
Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Epidermis
3. Overview of cSCC Carcinogenesis
4. The PI3K Pathway in Epidermal Homeostasis
5. PI3Ks-Dependent Pathways upon UV Irradiation
6. PI3Ks-Dependent Pathways in cSCC
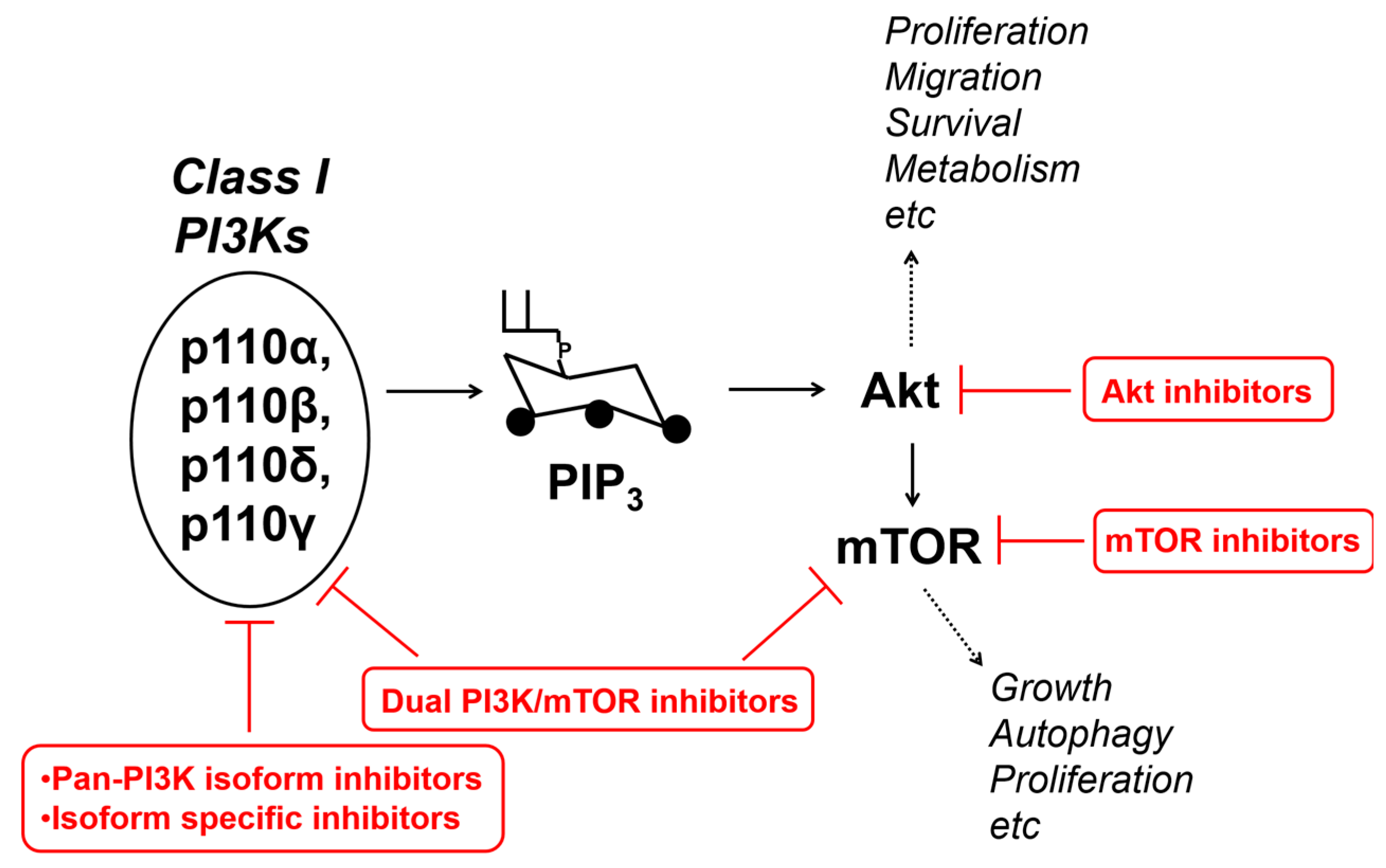
7. Targeting PI3Ks-Dependent Pathways in cSCC
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Surdu, S. Non-melanoma skin cancer: Occupational risk from UV light and arsenic exposure. Rev. Environ. Health 2014, 29, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous squamous cell carcinoma: A review of high-risk and metastatic disease. Am. J. Clin. Dermatol. 2016, 17, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Leigh, I.M. Progress in skin cancer: The U.K. experience. Br. J. Dermatol. 2014, 171, 443–445. [Google Scholar] [CrossRef] [PubMed]
- Skin Cancer Incidence Statistics. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/skin-cancer/incidence#ref-10 (accessed on 4 July 2017).
