1. Introduction
The Findability, Accessibility, Interoperability, and Reusability (FAIR) [
1] of research data is becoming increasingly more important, not only for making data more interoperable for machine learning processes but also to accelerate research processes in general. One of the limiting factors in the FAIRness is the availability and quality of metadata. With terminologies like the IUPAC Goldbook [
2], National Cancer Institute Thesaurus (NCIT) [
3], and Voc4Cat [
4], metadata aspects such as the gas hourly space velocity of a component used in a chemical experiment can be addressed. Nonetheless, the usefulness of such a metadata representation can be questionable. Detailed information is, of course, highly valuable, specifically for reevaluating experiments. However, it also increases the required querying capabilities of a researcher if they opt for simpler, more generic queries, such as inferring knowledge from existing metadata and transferring it into a representation more closely aligned with common terminology. The Reac4Cat ontology [
5] established a generic data pattern for modeling reactions and enabling the classification of reactions occurring in a reaction experiment. This approach is further extended here, as challenges with having a common data representation and logic integration have been addressed. To extract and generate a common knowledge representation, pipelines for knowledge insertion are presented, along with an approach for extracting and further enriching knowledge for reaction classification. Specifically, the decomposition of a chemical entity into its functional subgroups and their representation in an ontology-aligned knowledge graph are presented in detail. The classification of chemical reactions is based on the terminology of the Reac4Cat ontology. However, a pipeline is presented for introducing reference reactions and a new classification technique.
The concept of classifying reactions and, more specifically, experiments performed in the lab, with possibly unknown side reactions, is not a novel approach. Databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) [
6] or the Rhea curated reaction database [
7] are approaches to documenting a collection of reactions that already implement a semantic background such that, for example, reactants can be commonly addressed via terminology provided via the Chemical Entities of Biological Interest (ChEBI) ontology [
8]. Because there is interest in maintaining a curated and therefore validated list of reactions, simple experiments that do not meet a certain standard of documentation are not necessarily suitable for inclusion in such a database. As research continually progresses, it is essential to classify, document, and occasionally publish new data through a knowledge graph. Therefore, the presented approach focuses more on immediate interaction with new data. A somewhat similar approach to modeling reactions and, more specifically, reaction networks can be found in the tool set surrounding OntoRXN [
9]. OntoRXN is designed to calculate the specific reaction network associated with a particular reaction. Since this approach is not only more detailed and thereby more complex for querying purposes but also requires more knowledge of the process than often provided via, for example, an electronic lab notebook (ELN), this approach is regarded as promising as an extension for mature enough research data management pipelines.
2. Materials and Methods
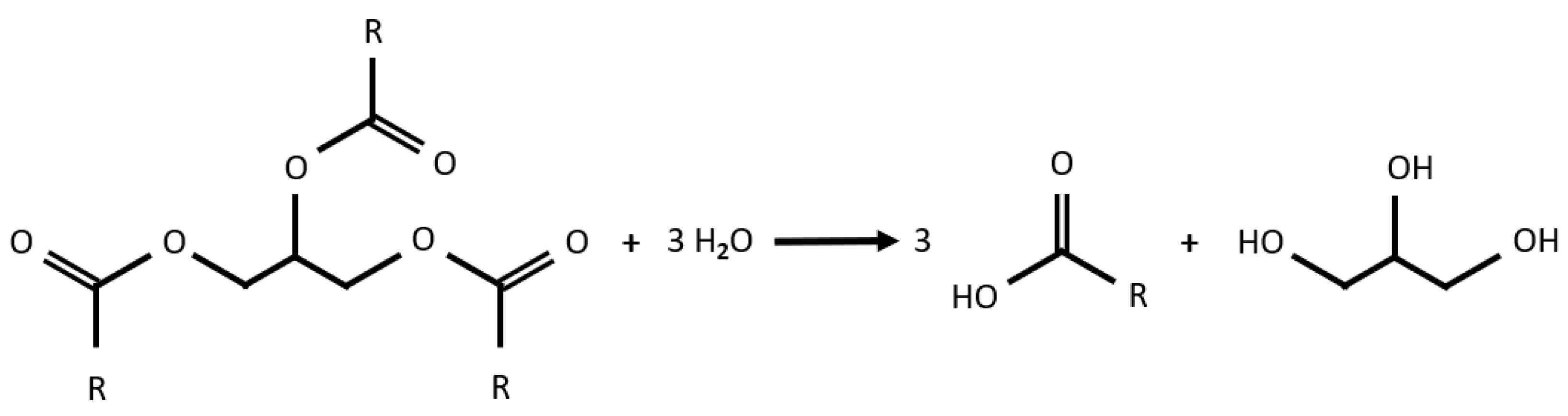
To illustrate how the pipeline works, two example reactions are discussed that are referenced throughout. The first is a fat cleavage (hydrolysis) into respective fatty acids and trivalent glycol alcohol. This is used to examine flexibility and axiomatization in greater detail. The general formula can be seen in
Figure 1.
