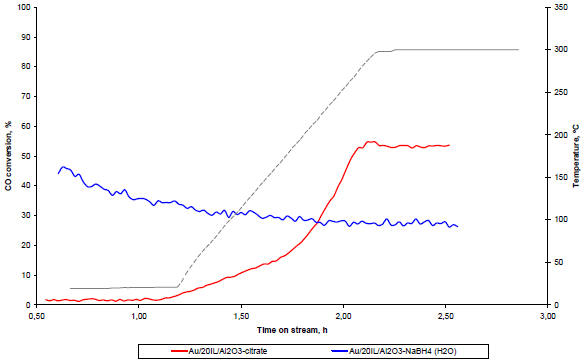
The most interesting results have been obtained when submitting the catalysts on CO oxidation reaction (Figure 7). The sample prepared in acetone solution does not show any activity in the employed range of temperatures (from room temperature to 300 °C). As observed, the gold particle size was rather big, but still not enough to explain this result, so there should be another reason for the complete inactivity of this sample, no matter the temperature. The dark field transmission study on this sample presented in Figure 5(D), shows that the reduced gold particles are defect free resulting in a perfect crystal. The absence of defects, i.e., no active sites for oxygen activation and CO oxidation, could explain the absence of activity for this sample [37,38]. This interesting result also confirms the importance of the presence of water in the preparation of the gold catalyst and gives the opportunity to hypothesize that the gold active sites are formed and stabilized not only by the presence of support but as well by the complicated network of hydrogen bonding. Actually, this result could explain why the small quantities of water are always beneficial for CO oxidation, as reported previously [39,40], not only because of the participation of water in the CO oxidation mechanism, but because of the active sites electronic distribution stabilization.
The samples prepared in aqueous solutions show a good activity in CO oxidation and high dependence on gold reduction conditions. When milder reduction conditions are applied, (Au/20IL/Al2O3-citrate) the catalyst shows a normal behavior, an increase of the activity with the increase of the temperature. No room temperature activity for this sample was observed. However, the catalytic behavior changes dramatically when rapid reduction was applied (NaBH4 instead of citrate). As described above, a stabilization of the gold particles by the ionic liquid could be envisaged. The catalyst was not influenced by temperature and shows an important activity in the whole temperature range.
This big difference in the catalytic comportment of the two samples prepared in aqueous media, very similar in terms of gold particle size, leads to the conclusion that the mode of stabilization of the gold active sites for the two samples does not even match. When the milder conditions of reduction are applied, the stabilization of the gold particles was prioritized on the surface of the support free of ionic liquid, that is why the catalyst presents a “normal” light off curve already observed for gold catalyst supported on alumina [41,42].
For the rapid reduction sample, a different mode of stabilization of the gold particles can be imagined. Gold can be stabilized, at the moment of the colloid nucleation and formation, by the creation of micelle-like aggregations in the low concentrated ionic liquid aqueous solutions; such formation being eminent and easy [43]. In this case, the as produced IL stabilized gold colloids will not interact directly with the support but through the imidazolium ring. Thus the delocalization of the electrons of gold on the imidazolium ring could allow a different mode of activation of oxygen (step limiting stage) not influenced by the temperature, which could explain the unusual catalytic behavior. However, this can be applied only for the so called “inactive” supports [44] for which the interaction gold-support is very weak. However, our recent study on the IL templated titania–gold systems also shows a different catalytic behavior, for which the presence of the ionic liquid was strongly responsible [12].
However, every excess of this IL (1-butyl-3-methylimidazolium methanesulfonate) could be detrimental for the catalytic activity, because of the preferential deposition of the imidazolium ring in the plane [45], thus completely covering the gold active sites. The latter can provide an additional explication for the complete inactivity of the acetone prepared samples in which the quantity of the IL is higher. Nevertheless, this inconvenience can be avoided by the application of IL with another counter ion, like Cl−, BF4−, etc. for which the geometry of deposition is different.