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Athas, W.F.; Hunt, W.C.; Key, C.R. Changes in nonmelanoma skin cancer incidence between 1977–1978 and 1998–1999 in northcentral New Mexico. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1105–1108. [Google Scholar]
- Gray, D.T.; Suman, V.J.; Su, W.P.D.; Clay, R.P.; Harmsen, W.S.; Roenigh, R.K. Trends in the population-based incidence of squamous cell carcinoma of the skin first diagnosed between 1984 and 1992. Arch. Dermatol. 1997, 133, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Apalla, Z.; Nashan, D.; Weller, R.B.; Castellsagué, X. Skin cancer: Epidemiology, disease burden, pathophysiology, diagnosis, and therapeutic approaches. Dermatol. Ther. 2017, 7, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Hannuksela-Svahn, A.; Pukkala, E.; Karvonen, J. Basal cell skin carcinoma and other nonmelanoma skin cancers in Finland from 1956 through 1995. Arch. Dermatol. 1999, 135, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Wassberg, C.; Thorn, M.; Johansson, A.; Bergstrom, R.; Berne, B.; Ringborg, U. Increasing incidence rates of squamous cell carcinoma of the skin in Sweden. Acta Derm. Venereol. 2001, 81, 268–272. [Google Scholar] [CrossRef] [PubMed]
- John, S.M.; Trakatelli, M.; Gehring, R.; Finlay, K.; Fionda, C.; Wittlich, M.; Augustin, M.; Hilpert, G.; Barroso Dias, J.M.; Ulrich, C.; et al. CONSENSUS REPORT: Recognizing non-melanoma skin cancer, including actinic keratosis, as an occupational disease—A call to action. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Torres, L.; Morris, S.; Kinge, J.M.; Poirier, V.; Verne, J. Measuring current and future cost of skin cancer in England. J. Public Health 2014, 36, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Katalinic, A.; Kunze, U.; Schäfer, T. Epidemiology of cutaneous melanoma and non-melanoma skin cancer in Schleswig-Holstein, Germany: Incidence, clinical subtypes, tumour stages and localization (epidemiology of skin cancer). Br. J. Dermatol. 2003, 149, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Aasi, S.Z.; Hollmig, S.T. Management of high-risk squamous cell carcinoma of the skin. Curr. Treat. Options Oncol. 2016, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Gurney, B.; Newlands, C. Management of regional metastatic disease in head and neck cutaneous malignancy. 1 cutaneous squamous cell carcinoma. Br. J. Oral Maxillofac. Surg. 2014, 52, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.C.; Otley, C.C.; Stasko, T.; Euvrard, S.; Brown, C.; Schanbacher, C.F.; Weaver, A.L.; Transplant-Skin Cancer Collaborative. Defining the clinical course of metastatic skin cancer in organ transplant recipients: A multicenter collaborative study. Arch. Dermatol. 2003, 139, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Carucci, J.A. Press for an underestimated nemesis. JAMA Dermatol. 2013, 149, 1147–1148. [Google Scholar] [CrossRef] [PubMed]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Jambusaria-Pahlajani, A.; Kanetsky, P.; Karia, P.S.; Hwang, W.; Gelfand, J.M.; Whalen, F.M.; Elenitsas, R.; Xu, X.; Schmults, C.D. Evaluation of AJCC tumor (T) staging for cutaneous squamous cell carcinoma and a proposed alternative tumor staging system. JAMA Dermatol. 2013, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.K.; Kelley, B.F.; Prokop, L.J.; Murad, M.H.; Baum, C.L. Risk factors for cutaneous squamous cell carcinoma recurrence, metastasis, and disease-specific death a systematic review and meta-analysis. JAMA Dermatol. 2016, 152, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Baroni, A.; Buommino, E.; De Gregorio, V.; Ruocco, E.; Ruocco, V.; Wolf, R. Structure and function of the epidermis related to barrier properties. Clin. Dermatol. 