The other reaction is an exemplary chemical reaction in which the substance quantities of the reactants and the products are hidden behind unexpressive data, such as the gas hourly space velocity or the liquid volume flows. Furthermore, it is a two-stage reaction, with the main reaction and a subsequent reaction that produces by-products. This reaction involves the hydrolysis of isoamyl acetate, followed by the oxidation of the resulting isoamyl alcohol with oxygen to form the respective isoamylaldehyde. The reaction is shown in
Figure 2. This reaction is characteristically used to identify partial reactions or the reactants from a mixture that are required for classification as a specific reaction, in which other chemical reactions are present. Hence, the reaction in
Figure 2 is also shown in its individual reaction steps. Since the decomposition into individual reaction steps and chemical activity states can be modeled in different levels of detail (individual quantum states, other steric arrangements), only the schematic procedure is presented here. It is up to the use case/user to choose the desired level of detail. Characteristically,
Figure 2 shows a reaction in which the second reaction consumes the products of the first reaction. The specification of a stoichiometry that assumes a complete conversion would be critical in the case of an experimental reaction since such a complete conversion does not have to be achieved, e.g., due to reactant deficiency. Since the subsequent reaction requires reactants from the first reaction, the reactants of the second reaction are to be found both among the reactants and among the products of the first reaction. The implications for modeling and evaluation will be discussed in more detail later. However, it is worth noting here that real chemical experiments cannot always be reproduced using the idealized representation.
As a base for the description of chemical reactions, both colloquial chemical names and IUPAC designations, as well as the description using the International Chemical Identifier (InChI) [
10], the InChI key (version of the respective InChI limited to 27 characters), and the Simplified Molecular Input Line Entry System (SMILES) [
11] notation, as well as the associated SMILES arbitrary target specification (SMARTS) [
12] notation, are used. In comparison, the colloquial chemical names are less precise than the IUPAC name, the SMILES notation, or InChi/InChi-key. However, the colloquial name remains in common use in many databases. IUPAC designation, SMILES notation, and InChI/InChI-key all have slightly different areas of application, where the informative value increases in respective order and the complexity of the description also increases. The SMILES notation remains the focus, as its semantics directly convey information about the structural composition. Nevertheless, this structural information is more readily interpretable than InChI Semantic, and there is therefore a plentiful supply of tools capable of analyzing the SMILES notation. A tool, which is intended for the evaluation of SMILES notations, is the SMARTS notation. It goes beyond conventional pattern matching, such as regular expressions, and can be used, for example, to restrict the neighborhood of a functional group described using SMILES notation. Examples of the syntax of the different formats are given in
Table 1. An example of pattern matching via regular expression and SMARTS notation is shown in
Table 2. It can be seen here that the SMARTS notation also examines the neighborhood (marked by underlining in
Table 2).