3. Experimental Section
3.1. CO Oxidation Reaction Conditions
The activity measurements, as described elsewhere [37,38] were carried out in a flow U-shape glass reactor at atmospheric pressure. The catalyst (80 mg, φ < 200 μm) was placed into the glass reactor and a reactive flow (3.4% CO and 21% O2 balanced by helium) was passed through it. The total flow was 42 mL min−1 and the quantitative analysis was carried out with a Balzers Omnistar Bentchop mass spectrometer. The catalyst was tested in the flow during 1 h at room temperature. Then, the system was heated to 300 °C and stabilized at this temperature for 30 min.
In the reaction of carbon monoxide oxidation the samples were employed after drying without any additional treatment.
3.2. Characterizations
X-ray diffraction (XRD) analysis was performed on a Siemens diffractometer D500. Diffraction patterns were recorded with Cu Ka radiation (40 mA, 40 kV) over a 2θ-range of 10 to 70° and a position-sensitive detector using a step size of 0.05° and a step time of 1 s.
The point of zero charge (PZC) of the support was determined by measuring (Malven Zetamaster) electrophoretic mobilities of aqueous dispersions as a function of pH, at constant ionic strength (10−2 mol L−1 NaCl). The pH was varied by adding adequate amounts of 0.01 mol L−1 HCl or NaOH.
The textural properties were studied by N2 adsorption measurements at liquid nitrogen temperature. The experiments were carried out on a Micromeritics ASAP 2010 equipment. Before analysis, the samples were degassed for 2 h at 150 °C in vacuum.
Scanning electron micrographs (SEM) were taken using a JEOL JSM-5400 electron microprobe and Link energy dispersive X-ray (EDX) analyser.
Transmission electron microscopy (TEM) observations were carried out in a Philips CM200 microscope operating at 200 kV. The samples were dispersed in ethanol by sonication and dropped on a copper grid coated with a carbon film.
The IR spectra were recorded on a Nicolet 380 Spectrometer at a spectral resolution of 2 cm−1 and accumulation of 100 scans. The pellets were prepared from the dried sample powder and KBr (1/20).
The Raman spectra were recorded on a dispersive Horiba Jobin Yvon LabRam HR800 microscope, with a 300 mW He-Ne red laser (784.56 nm) without filter and with a 600 g mm−1 grating, 15 acquisitions (10 s/scan). The microscope used a 50× objective with a confocal pinhole of 500 μm.
The elemental analysis of gold was performed by X-Ray Fluorescence in a Panalytical AXIOS PW4400 sequential spectrophotometer with Rh tube as source of radiation. The measurements were performed onto pressed pellets containing 6 wt% of wax.
4. Conclusions
Evidence of the ionic liquid attraction through the imidazolium ring to the modified support is provided. However, the deposition goes through a strongly electrostatic bond which shows prioritizing of the gold complexes in aqueous solutions.
Well dispersed gold nanoparticles on homogeneous ionic liquid layer supported on a mineral carrier have been successfully prepared. The size of the deposited gold particles seems to depend strongly on the solvent employed and less so on the reducing agent. When acetone is used as a solvent, the resulting gold nanoparticles show a defect free highly crystalline structure and are completely inactive in the oxidation of CO, which confirms the importance of the presence of active sites with structural defects.
The use of water as a solvent confirms its essential role, especially in the preparation resulting in the production of gold active sites stabilized not only by the presence of support, but also by the presence of the ionic liquid through the IL micelle formation.
A high influence of the reducing agent and ionic liquid presence are observed in the oxidation of CO for the samples prepared in aqueous media, resulting in samples with temperature independent activity, suggesting a different mode of stabilization of the active sites responsible for a different mode of oxygen activation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | Specific Surface Area, m2 g−1 | Pore Volume, cm3 g−1 | PZC, pH |
|---|---|---|---|
| γ-Al2O3 | 251 | 0.47 | 8.0 |
| γ-Al2O3 MOD | 195 | 0.59 | 5.4 |
| 20IL/γ-Al2O3 MOD | 19 | 0.09 | - |
| Au/20IL/γ-Al2O3 MOD (citrate) | 252 | 0.51 | - |
| Au/20IL/γ-Al2O3 MOD (NaBH4) | 131 | 0.45 | - |
| Au/20IL/γ-Al2O3 MOD (NaBH4-acetone) | 84 | 0.29 |
Acknowledgments
L. Bobadilla acknowledges Junta de Andalucía (Project PDG-TEP01965). S. Ivanova and A. Penkova acknowledge MEC for their contracts Ramon y Cajal and Juan de la Cierva.