2012, 30, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Rorke, E.A. Molecular biology of keratinocyte differentiation. Environ. Health Perspect. 1989, 80, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Radoja, N.; Gazel, A.; Banno, T.; Yano, S.; Blumenberg, M. Transcriptional profiling of epidermal differentiation. Physiol. Genom. 2006, 27, 65–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raymond, A.A.; Gonzalez de Peredo, A.; Stella, A.; Ishida-Yamamoto, A.; Bouyssie, D.; Serre, G.; Monsarrat, B.; Simon, M. Lamellar bodies of human epidermis: Proteomics characterization by high throughput mass spectrometry and possible involvement of CLIP-170 in their trafficking/secretion. Mol. Cell Proteomics 2008, 7, 2151–2175. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.A.; Nitoiu, D.; Kelsell, D.P. Cell-cell connectivity: Desmosomes and disease. J. Pathol. 2012, 226, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Najor, N.A.; Green, K.J. Desmosomes: Regulators of cellular signalling and adhesion in epidermal health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015297. [Google Scholar] [CrossRef] [PubMed]
- Neel, V.A.; Sober, A.J. Tumors arising from the epidermis. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.; Griffith, K.; Moon, T.E. Trends in the incidence of nonmelanoma skin cancers in south eastern Arizona, 1985–1996. J. Am. Acad. Dermatol. 2001, 45, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Criscione, V.D.; Weinstock, M.A.; Naylor, M.F.; Luque, C.; Eide, M.J.; Bingham, S.F.; Department of Veteran Affairs Topical Tretinoin Chemoprevention Trial Group. Actinic keratoses: Natural history and risk of malignant transformation in the veterans affairs topical tretinoin chemoprevention trial. Cancer 2009, 115, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Harvey, I.; Frankel, S.; Marks, R.; Shalom, D.; Nolan-Farrell, M. Non-melanoma skin cancer and solar keratoses II analytical results of the south wales skin cancer study. Br. J. Cancer 1996, 74, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.N.; Sammain, A.; Erdmann, R.; Hartmann, V.; Stockfleth, E.; Nast, A. The natural history of actinic keratosis: A systematic review. Br. J. Dermatol. 2013, 169, 502–518. [Google Scholar] [CrossRef] [PubMed]
- Salasche, S.J. Epidemiology of actinic keratoses and squamous cell carcinoma. J. Am. Acad. Dermatol. 2000, 42, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Purdie, K.J.; Harwood, C.A.; Gulati, A.; Chaplin, T.; Lambert, S.R.; Cerio, R.; Kelly, G.P.; Cazier, J.B.; Young, B.D.; Leigh, I.M.; et al. Single nucleotide polymorphism array analysis defines a specific genetic fingerprint for well-differentiated cutaneous SCCs. J. Investig. Dermatol. 2009, 129, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Purdie, K.J.; Lambert, S.R.; Teh, M.T.; Chaplin, T.; Molloy, G.; Raghavan, M.; Kelsell, D.P.; Leigh, I.M.; Harwood, C.A.; Proby, C.M.; et al. Allelic imbalances and microdeletions affecting the PTPRD gene in cutaneous squamous cell carcinomas detected using single nucleotide polymorphism microarray analysis. Genes Chromosom. Cancer 2007, 46, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Proby, C.M.; Inman, G.J.; Leigh, I.M. The promise of genomics and the development of targeted therapies for cutaneous squamous cell carcinoma. Acta Derm. Venereol. 