The data basis for the tools presented here is provided by four pillars: manual data entry, uploading using forms, data exchange with databases, and the knowledge pre-modeled in ontologies. Since both manual data entry methods and form-based incorporation are tools adapted to the methodology presented here, existing knowledge sources are discussed first.
In this case, PubChem [
14] and KEGG represent the knowledge sources addressed via Application Programming Interface (API). The KEGG is a database of chemical, genomic, health, and reaction network (self-described as “systems”) information published in 1995. The 12,306 reactions with 3202 reaction classes paired with 19,508 chemical substances form a good basis as a reference database, which is manually curated by the Kanehisa Laboratory at Kyoto University [
6]. The KEGG utilizes its proprietary chemical identifiers, as well as identifiers from PubChem and ChEBI. One of the reactions can be seen in the sketch in
Figure 3, which only shows some of the information provided from the KEGG database.
PubChem is a database provided by the National Library of Medicine and it covers an even larger number of chemicals, with over 121 million curated and 334 million community-derived entries [
14]. Its description of a chemical and references to other databases are typically more detailed than those of KEGG. The primary advantage here is that all curated PubChem entries have both a SMILES and an InChI ID, which can also be queried via the API. In the following method, it serves as the basis for describing and referencing chemical entities in the knowledge graphs, thanks to its broader scope of chemical entities.
ChEBI is used to provide a standardized basis for describing chemical entities. Due to its existing structure, in the form of its taxonomy and hierarchy, it gives a good basis for describing chemicals. Since both the KEGG and PubChem lists the ChEBI class ID as a reference for their chemicals; instantiating the chemicals is also simplified.
In addition to using the ChEBI ontology, various ontologies and fragments thereof are reused, all of which come from the collection of Open Biological and Biomedical Ontologies (OBO) Foundry [
16] ontologies. The ontology basis or top level is the BFO [
17], but, due to limiting the number of mentioned reused ontologies, only parts from sub-level ontologies are further described. Important to note is the RXNO [
18], which provides some of the main terminology for classifying reactions based on a named reaction, the MOP [
19], which is somewhat of a predecessor to the RXNO, by refining, adding, and also extending the reaction description with more molecular processes, and the Reac4Cat ontology, which itself is not an OBO collection ontology but is aligned with the OBO principals [
16] and serves as the main terminology for describing and modeling reaction data of an experiment.
3. Results
A brief overview of the method presented here is shown in
Figure 4.
As this is a multi-step procedure, each significant step will be presented in the following sub-chapters. The user input, which marks the start of data processing, is not discussed first, as the knowledge graph represents the core structure. The steps Data Extraction and Data Normalization (
Section 3.2) and Data Normalization (
Section 3.3) thus represent the preparation of the data for populating this graph, followed by Semantic Enrichment via Structure Analysis (
Section 3.4). Accordingly, this section presents the changes and additions made in the modeling of ChEBI, RXNO, MOP, and the Reac4Cat ontology. Since parts of the modeling are based on the automated data evaluation in the
Section 3.4, the general procedure is presented here. After processing the ontology, the eponymous procedure is described in the
Section 3.3. The aim here is to prepare the reaction data so that it can be modeled in Reac4Cat. However, the reactants and products have also been converted into their SMILES notation, allowing for their evaluation. The previously introduced databases are used as a look-up service and to normalize the different chemical identifiers. The
Section 3.4 describes the evaluation of chemical substances based on functional groups via SMILES and SMARTS notation. As described above, the decomposition also serves as a basis for new classes, leading to the extended knowledge graph described in the
Section 3.1. Since an alternative logic to the original axiomatization from Reac4Cat is used, which is firmly based on SHACL [
20] and SPARQL [
21], this and the data processing step based on it are presented in the last section,
Section 3.4. Both standard inferencing in OWL [
22] and external inferencing using SPARQL are presented here.