References
- Mikkola, J.-P.T.; Virtanen, P.P.; Kordas, K.; Karhu, H.; Salmi, T.O. SILCA-supported ionic liquid catalysts for fine chemicals. Appl. Catal. A Gen. 2007, 328, 68–76. [Google Scholar]
- Dupont, J.; de Souza, R.; Suarez, P.A.Z. Ionic liquid (molten salt) phase organometallic catalysis. Chem. Rev. 2002, 102, 3667–3692. [Google Scholar]
- Zhang, J.L.; Zhang, Q.; Li, J. Electrochemical behaviors and spectral studies of ionic liquid (1-butyl-3-methylimidazolium tetrafluoroborate) based sol-gel electrode. J. Electroanal. Chem. 2007, 603, 243–248. [Google Scholar]
- Yoshizawa, M.; Hirao, M.; Ito-Akita, K.; Ohno, H. Ion conduction in zwitterionic-type moltensalts and their polymers. J. Mater. Chem. 2001, 11, 1057–1062. [Google Scholar]
- Carlin, R.T.; Wilkes, J.S. Advances in Nonaqueous Chemistry; WILEY-VCH: New York, NY, USA, 1994. [Google Scholar]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar]
- Wasserscheid, P.; Keim, W. Ionic liquids—New “solutions” for transition metal catalysis. Angew. Chem. Int. Ed. 2000, 39, 3772–3789. [Google Scholar]
- Sheldon, R. Catalytic reactions in ionic liquids. Chem. Commun. 2001, 23, 2399–2407. [Google Scholar]
- De Castro, C.; Sauvage, E.; Valkenberg, M.H.; Holderich, W.F. Immobilised ionic liquids as lewis acid catalysts for the alkylation of aromatic compounds with dodecene. J. Catal. 2000, 196, 86–94. [Google Scholar]
- Zhou, Y. Recent advances in ionic liquids for synthesis of inorganic nanomaterials. Curr. Nanosci. 2005, 1, 35–42. [Google Scholar]
- Fonseca, G.S.; Umpierre, A.P.; Fichtner, P.F.P.; Texeira, S.R.; Dupont, J. The use of imidazolium ionic liquids for the formation and stabilization of Ir0 and Rh0 nanoparticles: Efficient catalysts for the hydrogenation of arenes. Chem. Eur. J. 2003, 9, 3263–3269. [Google Scholar]
- Avellaneda, R.S.; Ivanova, S.; Sanz, O.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Ionic liquid templated TiO2 nanoparticles as a support in gold environmental catalysis. Appl. Catal. B Environ. 2009, 93, 140–148. [Google Scholar]
- Migowski, P.; Dupont, J. Catalytic applications of metal nanoparticles in imidazolium ionic liquids. Chem. Eur. J. 2007, 13, 32–39. [Google Scholar]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 2, 405–408. [Google Scholar]
- Schmid, G.; Corain, B. Nanoparticulated gold: Syntheses, structures, electronics, and reactivities. Eur. J. Inorg. Chem. 2003, 17, 3081–3098. [Google Scholar]
- Sau, T.K.; Murphy, C.J. Room temperature, high-yield synthesis of multiple shapes of gold nanoparticles in aqueous solution. J. Am. Chem. Soc. 2004, 126, 8648–8649. [Google Scholar]
- Ivanova, S.; Pitchon, V.; Zimmermann, Y.; Petit, C. Preparation of alumina supported gold catalysts: Influence of washing procedures, mechanism of particles size growth. Appl. Catal. A Gen. 2006, 298, 57–64. [Google Scholar]
- Tsunoyama, H.; Sakurai, H.; Negishi, Y.; Tsukuda, T. Size-specific catalytic activity of polymer-stabilized gold nanoclusters for aerobic alcohol oxidation in water. J. Am. Chem. Soc. 2005, 127, 9374–9375. [Google Scholar]