2016, 96, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Engelhardt, C.; Morgan, S.S. Treatment of unresectable and metastatic cutaneous squamous cell carcinoma. Oncologist 2010, 15, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Einspahr, J.G.; Calvert, V.; Alberts, D.S.; Curiel-Lewandrowski, C.; Warneke, J.; Krouse, R.; Stratton, S.P.; Liotta, L.; Longo, C.; Pellacani, G.; et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev. Res. 2012, 5, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.R.; Mladkova, N.; Gulati, A.; Hamoudi, R.; Purdie, K.; Cerio, R.; Leigh, I.; Proby, C.; Harwood, C.A. Key differences identified between actinic keratosis and cutaneous squamous cell carcinoma by transcriptome profiling. Br. J. Cancer 2014, 110, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Adelmann, C.H.; Truong, K.A.; Liang, R.J.; Bansal, V.; Gandee, L.; Saporito, R.C.; Lee, W.; Du, L.; Nicholas, C.; Napoli, M.; et al. MEK is a therapeutic and chemopreventative target in squamous cell carcinoma. J. Investig. Dermatol. 2016, 136, 1920–1924. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T. An introduction to phosphoinositides. Curr. Top. Microbiol. Immunol. 2012, 362, 1–42. [Google Scholar] [PubMed]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion blocks apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.R.; Toker, A. Signalling specificity in the Akt pathway in breast cancer. Biochem. Soc. Trans. 2014, 42, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Manning, B.D. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 2011, 22, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Sayama, K.; Yamasaki, K.; Hanakawa, Y.; Shirakata, Y.; Tokumaru, S.; Ijuin, T.; Takenawa, T.; Hashimoto, K. Phosphatidylinositol 3-kinase is a key regulator of early phase differentiation in keratinocytes. J. Biol. Chem. 2002, 277, 40390–40396. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Klippel, A.; Werner, S. Regulation of epidermal homeostasis and repair by phosphoinositide 3-kinase. J. Cell Sci. 2006, 119, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, R.F.; Welti, J.C.; Cooke, J.C.; Avilion, A.A.; Monks, B.; Birnbaum, M.J.; Byrne, C. AKT-dependent HspB1 (Hsp27) activity in epidermal differentiation. J. Biol. Chem. 2007, 282, 17297–17305. [Google Scholar] [CrossRef] [PubMed]
- Sully, K.; Akinduro, O.; Philpott, M.P.; Naeem, A.S.; Harwood, C.A.; Reeve, V.E.; O’Shaughnessy, R.F.; Byrne, C. The mTOR inhibitor rapamycin opposes carcinogenic changes to epidermal Akt1/PKBα isoform signaling. Oncogene 2013, 32, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.S.; Zhu, Y.; Di, W.L.; Marmiroli, S.; O’Shaughnessy, R.F. AKT1-mediated Lamin A/C degradation is required for nuclear degradation and normal epidermal terminal differentiation. Cell Death Differ. 2015, 22, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Itami, S.; Ohishi, M.; Hamada, K.; Inoue, T.; Komazawa, N.; Senoo, H.; Sasaki, T.; Takeda, J.; Manabe, M.; et al. Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res. 2003, 63, 674–681. [Google Scholar] [PubMed]
- Backman, S.A.; Ghazarian, D.; So, K.; Sanchez, O.; Wagner, K.U.; Hennighausen, L.; Suzuki, A.; Tsao, M.S.; Chapman, W.B.; Stambolic, V.; et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc. Natl. Acad. Sci. USA 2004, 101, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Owens, D.M.; Ghosh, S.; Farber, D.L. Conditional PDK1 ablation promotes epidermal and T-Cell-mediated dysfunctions leading to inflammatory skin disease. J. Investig. Dermatol. 