3.1. Base Ontology Modifications
Modifying an ontology requires adherence to quality standards and best practices. In light of the challenges associated with designing automated modification steps, it is recommended that the ontology be developed and curated by a human expert. As such, some parts of the modifications to the ontology are currently hard-coded (changes to the code would be required to alter these modifications) into the script, which is intentionally done to limit the ease and thereby the chance of undesirable modifications. Only the general approach of extending the ontology is described here. For best practices, it is recommended to either have the ontology prepared by an expert or set up once, considering the use case-specific limitations, to facilitate an automatic extension script.
Reac4Cat describes the core ontological model for describing an experiment. The classification into reaction types is performed primarily through the design pattern of the RXNO/MOP, which is a slight modification from the Reac4Cat ontologies approach of using general class axioms or left-hand-side logic. Finally, the chemical entity modeling is handled via ChEBI. Starting with the chemical entity modeling, ChEBI is used in its ontological design, rather than as a database. In other words, while the classes of the ChEBI ontologies are used, representing a chemical entity involves referencing a real-world chemical entity to a digital counterpart representation, which, in this case, utilizes the PubChem database entries.
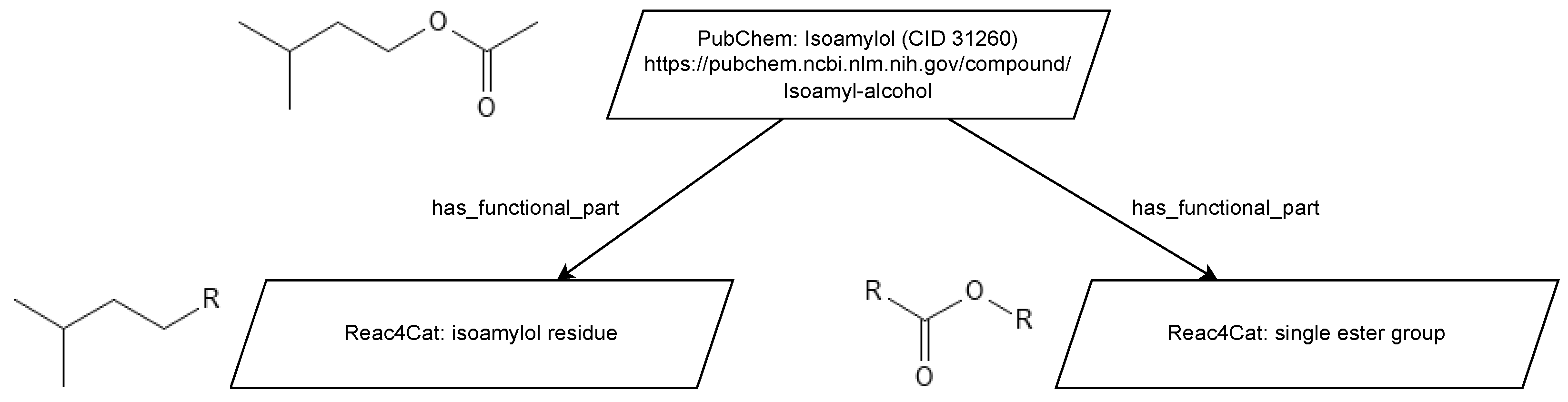
A representation of this can be seen in
Figure 5, where ontology classes are shown as ellipses and data instances are depicted via the trapezoidal shape. Where new classes are needed, they must be added to incorporate new chemical entities represented in the experimental data. Examples of this include the isoamylol class, which can be reused from the ChEBI ontology in this case. Such an addition of classes would occur, for example, for the permutation of various esters that have variations in their alkyl groups. For the data instance, the direct link to the PubChem website is used. This, in turn, helps with digital agents, which can, for example, utilize the PubChem API to extract more information, such as physical properties like density. While this information could also be imported into the knowledge graph, limiting the knowledge to a required minimum is beneficial for reasoning and querying operations.