- Zhang, Z.; Berg, A.; Levanon, H.; Fessenden, R.W.; Meisel, D. On the interactions of free radicals with gold nanoparticles. J. Am. Chem. Soc. 2003, 125, 7959–7663. [Google Scholar]
- Lei, G.-T.; Wang, Z.; Lee, C.-Y.; Yang, H.-Y.; Wang, C.R.C. Aqueous-organic phase transfer of gold nanoparticles and gold nanorods using an ionic liquid. J. Am. Chem. Soc. 2004, 126, 5036–5037. [Google Scholar]
- Schrekker, H.S.; Gelesky, M.A.; Stracke, M.P.; Schrekker, C.M.L.; Machado, G.; Teixeira, S.R.; Rubim, J.C.; Dupont, J. Disclosure of the imidazolium cation coordination and stabilization mode in ionic liquid stabilized gold(0) nanoparticles. J. Colloid Interface Sci. 2007, 316, 189–195. [Google Scholar]
- Cassol, C.C.; Ebeling, G.; Ferrera, B.; Dupont, J. A simple and practical method for the preparation and purity determination of halide-free imidazolium ionic liquids. Adv. Synth. Catal. 2006, 348, 243–248. [Google Scholar]
- Francisco, M.S.P.; Cardoso, W.S.; Gushikem, Y.; Landers, R.; Kholin, Y.V. Surface modification with phosphoric acid of SiO2/Nb2O5 prepared by the sol-gel method: Structural-textural and acid sites studies and an ion exchange model. Langmuir 2004, 20, 8707–8714. [Google Scholar]
- Turkevich, J. Colloidal Gold part I. Historical and preparative aspects, morphology and structure. Gold Bull. 1985, 18, 86–91. [Google Scholar]
- Douglas, F.; Yañez, R.; Ros, J.; Marín, S.; de la Escosura-Muñiz, A.; Alegret, S.; Merkoci, A. Silver, gold and the corresponding core shell nanoparticles: Synthesis and characterization. J. Nanopart. Res. 2008, 10, 97–106. [Google Scholar]
- Nawrocki, J.; Dunlap, C.; McCormick, A.; Carr, P.W. Part II. Chromatography using ultra-stable metal oxide-based stationary phases for HPLC. J. Chromatogr. A 2004, 1028, 1–30. [Google Scholar]
- Mekhemer, G.A.H.; Nohman, A.K.H.; Fouad, N.E.; Khalaf, N.E. Surface to bulk characterization of phosphate modified aluminas. Colloids Surf. A Physicochem. Eng. Aspects 2000, 161, 439–446. [Google Scholar]
- Gu, W.; Shen, M.; Chang, X.; Wang, Y.; Wang, J. Gelification process to prepare phosphate modified alumina: Study on structure and surface properties. J. Alloys Compd. 2007, 441, 311–316. [Google Scholar]
- Kasprzyk-Hordern, B. Chemistry of alumina, reactions in aqueous solution and its application in water treatment. Adv. Colloid Interface Sci. 2004, 110, 19–48. [Google Scholar]
- Busca, G.; Ramis, G.; Lorenzelli, V.; Rossi, P.F.; La Ginestra, A.; Patrono, P. Phosphoric acid on oxide carriers. 1. Characterization of silica, alumina, and titania impregnated by phosphoric acid. Langmuir 1989, 5, 911–916. [Google Scholar]
- Mehnert, C.P. Supported ionic liquid catalysts. Chem. Eur. J. 2005, 11, 50–56. [Google Scholar]
- De Yu, L.; O'Connor, B.H.; Roach, G.I.D.; Cornell, J.B. Crystal structure communication. Acta Crystallogr. A Found. Crystallogr. 1990, 46, C61. [Google Scholar]
- Heimer, N.E.; Del Sesto, R.E.; Meng, Z.; Wilkes, J.S.; Carper, W.R. Vibrational spectra of imidazolium tetrafluoroborate ionic liquids. J. Mol. Liq. 2006, 124, 84–95. [Google Scholar]
- Dieter, K.M.; Dymek, C.J.; Heimer, N.E.; Rovang, J.W.; Wilkes, J.S. Ionic structure and interactions in 1-methyl-3-ethylimidazolium chloride-aluminum chloride molten salts. J. Am. Chem. Soc. 1988, 110, 2722–2726. [Google Scholar]