2015, 135, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Haydenm, M.S.; Park, S.G.; Oh, H.; Seeley, J.J.; Grinberg-Bleyer, Y.; Beck, K.M.; Miyachi, Y.; Kabashima, K.; Hashimoto, T.; et al. PDK1 is a regulator of epidermal differentiation that activates and organizes asymmetric cell division. Cell Rep. 2016, 15, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Hobert, J.A.; Eng, C. PTEN hamartoma tumor syndrome: An overview. Genet. Med. 2009, 11, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Von, T.; Bronson, R.; Ruan, M.; Mu, W.; Huang, A.; Maira, S.M.; Zhao, J.J. Spatially distinct role of class Ia PI3K isoforms in the development and maintenance of PTEN hamartoma tumor syndrome. Genes Dev. 2013, 27, 1568–1580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Maffucci, T. Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans. 2007, 35, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Ali, K.; Bilancio, A.; Geering, B.; Foukas, L.C. Signalling by PI3K isoforms: Insights from gene-targeted mice. Trends Biochem. Sci. 2005, 30, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T.; Falasca, M. New insight into the intracellular roles of class II phosphoinositide 3-kinases. Biochem. Soc. Trans. 2014, 42, 1378–1382. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Truong, A.B.; Cai, T.; Khavari, P.A. The class II phosphoinositide 3-kinase C2beta is not essential for epidermal differentiation. Mol. Cell. Biol. 2005, 25, 11122–11130. [Google Scholar] [CrossRef] [PubMed]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C.; et al. Constitutive autophagy and nucleophagy during epidermal differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The regulation and function of class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Singleton, P.A.; Bourguignon, L.Y.; Bikle, D.D. Calcium-induced human keratinocyte differentiation requires src- and fyn-mediated phosphatidylinositol 3-kinase-dependent activation of phospholipase C-gamma1. Mol. Biol. Cell 2005, 16, 3236–3346. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Ikehata, H.; Kawai, K.; Komura, J.; Sakatsume, K.; Wang, L.; Imai, M.; Higashi, S.; Nikaido, O.; Yamamoto, K.; Hieda, K.; et al. UVA1 genotoxicity is mediated not by oxidative damage but by cyclobutane pyrimidine dimers in normal mouse skin. J. Investig. Dermatol. 2008, 128, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, F.R.; Rebel, H. Early events in UV carcinogenesis-DNA damage, target cells and mutant p53 foci. Photochem. Photobiol. 2008, 84, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, D.; Alas, L.G.; Yee, V.; Oberyszyn, A.; Kibitel, J.T.; Mitchell, D.; Rosenstein, R.; Spinowitz, A.; Citron, M. Pyrimidine dimer removal enhanced by DNA repair liposomes reduces the incidence of UV skin cancer in mice. Cancer Res. 1992, 52, 4227–4231. [Google Scholar] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; He, Y.Y. PTEN in DNA damage repair. Cancer Lett. 2012, 319, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Feng, L.; Shea, C.R.; Soltani, K.; Zhao, B.; Han, W.; Smart, R.C.; Trempus, C.S.; He, Y.Y. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011, 71, 5287–5295. [Google Scholar] [CrossRef] [PubMed]
- Hocker, T.; Tsao, H. Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Hum. Mutat. 2007, 28, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; He, Y.Y. PTEN: New insights into its regulation and function in skin cancer. J. Investig. Dermatol. 2009, 129, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Y.; Pi, J.; Huang, J.L.; Diwan, B.A.; Waalkes, M.P.; Chignell, C.F. Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance. Oncogene 2006, 25, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Han, W.; Maddox, J.; Soltani, K.; Shea, C.R.; Freeman, D.M.; He, Y.Y. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene 2010, 29, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Willems, E.; Singh, A.; Ong, I.M.; Verma, A.K. Ultraviolet radiation-induced differential microRNA expression in the skin of hairless SKH1 mice, a widely used mouse model for dermatology research. Oncotarget 2016, 7, 84924–84937. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; He, Y.Y. Requirement for metalloproteinase-dependent ERK and AKT activation in UVB-induced G1-S cell cycle progression of human keratinocytes. Photochem. Photobiol. 2009, 85, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.D.; DiGiovanni, J.; Lynch, C.J.; Shantz, L.M. Inhibition of mTOR suppresses UVB-induced keratinocyte proliferation and survival. Cancer Prev. Res. 2012, 5, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.E.; Janda, J.; Criswell, J.; Blohm-Mangone, K.; Olson, E.R.; Liu, Z.; Barber, C.; Petricoin, E.F.; Calvert, V.S.; Einspahr, J.; et al. Inhibition of Akt enhances the chemopreventive effects of topical rapamycin in mouse skin. Cancer Prev. Res. 2016, 9, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, Y.; Stratton, S.P.; Curiel-Lewandrowski, C.; Warneke, J.; Hu, C.; Bowden, G.T.; Dickinson, S.E.; Dong, Z.; Bode, A.M.; Saboda, K.; et al. Activation of the PI3K/Akt/mTOR and MAPK signaling pathways in response to acute solar-simulated light exposure of human skin. Cancer Prev. Res. 2015, 8, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Afaq, F.; Mukhtar, H. Differential activation of signaling pathways by UVA and UVB radiation in normal human epidermal keratinocytes. Photochem. Photobiol. 2012, 88, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Vogt, P.K.; Rommel, C. PI3K: From the bench to the clinic and back. Curr. Top. Microbiol. Immunol. 2010, 347, 1–19. [Google Scholar] [PubMed]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Nakahara, T.; Takahara, M.; Kido, M.; Dugu, L.; Uchi, H.; Takeuchi, S.; Tu, Y.T.; Moroi, Y.; Furue, M. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br. J. Dermatol. 2009, 160, 442–445. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, R.F.; Akgũl, B.; Storey, A.; Pfister, H.; Harwood, C.A.; Byrne, C. Cutaneous human papillomaviruses down-regulate AKT1, whereas AKT2 up-regulation and activation associates with tumors. Cancer Res. 2007, 67, 8207–8215. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Landthaler, M.; Vogt, T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp. Dermatol. 2010, 19, e222–e227. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.W.; Lam, E.T.; Chu, C.; et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Quinn, A.G.; Sikkink, S.; Rees, J.L. Basal cell carcinomas and squamous cell carcinomas of human skin show distinct patterns of chromosome loss. Cancer Res. 1994, 54, 4756–4759. [Google Scholar] [PubMed]
- Kubo, Y.; Urano, Y.; Hida, Y.; Arase, S. Lack of somatic mutation in the PTEN gene in squamous cell carcinomas of human skin. J. Dermatol. Sci. 1999, 19, 199–201. [Google Scholar] [CrossRef]
- Murao, K.; Kubo, Y.; Ohtani, N.; Hara, E.; Arase, S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: Frequent inactivation of the RB1/p16 and p53 pathways. Br. J. Dermatol. 2006, 155, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Hertzler-Schaefer, K.; Mathew, G.; Somani, A.K.; Tholpady, S.; Kadakia, M.P.; Chen, Y.; Spandau, D.F.; Zhang, X. Pten loss induces autocrine FGF signaling to promote skin tumorigenesis. Cell Rep. 2014, 6, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Marshall, C.; Griffin, T.; Hanson, M.; Hick, R.; Dentchev, T.; Williams, E.; Werth, A.; Miller, C.; et al. Srcasm inhibits Fyn-induced cutaneous carcinogenesis with modulation of Notch1 and p53. Cancer Res. 2009, 69, 9439–9447. [Google Scholar] [CrossRef] [PubMed]