As the ChEBI database does not include as many chemical substances as the PubChem database, manually adding chemical entity representations, such as the isoamylol class, to the used ontology is recommended for curation purposes, provided there is no complete overlap between the two databases.
While the ChEBI ontology classifies chemical entities via characteristic functionalities, such as a chemical entity being a carbonyl compound (CHEBI:35701 [
23]), modeling reaction networks more holistically requires this description to be more specific. For example, a chemical in the group of carbonyl compounds can be an aldehyde, a ketone, or even both. As such, functional group modeling is introduced, whereby a chemical entity such as isoamyl acetate is assigned multiple functional groups via the has_functional_group relation. This can be seen in
Figure 6. The functional group instances are, by intention, not depicted with their respective Uniform Resource Identifier (URI) as, due to the number of possible functional groups, no terminology service currently provides them with a fixed URI. As such, local reference URIs or RDF blind notes are used. Here, for example,
https://github.com/AleSteB/Reac4Cat/blob/main/reac4cat_with_examples.owl#Isoamylol_residue is used, which, by design, does not resolve correctly. These functional groups are also assigned to classes, which are designed with the general ChEBI design pattern for chemical entities.
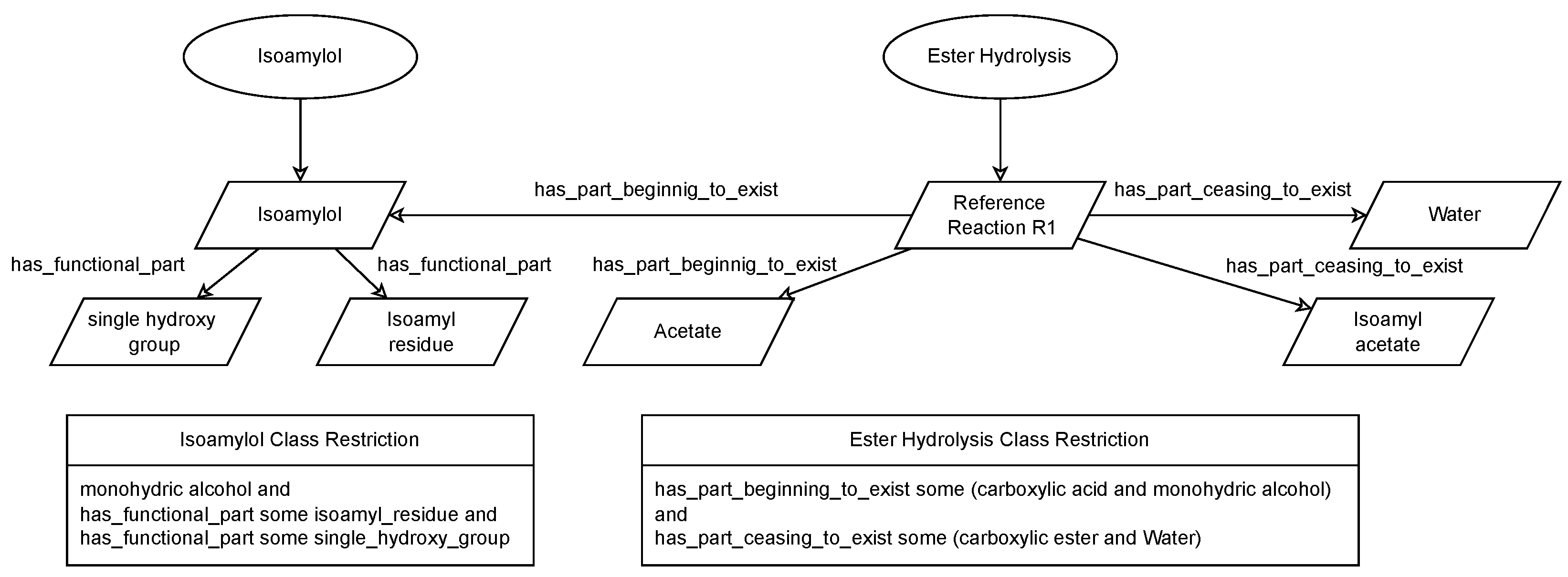
To differentiate between reference reactions and experimental data, two distinct data patterns are employed.
