
Epithelial-to-Pericyte Transition in Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Overview of Epithelial-to-Mesenchymal Transition (EMT) in Cancer
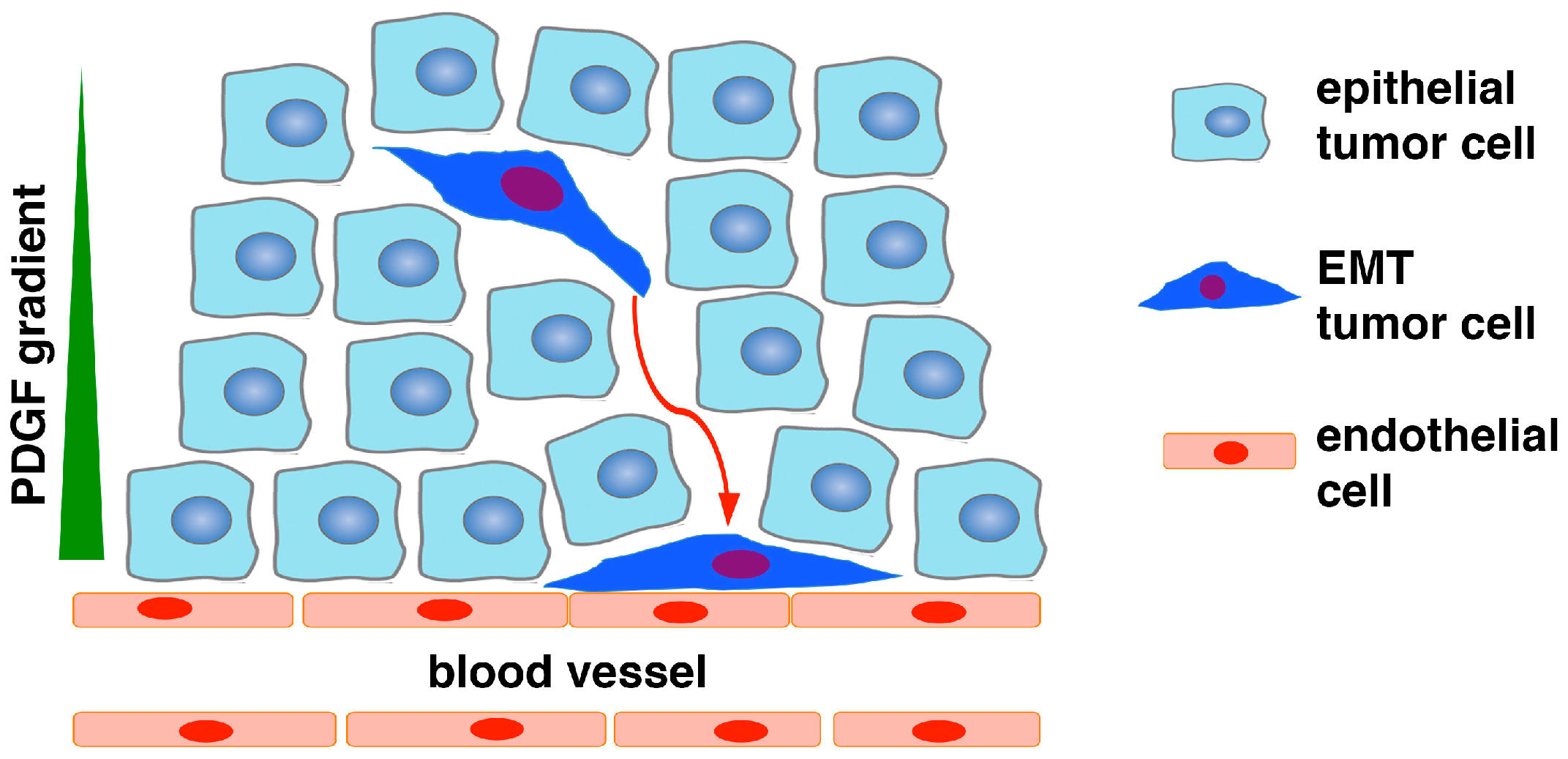
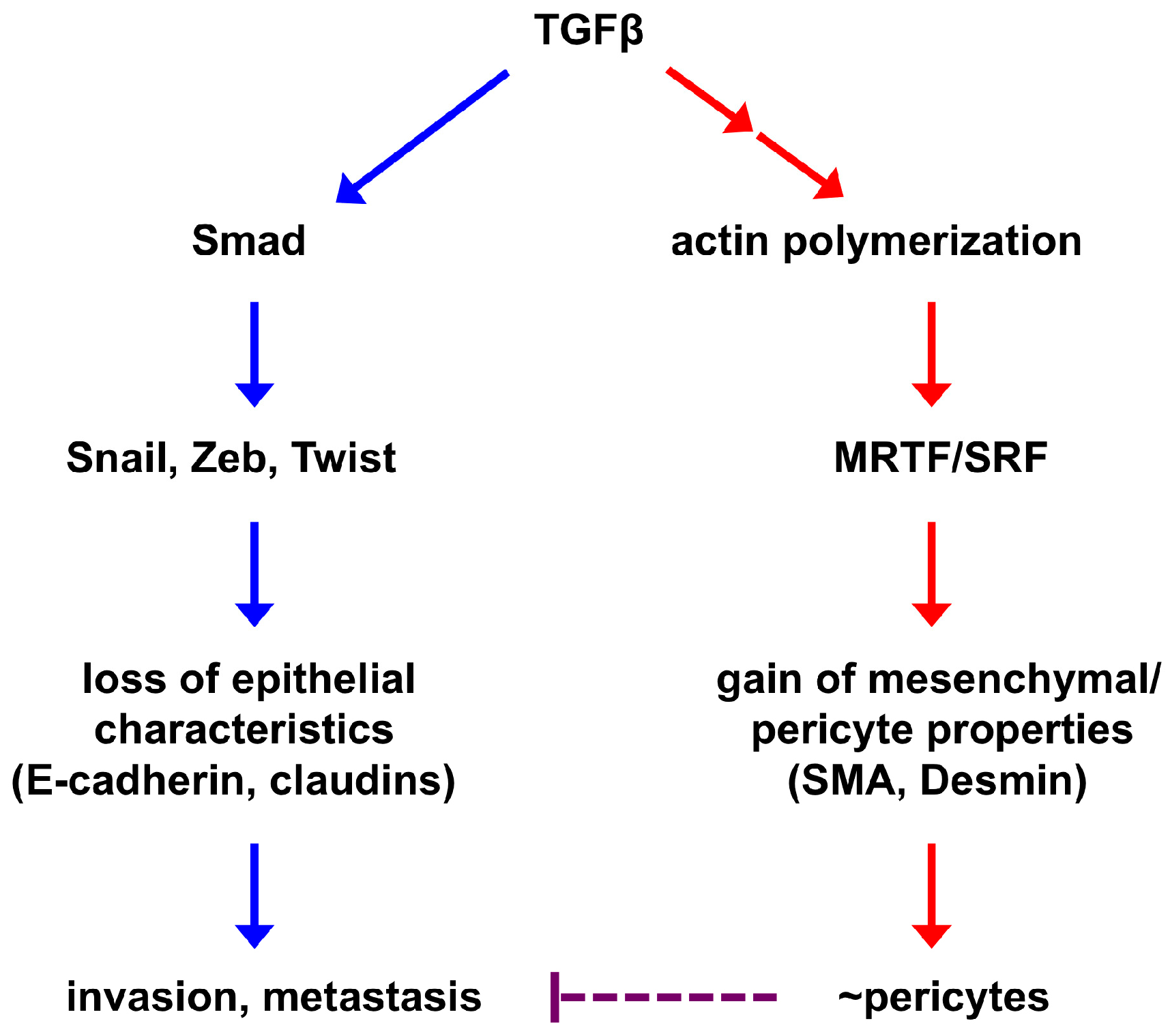
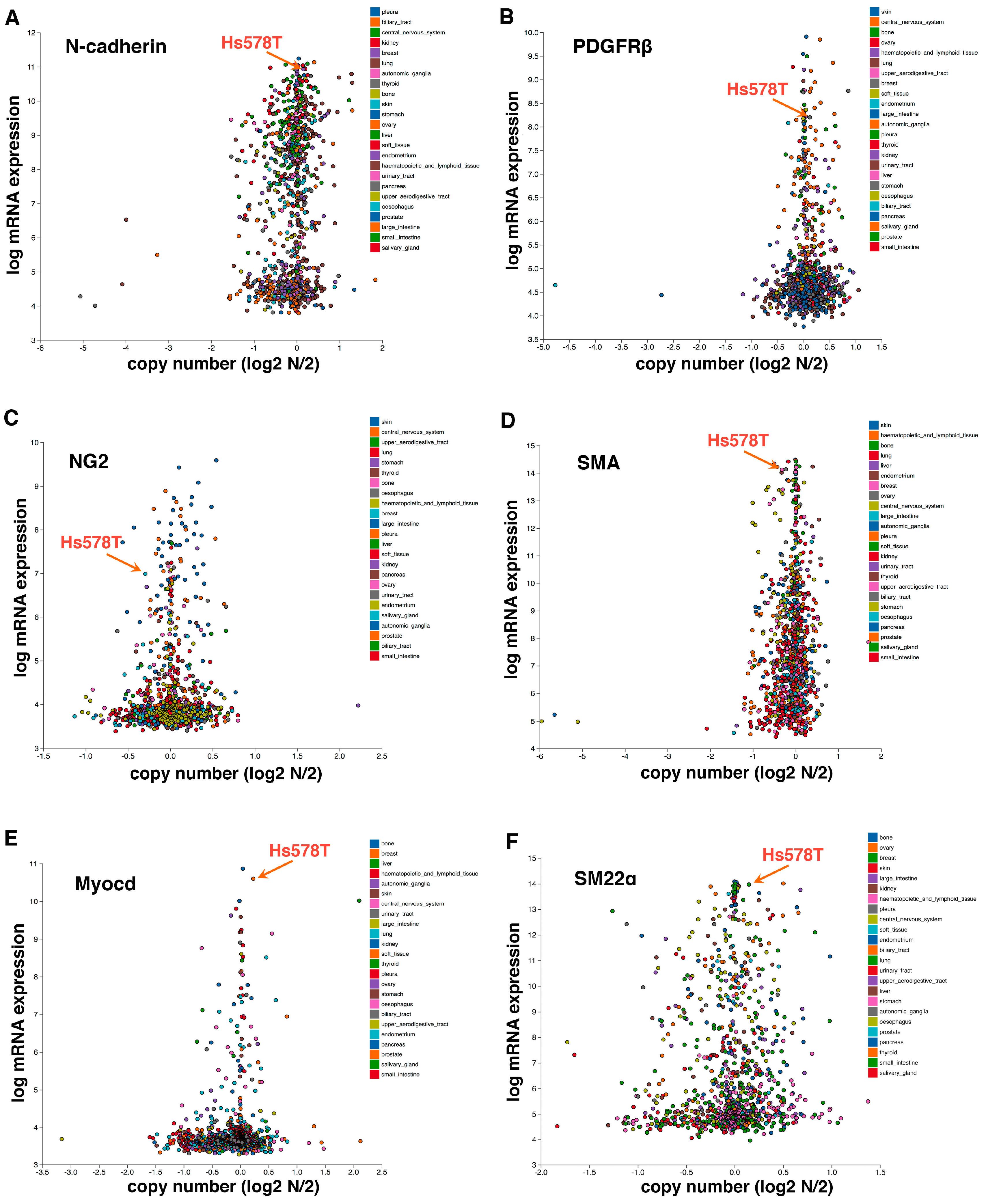
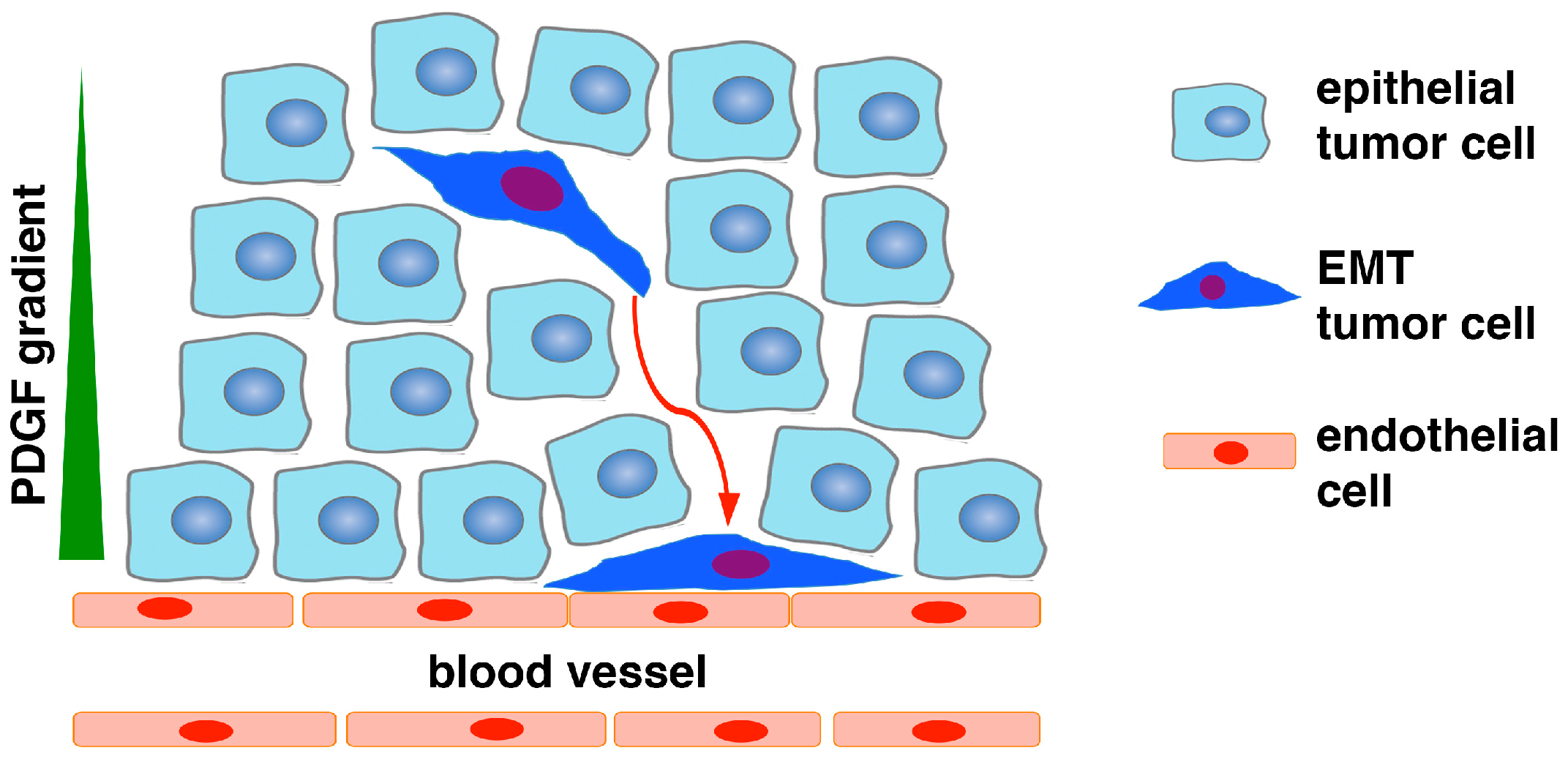
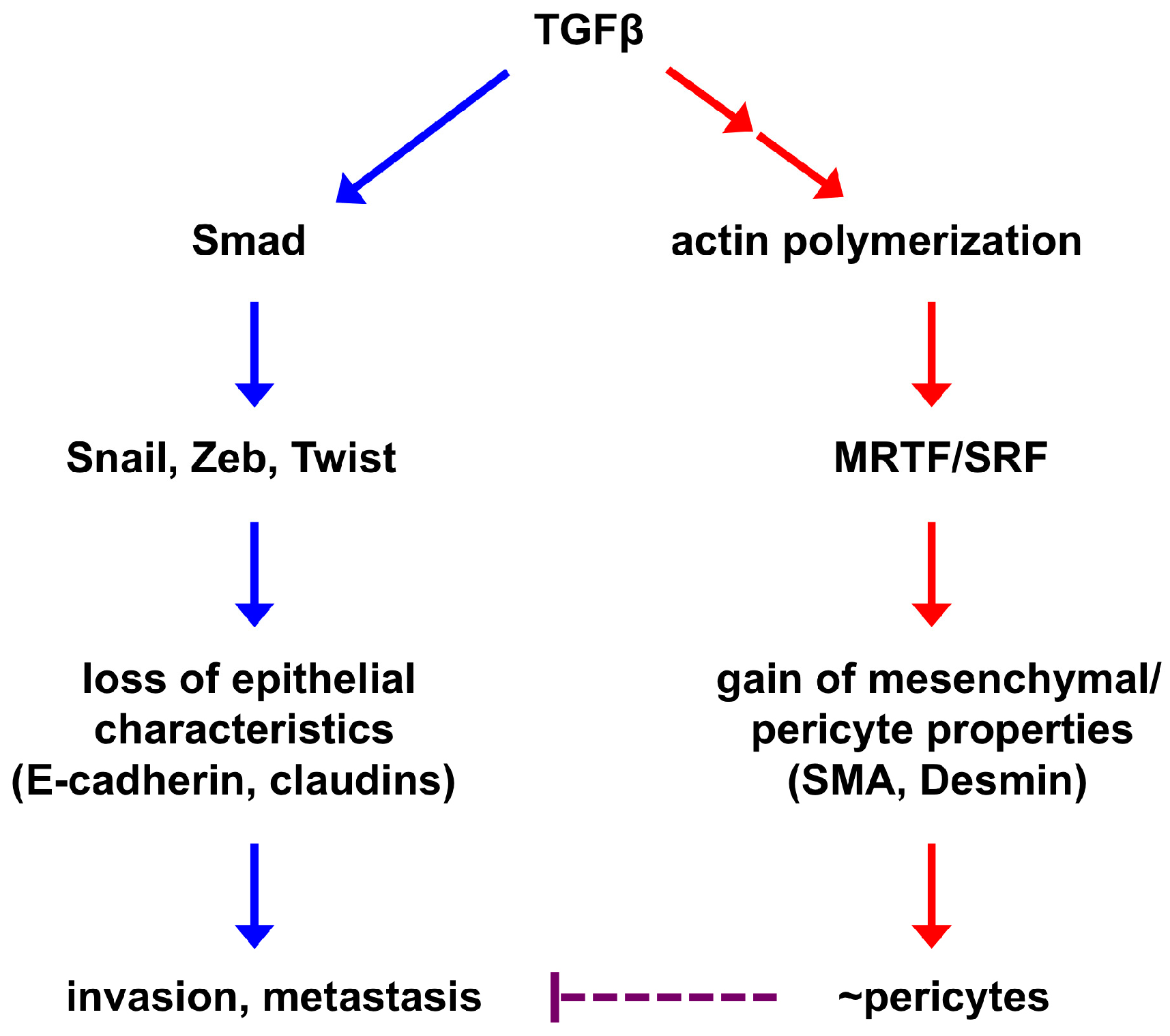
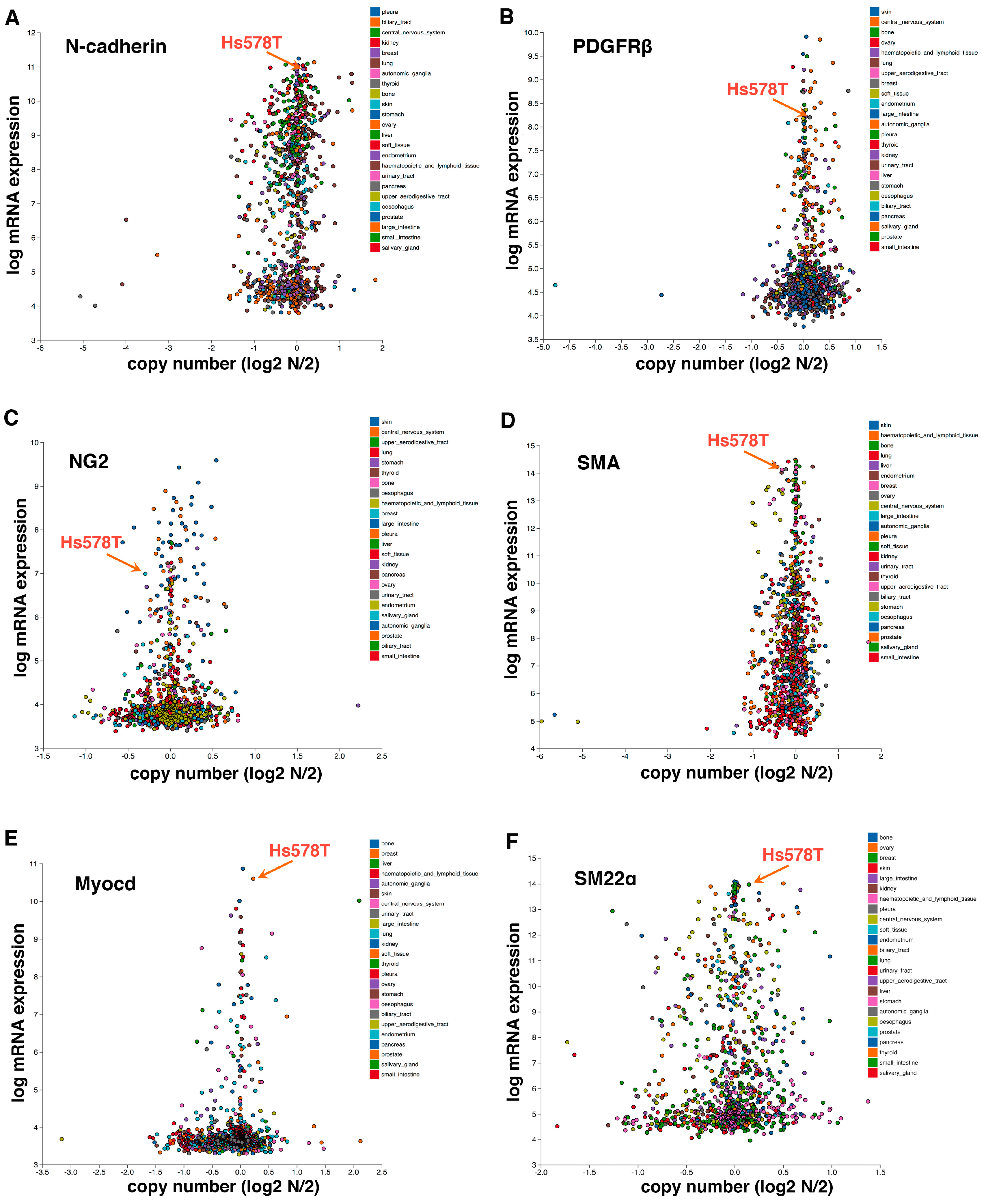
2. Epithelial-to-Pericyte Transition (EPT)
3. SRF as a Potential Key Regulator of EPT
4. EPT in Development and Cancer
5. Prospective Significance of EPT in Cancer
5.1. EPT in Tumor Vascularization and Growth
5.2. EPT in Resistance to Anti-Angiogenesis Therapy
5.3. EPT in Resistance to Chemotherapy and Targeted Therapy
6. Conclusions
Conflicts of Interest
References
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell. Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.T.; Li, F.H.; Wong, S.T.C.; Choi, H.; Rayes, T.E.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.K.; Jin, Y.; Luo, H.C.; Tang, M.; Pampo, C.; Shao, R.; Siemann, D.W.; Wu, L.Z.; Heldermon, C.D.; Law, B.K.; et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J. Clin. Investig. 2016, 126, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Ozerdem, U.; Grako, K.A.; Dahlin-Huppe, K.; Monosov, E.; Stallcup, W.B. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev. Dyn. 2001, 222, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Ozerdem, U.; Stallcup, W.B. Pathological angiogenesis is reduced by targeting pericytes via the NG2 proteoglycan. Angiogenesis 2004, 7, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.J.; Youa, W.K.; Bonaldob, P.; Seyfriedc, T.N.; Pasqualea, E.B.; Stallcupa, W.B. Pericyte deficiencies lead to aberrant tumor vascularizaton in the brain of the NG2 null mouse. Dev. Biol. 2010, 344, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, L.; Gutierrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Diaz-Flores, L., Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yu, L.; Wang, Y.L.; Wang, J.; Yang, G.; Meng, F.W.; Han, H.; Meng, A.; Wang, Y.P.; Yang, X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 2011, 20, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Wolburg, H.; Redies, C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev. Dyn. 2000, 218, 472–479. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [PubMed]
- Jahn, S.C.; Law, M.E.; Corsino, P.E.; Parker, N.N.; Pham, K.; Davis, B.J.; Lu, J.R.; Law, B.K. An in vivo model of epithelial to mesenchymal transition reveals a mitogenic switch. Cancer Lett. 2012, 326, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.I.; Moorehead, R.A. Mammary tumors that become independent of the type I insulin-like growth factor receptor express elevated levels of platelet-derived growth factor receptors. BMC Cancer 2011, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Jechlinger, M.; Grunert, S.; Tamir, I.H.; Janda, E.; Ludemann, S.; Waerner, T.; Seither, P.; Weith, A.; Beug, H.; Kraut, N. Expression profiling of epithelial plasticity in tumor progression. Oncogene 2003, 22, 7155–7169. [Google Scholar] [CrossRef] [PubMed]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Steller, E.J.; Raats, D.A.; Koster, J.; Rutten, B.; Govaert, K.M.; Emmink, B.L.; Snoeren, N.; van Hooff, S.R.; Holstege, F.C.; Maas, C.; et al. PDGFRB promotes liver metastasis formation of mesenchymal-like colorectal tumor cells. Neoplasia 2013, 15, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 25, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Lee, P.; Kocieniewski, P.; Azhar, M.; Hosokawa, R.; Chai, Y.; Fuchs, E. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS Biol. 2005, 3, e11. [Google Scholar]
- Du, F.; Nakamura, Y.; Tan, T.L.; Lee, P.; Lee, R.; Yu, B.; Jamoraet, C. Expression of snail in epidermal keratinocytes promotes cutaneous inflammation and hyperplasia conducive to tumor formation. Cancer Res. 2010, 70, 10080–10089. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shen, M.; Zha, Y.L.; Li, W.Y.; Wei, Y.; Blanco, M.A.; Ren, G.W.; Zhou, T.H.; Storz, P.; Wang, H.Y.; et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Shenoy, A.K.; Doernberg, S.; Chen, H.; Luo, H.C.; Shen, H.X.; Lin, T.; Tarrash, M.; Cai, Q.S.; Hu, X.; et al. FBXO11 promotes ubiquitination of the Snail family of transcription factors in cancer progression and epidermal development. Cancer Lett. 2015, 362, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Shen, H.; Jin, Y.; Lin, T.; Cai, Q.; Pinard, M.A.; Biswas, S.; Tran, Q.; Li, G.; Shenoy, A.K.; et al. The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition. J. Biol. Chem. 2013, 288, 27680–27691. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Olson, E.N. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr. Opin. Genet. Dev. 2004, 14, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Parmacek, M.S. Myocardin-related transcription factors: Critical coactivators regulating cardiovascular development and adaptation. Circ. Res. 2007, 100, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Mericskay, M.; Parlakian, A.; Porteu, A.; Dandre, F.; Bonnet, J.; Paulin, D.; Li, Z.L. An overlapping CArG/octamer element is required for regulation of desmin gene transcription in arterial smooth muscle cells. Dev. Biol. 2000, 226, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Psichari, E.; Balmain, A.; Plows, D.; Zoumpourlis, V.; Pintzas, A. High activity of serum response factor in the mesenchymal transition of epithelial tumor cells is regulated by RhoA signaling. J. Biol. Chem. 2002, 277, 29490–29495. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sebe, A.; Peterfi, Z.; Masszi, A.; Thirone, A.C.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szaszi, K.; Mucsi, I.; et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol. Biol. Cell 2007, 18, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Descot, A.; Julien, S.; Genth, H.; Posern, G. Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-MAL signalling. J. Cell Sci. 2008, 121, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, G.; Manabe, I.; Tsushima, K.; Fujiu, K.; Oishi, Y.; Imai, Y.; Maemura, K.; Miyagishi, M.; Higashi, Y.; Kondoh, H.; et al. DeltaEF1 mediates TGF-beta signaling in vascular smooth muscle cell differentiation. Dev. Cell. 2006, 11, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Trost, A.; Schroedl, F.; Lange, S.; Rivera, F.J.; Tempfer, H.; Korntner, S.; Stolt, C.C.; Wegner, M.; Bogner, B.; Kaser-Eichberger, A.; et al. Neural crest origin of retinal and choroidal pericytes. Invest. Ophthalmol. Vis. Sci. 2013, 54, 7910–7921. [Google Scholar] [CrossRef] [PubMed]
- Wilm, B.; Ipenberg, A.; Hastie, N.D.; Burch, J.B.; Bader, D.M. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 2005, 132, 5317–5328. [Google Scholar] [CrossRef] [PubMed]