- Waterman, E.A.; Sakai, N.; Nguyen, N.T.; Horst, B.A.; Veitch, D.P.; Dey, C.N.; Ortiz-Urda, S.; Khavari, P.A.; Marinkovich, M.P. A laminin-collagen complex drives human epidermal carcinogenesis through phosphoinositol-3-kinase activation. Cancer Res. 2007, 67, 4264–4270. [Google Scholar] [CrossRef] [PubMed]
- Segrelles, C.; Ruiz, S.; Perez, P.; Murga, C.; Santos, M.; Budunova, I.V.; Martínez, J.; Larcher, F.; Slaga, T.J.; Gutkind, J.S.; et al. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene 2002, 21, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Wilker, E.; Lu, J.; Rho, O.; Carbajal, S.; Beltrán, L.; DiGiovanni, J. Role of PI3K/Akt signaling in insulin-like growth factor-1 (IGF-1) skin tumor promotion. Mol. Carcinog. 2005, 44, 137–145. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanni, J.; Bol, D.K.; Wilker, E.; Beltrán, L.; Carbajal, S.; Moats, S.; Ramirez, A.; Jorcano, J.; Kiguchi, K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000, 60, 1561–1570. [Google Scholar] [PubMed]
- Segrelles, C.; Lu, J.; Hammann, B.; Santos, M.; Moral, M.; Cascallana, J.L.; Lara, M.F.; Rho, O.; Carbajal, S.; Traag, J.; et al. Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res. 2007, 67, 10879–10888. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, E.S.; Cichoń, M.A.; Vyas, J.J.; Patel, N.; Ghali, L.; Cerio, R.; Storey, A.; O’Toole, E.A. Axl promotes cutaneous squamous cell carcinoma survival through negative regulation of pro-apoptotic Bcl-2 family members. J. Investig. Dermatol. 2011, 131, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Williams, R.; Hawkins, P. Phosphoinositide 3-kinases as drug targets in cancer. Curr. Opin. Pharmacol. 2005, 5, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M. PI3K/Akt signalling pathway specific inhibitors: A novel strategy to sensitize cancer cells to anti-cancer drugs. Curr. Pharm. Des. 2010, 16, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Bjerke, L.; Clarke, P.A.; Workman, P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [PubMed]
- Ciraolo, E.; Iezzi, M.; Marone, R.; Marengo, S.; Curcio, C.; Costa, C.; Azzolino, O.; Gonella, C.; Rubinetto, C.; Wu, H.; et al. Phosphoinositide 3-kinase p110beta activity: Key role in metabolism and mammary gland cancer but not development. Sci. Signal. 2008, 1, ra3. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [PubMed]
- Torbett, N.E.; Luna-Moran, A.; Knight, Z.A.; Houk, A.; Moasser, M.; Weiss, W.; Shokat, K.M.; Stokoe, D. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem. J. 2008, 415, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Youn, H.; Tang, J.; Tawfik, O.; Dennis, K.; Terranova, P.F.; Du, J.; Raynal, P.; Thrasher, J.B.; Li, B. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene 2008, 27, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dodhia, S.; Su, G.H. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef] [PubMed]
- Balagula, Y.; Kang, S.; Patel, M.J. Synergism between mTOR pathway and ultraviolet radiation in the pathogenesis of squamous cell carcinoma and its implication for solid-organ transplant recipients. Photodermatol. Photoimmunol. Photomed. 2015, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, F.R.; Koehl, G.E.; Voskamp, P.; Strik, A.; Rebel, H.G.; Gaumann, A.; de Fijter, J.W.; Tensen, C.P.; Bavinck, J.N.; Geissler, E.K. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int. J. Cancer 2010, 127, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Wulff, B.C.; Kusewitt, D.F.; VanBuskirk, A.M.; Thomas-Ahner, J.M.; Duncan, F.J.; Oberyszyn, T.M. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J. Investig. Dermatol. 2008, 128, 2467–2473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rho, O.; Kiguchi, K.; Jiang, G.; DiGiovanni, J. Impact of mTORC1 inhibition on keratinocyte proliferation during skin tumor promotion in wild-type and BK5.AktWT mice. Mol. Carcinog. 2014, 53, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Amornphimoltham, P.; Leelahavanichkul, K.; Molinolo, A.; Patel, V.; Gutkind, J.S. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin. Cancer Res. 2008, 14, 8094–8101. [Google Scholar] [CrossRef] [PubMed]