Figure 7 represents the data pattern for reference reactions, where new reference data should be classified via axioms of the respective reaction classes. This enables various permutations of a reaction to be introduced and still classified as the same reaction. Specifically, permutations of chemical entities (e.g., isoamylol, butanol) are presented by the axioms here. However, the adoption of more advanced axiomatic systems has the potential to impose restrictions not only on chemical entities but also on environmental factors. Due to the modeling of functional groups, as demonstrated by the isoamylol and isoamylol class restrictions, more complex yet refined axioms can be designed for the main reaction class.
Figure 8 presents the data pattern used for experimental reactions. This model is derived from the Reac4Cat ontology [
5] and is intentionally different from the reference data pattern. For purposes of simplicity,
Figure 8 only sketches the design pattern. Please refer to the original publication for the in-depth description. The primary objective of this differentiation is to prevent the drawing of inferences that should not be drawn. For reference reactions, it can be assumed that all data are available. However, experimental data may be incomplete. Reasoning based on such incomplete information may cause inconsistencies and should, therefore, be avoided. By using different data patterns and thereby different relations between instances describing reference datasets and experimental datasets, the axioms and thus the logic can differentiate between different data patterns.
With an ontology generated through this approach, the data can be classified as described in
Section 3.4.
3.2. Data Extraction and Data Normalization
To introduce experimental data and reference data into the data processing pipeline, a graphical user interface (GUI) is developed, allowing researchers to perform these evaluations independently, rather than relying on a hosted service. What is presented here could, of course, be established as a web service. As the current method still has some limitations that should be addressed first, no details of hosting this as a web service are discussed in this work. Work in this field is, however, still ongoing and will be part of future work.
As shown in
Figure 9, the GUI has three primary data import methods: manual entry, by EnzymeML data sheets [
24], and by using the “Upload Manually” button. Additionally, data can be imported by searching for a KEGG reaction in the search bar. KEGG entries are considered as reference reactions, while imports over the manual route and the EnzymeML data sheet are considered as experimental data. For manual entry, the data is processed directly. In contrast, for the EnzymeML sheet, the data is extracted by the pyEnzymeML package [
25] and then processed. Both, the manual route and the EnzymeML data sheets, can directly provide SMILES notations, which can be used to find the respective entries in the PubChem database and perform SMARTS pattern analysis. If the SMILES are not provided, a manual search using the colloquial name is performed to retrieve the SMILES notation from PubChem for SMARTS pattern analysis. For the import of KEGG reactions, only the reaction identifier is needed, such as ‘R12516’; the reaction entry is shown in
Figure 3. The respective PubChem Entry and SMILES notation are retrieved through the KEGG API.
The missing PubChem entries and SMILES notations are all collected by the PubChem API, and the PubChem entries (URIs) are instantiated in the knowledge graph. For normalization purposes, a SMILES notation is required to perform the SMARTS pattern analysis. As such, entries with only InChI notation are converted into their respective SMILES notation through a search over the PubChem API.
3.3. Semantic Enrichment via Structure Analysis
For semantic enrichment of the data, functional groups are extracted from the chemical entities and modeled in the knowledge graph. This approach offers the advantage that class axioms for general reactions do not necessitate exact chemical entity matching. Instead, one can rely on the characteristics of the functional groups of a specific chemical entity for classification. This enables the further generalization of the axioms used for describing chemical reactions. Since decomposing complex structures, such as the isoamyl residue of isoamyl acetate, into further functional groups, such as a methyl residue, is possible, one needs to decide to what level this decomposition is reasonable. Since, to the authors’ knowledge, there are no ontologies/RDF terminologies that model the hierarchical structure of such function groups. These hierarchies, which are necessary for the respective modeling purposes, are not pre-specified. It is up to the user of this method to describe the required hierarchy. To make the functional groups more accessible for the reasoning purposes of an ontology, the classes of these functional groups should be modeled when a decision is made on which SMARTS patterns to use for pattern matching. The matching process, which is performed using the SMARTS pattern and RDKit (Release_2025_03_4) [
26], the respective Python 3.13 package for evaluating these patterns, is then used to model the respective functional groups in the knowledge graph.