- Que, J.; Wilm, B.; Hasegawa, H.; Wang, F.; Bader, D.; Hogan, B.L.M. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc. Natl. Acad. Sci. USA 2008, 105, 16626–16630. [Google Scholar] [CrossRef] [PubMed]
- Asahina, K.; Zhou, B.; Pu, W.T.; Tsukamoto, H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 2011, 53, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H.; et al. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, C.; Haroun, R.I.; Brem, H.; Tyler, B.M.; Jones, R.V.; Fernandez, P.M.; Patierno, S.R.; Kleinman, H.K.; Barnhill, R.L. Pericytic-like angiotropism of glioma and melanoma cells. Am. J. Dermatopathol. 2002, 24, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, C.; Peault, B.; Wadehra, M.; Kleinman, H.K.; Barnhill, R.L. Could pericytic mimicry represent another type of melanoma cell plasticity with embryonic properties? Pigment. Cell Melanoma. Res. 2013, 26, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Taylor, S.L.; Oh, D.S.; Schwartz, L.M. Vascular heterogeneity and targeting: The role of YKL-40 in glioblastoma vascularization. Oncotarget 2015, 6, 40507–40518. [Google Scholar] [PubMed]
- Song, N.; Huang, Y.; Shi, H.; Yuan, S.; Ding, Y.; Song, X.; Fu, Y.; Luo, Y. Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1alpha/CXCR4 axis. Cancer Res. 2009, 69, 6057–6064. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shrestha, S.; Rao, P.N.; Asatrian, G.; Scott, M.A.; Nguyen, V.; Giacomelli, P.; Soo, C.; Ting, K.; Eilber, F.C.; et al. Pericytic mimicry in well-differentiated liposarcoma/atypical lipomatous tumor. Hum. Pathol. 2016, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shrestha, S.; Yen, Y.H.; Asatrian, G.; Mravic, M.; Soo, C.; Ting, K.; Dry, S.M.; Peault, B.; James, A.W. Pericyte Antigens in Perivascular Soft Tissue Tumors. Int. J. Surg. Pathol. 2015, 23, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; de Sampaio, P.C.; Lundy, D.M.; Peng, Q.; Evans, K.W.; Sugimoto, H.; Gagea, M.; Kienast, Y.; do Amaral, N.S.; Rocha, R.M.; et al. Heterogeneous perivascular cell coverage affects breast cancer metastasis and response to chemotherapy. JCI Insight 2016, 1, e90733. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Semb, H. Pericytes: Gatekeepers in tumour cell metastasis? J. Mol. Med. (Berl.) 2008, 86, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug. Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Koul, S.; Lee, C.Y.; Zhang, Z.; Halmos, B. AXL kinase as a novel target for cancer therapy. Oncotarget 2014, 5, 9546–9563. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Corso, S.; Giordano, S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: From basic research to a clinical perspective. Cancer Discov. 2013, 3, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Behnam, A.B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Advances in understanding the leukaemia microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.L.; Chute, J.P. The vascular niche: Home for normal and malignant hematopoietic stem cells. Leukemia 2012, 26, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Laurance, S.; Lemarie, C.A.; Blostein, M.D. Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Adv. Nutr. 2012, 3, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.K.; Lu, J. Relevance of epithelial-to-pericyte transition in cancer. Mol. Cell. Oncol. 2017, 4, e1260672. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Shenoy, A.K. Epithelial-to-Pericyte Transition in Cancer. Cancers 2017, 9, 77. https://doi.org/10.3390/cancers9070077
Lu J, Shenoy AK. Epithelial-to-Pericyte Transition in Cancer. Cancers. 2017; 9(7):77. https://doi.org/10.3390/cancers9070077
Chicago/Turabian StyleLu, Jianrong, and Anitha K. Shenoy. 2017. "Epithelial-to-Pericyte Transition in Cancer" Cancers 9, no. 7: 77. https://doi.org/10.3390/cancers9070077
APA StyleLu, J., & Shenoy, A. K. (2017). Epithelial-to-Pericyte Transition in Cancer. Cancers, 9(7), 77. https://doi.org/10.3390/cancers9070077