- Talaty, E.R.; Raja, S.; Storhaug, V.J.; Dolle, A.; Carper, W.R. Raman and infrared spectra and ab initio calculations of C2–4MIM imidazolium hexafluorophosphate ionic liquids. J. Phys. Chem. B 2004, 108, 13177–13184. [Google Scholar]
- Katsyuba, S.A.; Zvereva, E.E.; Vidis, A.; Dyson, P.J. Application of density functional theory and vibrational spectroscopy toward the rational design of ionic liquids. J. Phys. Chem. A 2007, 111, 352–370. [Google Scholar]
- Romero-Sarria, F.; Penkova, A.; Martinez, L.M.; Centeno, M.A.; Hadjiivanov, K.; Odriozola, J.A. Role of water in the CO oxidation reaction on Au/CeO2: Modification of the surface properties. Appl. Catal. B Environ. 2008, 84, 119–124. [Google Scholar]
- Carriazo, G.; Martínez, L.M.; Odriozola, J.A.; Moreno, S.; Molina, R.; Centeno, M.A. Gold supported on Fe, Ce, Al, pillared bentonites for CO oxidation reaction. Appl. Catal. B Environ. 2007, 72, 157–165. [Google Scholar]
- Date, M.; Ichihashi, Y.; Yamashita, T.; Chiorino, A.; Boccuzzi, F.; Haruta, M. Performance of Au/TiO2 catalyst under ambient conditions. Catal. Today 2002, 72, 89–94. [Google Scholar]
- Date, M.; Haruta, M. Moisture effect on CO oxidation over Au/TiO2 catalyst. J. Catal. 2001, 201, 221–224. [Google Scholar]
- Ivanova, S.; Petit, C.; Pitchon, V. Application of alumina supported gold-based catalysts in total oxidation of CO and light hydrocarbons mixture. Catal. Today 2006, 113, 182–186. [Google Scholar]
- Ivanova, S.; Petit, C.; Pitchon, V. A new preparation method for the formation of gold nanoparticles on an oxide support. Appl. Catal. A Gen. 2004, 267, 191–201. [Google Scholar]
- Tsuchiya, H.; Imanari, M.; Ishihara, S.; Nakakoshi, M.; Nishikawa, K.; Seki, H.; Tashiro, M. NMR study for self-aggregation of 1-butyl-3-methylimidazolium bromide in aqueous solution. Anal. Sci. 2008, 24, 1369–1371. [Google Scholar]
- Schubert, M.M.; Hackenberg, S.; Van Veen, A.C.; Muhler, M.; Plzak, V.; Behm, R.J. CO oxidation over supported gold catalysts—“Inert” and “active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar]
- Santos, C.S.; Baldelli, S. Surface orientation of 1-methyl-, 1-ethyl-, and 1-butyl-3-methylimidazolium methyl sulfate as probed by sum-frequency generation vibrational spectroscopy. J. Phys. Chem. B 2007, 111, 4715–4723. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ivanova, S.; Bobadilla, L.F.; Penkova, A.; Sarria, F.R.; Centeno, M.A.; Odriozola, J.A. Gold Functionalized Supported Ionic Liquids Catalyst for CO Oxidation. Catalysts 2011, 1, 52-68. https://doi.org/10.3390/catal1010052
Ivanova S, Bobadilla LF, Penkova A, Sarria FR, Centeno MA, Odriozola JA. Gold Functionalized Supported Ionic Liquids Catalyst for CO Oxidation. Catalysts. 2011; 1(1):52-68. https://doi.org/10.3390/catal1010052
Chicago/Turabian StyleIvanova, Svetlana, Luis Francisco Bobadilla, Anna Penkova, Francisca Romero Sarria, Miguel Angel Centeno, and Jose Antonio Odriozola. 2011. "Gold Functionalized Supported Ionic Liquids Catalyst for CO Oxidation" Catalysts 1, no. 1: 52-68. https://doi.org/10.3390/catal1010052