3.4. Automated Semantic Analysis and Reasoning
Two different methods are used to classify the experimental reaction tests and the reference reactions. Since ontologies are used to describe data deterministically and also factually correctly, experimental reactions pose a challenge. Metadata is often found to be incomplete and relies on human interpretation. A guarantee of accuracy is not always possible, even by humans. In order not to burden the logic of an ontology with errors, only the reference reactions that are assumed to be factually correct are classified directly in the ontology. The classification of experimental data, which, in some cases, must be regarded as factually incorrect or incomplete, is only classified by comparison with the reference measurements. Two different data processing methods are chosen for this purpose. For the regular classification of reference reactions, the standard OWL logic, located in the ontology, is used. This is checked using a reasoner such as HermiT [
27], which is used to perform automatic reasoning. The general logic was already explained in
Figure 7.
A different procedure is used for the comparison and classification of experimentally recorded reactions. Apart from additional conditions that can be implemented, it is assumed that an experiment contains a defined reference reaction if the reactants and products of this reference reaction are present in the experiment. As this can still be subject to errors, only an annotated reference is generated, rather than performing the complete logical classification. This should only be carried out if a classification can be assumed to be error-free with sufficient certainty. As the number of possible rules exceeds the scope of the description here, the general procedure is described.
To clearly emphasize the difference between a reference reaction and an experiment, a distinction in the data patterns was chosen to describe the information, as in
Section 3.1 Base Ontology Modifications and
Figure 7 and
Figure 8. Applying the ontology logic only to the reference reactions, rather than it being erroneously applied to the experimental reactions, thereby reduces the potential for error. The comparison of whether a reference reaction is present in an experiment is now performed with a SPARQL query instead of the complex logic used in the first version of the Reac4Cat ontology. This offers some advantages, especially in terms of computational effort. Since the logic or query is only applied to an excerpt of the knowledge graph, rather than the entire knowledge graph, memory demand and, consequently, computational time are reduced.
To better capture the logic embedded in the SPARQL query [
21], SHACL [
20], especially the SHACL Advanced Feature set (SHACL-AF) [
28], is used to generate a fixed semantic artifact, which can be imported into the actual ontology by an import statement in the ontology file. To work with the advanced feature set, pySHACL is used as the Python package [
29]. The advantage of SHACL-AF is that a SPARQL query can be executed in a SHACL shape. Additionally, SHACL-AF assumes that modifying the actual knowledge graph with SHACL validation should be permitted. In contrast, previous SHACL specifications were intended only for validation, without any modification to the knowledge graph.
In the SPARQL query, which is applied outside the actual SHACL shape as part of the provided code, it is examined whether the reactants of the reference reaction match those of the experiment and also whether the products of the reference reaction match those of the experiment. The SPARQL query is given in
Figure 10.
The comparison conducted herein is executed through the utilization of the “FILTER NOT EXISTS” function terms. Consequently, a one-to-one matching of the reactants and products is not performed; instead, it is ensured that all reactants and products can be identified within the experiment. Once these are found, the experiment is linked to the reference reaction using the relation “has_predicted_reaction_part”. Further criteria and checks can, of course, be integrated into the SPARQL query, which increases the accuracy of the reaction classification. Thanks to the very generic terminology used in the SPARQL query, it is possible to write simple axioms, such as those found in an ontology. However, the logic of the actual ontology should not be compromised by errors in the data. Additionally, the preceding SHACL shape also enables the detection of general modeling errors, which, in turn, ensure the smooth functioning of the ontology logic’s reasoning and thereby simplify the use of more complex logics.