- Geissler, E.K. Skin cancer in solid organ transplant recipients: Are mTOR inhibitors a game changer? Transpl. Res. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, R.; Downing, C.; Tyring, S.K. Cutaneous squamous cell carcinomas in organ transplant recipients. J. Clin. Med. 2015, 4, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Overgaard, N.H.; Burke, M.T.; Isbel, N.; Frazer, I.H.; Simpson, F.; Wells, J.W. Does the nature of residual immune function explain the differential risk of non-melanoma skin cancer development in immunosuppressed organ transplant recipients? Int. J. Cancer 2016, 138, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, H.M.; Cherikh, W.S.; Cheng, Y.; Hanto, D.W.; Kahan, B.D. Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation 2005, 80, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, S.; Morelon, E.; Rostaing, L.; Goffin, E.; Brocard, A.; Tromme, I.; Broeders, N.; del Marmol, V.; Chatelet, V.; Dompmartin, A.; et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N. Engl. J. Med. 2012, 367, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.B.; Walker, R.; Tai, S.S.; Jian, Q.; Russ, G.R. Randomized controlled trial of sirolimus for renal transplant recipients at high risk of melanoma skin cancer. Am. J. Transpl. 2012, 12, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk-van den Akker, J.M.; Harden, P.N.; Hoitsma, A.J.; Proby, C.M.; Wolterbeek, R.; Bouwes Bavinck, J.N.; de Fijter, J.W. Two-year randomized controlled prospective trial converting treatment of stable renal transplant recipients with cutaneous invasive squamous cell carcinomas to sirolimus. J. Clin. Oncol. 2013, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Alberú, J.; Pascoe, M.D.; Campistol, J.M.; Schena, F.P.; Rial Mdel, C.; Polinsky, M.; Neylan, J.F.; Korth-Bradley, J.; Goldberg-Alberts, R.; Maller, E.S.; et al. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24 month results from the CONVERT trial. Transplantation 2011, 92, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Myers, A.P.; Cantley, L.C. What a tangled web we weave: Emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013, 3, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Mavrommati, I.; Maffucci, T. mTOR inhibitors: Facing new challenges ahead. Curr. Med. Chem. 2011, 18, 2743–2762. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janus, J.M.; O’Shaughnessy, R.F.L.; Harwood, C.A.; Maffucci, T. Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers 2017, 9, 86. https://doi.org/10.3390/cancers9070086
Janus JM, O’Shaughnessy RFL, Harwood CA, Maffucci T. Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers. 2017; 9(7):86. https://doi.org/10.3390/cancers9070086
Chicago/Turabian StyleJanus, Joanna M., Ryan F. L. O’Shaughnessy, Catherine A. Harwood, and Tania Maffucci. 2017. "Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas" Cancers 9, no. 7: 86. https://doi.org/10.3390/cancers9070086
APA StyleJanus, J. M., O’Shaughnessy, R. F. L., Harwood, C. A., & Maffucci, T. (2017). Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers, 9(7), 86. https://doi.org/10.3390/cancers9070086