The results of the SHACL validation, the OWL reasoning, and the SPARQL querying are displayed in the GUI, allowing them to be cross-checked and validated by the researcher.
4. Discussion
The presented technique enables the modeling of experimental reaction datasets with minimal metadata in greater detail within the Reac4Cat ontology by extracting functional groups, which also facilitates comparison with reference reactions. Referenced reactions are also represented in more detail as reaction classification is possible through the detailed description of the functional groups. By this increase in modeling depth, which comes without great effort for the curators and users, further restrictions on the reaction classification can be integrated more meaningfully.
Furthermore, the use of SPARQL queries, which are embedded in SHACL shapes thanks to the SHACL-AF, enables complex logic, such as that presented in the Reac4Cat ontology, to be performed outside the actual OWL reasoning. This saves resources precisely because the left-hand-side logic [
30] is very computationally expensive. In contrast, only small parts of the graph need to be queried and checked thanks to the SHACL-SPARQL query. The general class axiom approach of the Reac4Cat ontology was found to be quite demanding in terms of computational resources. Due to the complexity of the axioms on the left-hand side of logic, it was found that reasoning performed with HermiT resulted in reasoning times of up to one day, which often exceeded the stable runtime of the reasoner itself. As testing the cause of this time effect for the left-hand side logic was not feasible, the SHACL-AF was prioritized for use. Here, querying times of less than one second were found for datasets with up to forty reaction entities. As this appeared to be following regular SHACL validation and SPARQL query times, no further investigation was performed.
Combined with a simple user interface and the possibility of implementing the described method as a background service linked to a chemical database sample, this also provides a means for such methods as those used in reaction and catalysis research to be easily implemented in everyday research. Other databases that use similar metadata and APIs can also be easily integrated.
However, the quality of the reaction classification should also be discussed again. For the experiment in
Figure 2, the SPARQL query identifies the reaction of isoamyl acetate and water to form isoamyl alcohol and acetate. A reference is then established using the has_predicted_reaction_part relation. To identify the subsequent reaction of isoamylol with oxygen to form isoamylaldehyde and water, not only the reactants but also the products are searched for as possible reactants in the SPARQL query. However, the number of potential reactions that could be found here also increases considerably.
Due to the more detailed modeling of chemical entities using functional groups, a more complex classification is also possible. For example, a distinction can be made between simple ester cleavage and complete hydrolysis/saponification of a fatty molecule, where modeling functional groups is advantageous for describing glycerol. Other areas of application include, for example, the description of lactam ring cleavages, where rings of different chain lengths are depicted.
Several limitations still restrict automated data processing, particularly in terms of metadata. Firstly, there is a lack of and inconsistent metadata and metadata schema. For example, specifications such as SMILES notation or InChI notation are not always mandatory, even if the specification of chemical entities as metadata is required for reaction data sets. Inconsistent notations can quickly lead to mix-ups, not only in data sets but also in databases, for example, when the dissolved ionic form of acetate is intended but the notation does not indicate this. The existing ontologies also have weaknesses that will be corrected over time. For example, there are few alternatives to ChEBI, which, for many chemical substances, assumes the state found in the human body as the standard chemical state of a substance. It is also important to mention the limitations of the proposed approach here. Due to the lack of a terminology that can provide the functional group modeling required for the correct embedding of instances generated via the SMARTS pattern comparison, parts of the code rely on hardcoded fragments. These hardcoded aspects, like the SMARTS pattern for functional groups, new functional group instances and classes, and new axioms for reaction classification, should be optimized in the future. Therefore, the development of embedding these functionalities close to either the ontology or available for the SMARTS pattern matching, which can be considered a digital agent, is ongoing.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}