
Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer

Abstract
:1. Head and Neck Cancer
2. Epithelial-to-Mesenchymal Transition in Cancer
3. Epithelial-to-Mesenchymal Transition in HNSCC
4. The Transcription Factor SOX2
5. The Kallikrein-Related Peptidase 6
6. EMT-Like Phenotype in Mucosal Melanoma of the Head and Neck
7. EMT-Like Phenotype in Salivary Gland Malignancies
8. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Pai, S.I.; Westra, W.H. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus—Positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Hammond, T.H.; Young, G.S.; Avon, A.L.; Ozer, E.; Schuller, D.E. Factors affecting long-term survival in patients with recurrent head and neck cancer may help define the role of post-treatment surveillance. Laryngoscope 2009, 119, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Jungbluth, A.A.; Patel, S.G.; Iversen, K.; Hoshaw-Woodard, S.; Busam, K.J. Expression and significance of cancer testis antigens in primary mucosal melanoma of the head and neck. Head Neck 2004, 26, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Thierauf, J.; Veit, J.A.; Lennerz, J.K.; Weissinger, S.E.; Affolter, A.; Döscher, J.; Bergmann, C.; Knopf, A.; Grünow, J.; Grünmüller, L.; et al. Expression of kallikrein-related peptidase 6 in primary mucosal malignant melanoma of the head and neck. Head Neck Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Thierauf, J.; Veit, J.; Döscher, J.; Theodoraki, M.-N.; Greve, J.; Hoffmann, T. Schleimhautmelanome des Kopf-Hals-Bereichs. Laryngorhinootologie 2015, 94, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Coca-Pelaz, A.; Rodrigo, J.P.; Bradley, P.J.; Vander Poorten, V.; Triantafyllou, A.; Hunt, J.L.; Strojan, P.; Rinaldo, A.; Haigentz, M.; Takes, R.P.; et al. Adenoid cystic carcinoma of the head and neck—An update. Oral Oncol. 2015, 51, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; de Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Cancer theory faces doubts. Nature 2011, 472, 273. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Derycke, L.D.M.; Bracke, M.E. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004, 48, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Seo, B.R.; Fischbach, C.; Gourdon, D. Fibronectin mechanobiology regulates tumorigenesis. Cell Mol. Bioeng. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, C.S.; van Tubergen, E.A.; Inglehart, R.C.; D’Silva, N.J. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J. Dent. Res. 2013, 92, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (Fresolimumab): A human anti-transforming growth factor-beta (tgfβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Bhowmick, N. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Borgoño, C.A.; Diamandis, E.P. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 2004, 4, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Li, S.; He, Q.; Liu, Z.; Zhao, L.; Zhao, T.; Wang, A. Deregulation of Bmi-1 is associated with enhanced migration, invasion and poor prognosis in salivary adenoid cystic carcinoma. Biochim. Biophys. Acta 2014, 1840, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Wang, W.; Hu, Z.; Shan, C.; Wang, L.; Wu, B.; Yang, Z.; Yang, X.; Lei, D. BDNF mediated TrkB activation contributes to the EMT progression and the poor prognosis in human salivary adenoid cystic carcinoma. Oral Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, J.; Chandrashekar, C.; Radhakrishnan, R. Critical biomarkers of epithelial-mesenchymal transition in the head and neck cancers. J. Cancer Res. Ther. 2014, 10, 512–518. [Google Scholar] [CrossRef]
- De Cecco, L.; Nicolau, M.; Giannoccaro, M.; Grazia Daidone, M.; Bossi, P.; Locati, L.; Licitra, L.; Canevari, S. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget 2015, 6, 9627–9642. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Brägelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Bauml, J.; Seiwert, T.Y.; Pfister, D.G.; Worden, F.; Liu, S.V.; Gilbert, J.; Saba, N.F.; Weiss, J.; Wirth, L.; Sukari, A.; et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: Results from a single-arm, phase II study. J. Clin. Oncol. 2017, 35, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Wuebben, E.L.; Rizzino, A. The dark side of SOX2: Cancer—A comprehensive overview. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Bayo, P.; Jou, A.; Stenzinger, A.; Shao, C.; Gross, M.; Jensen, A.; Grabe, N.; Mende, C.H.; Rados, P.V.; Debus, J.; et al. Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Mol. Oncol. 2015, 9, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Goding, C.R.; Pei, D.; Lu, X. Cancer: Pathological nuclear reprogramming? Nat. Rev. Cancer 2014, 14, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Prassas, I.; Eissa, A.; Poda, G.; Diamandis, E.P. Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat. Rev. Drug Discov. 2015, 14, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Krenzer, S.; Peterziel, H.; Mauch, C.; Blaber, S.I.; Blaber, M.; Angel, P.; Hess, J. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Invest. Dermatol. 2011, 131, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Pampalakis, G.; Prosnikli, E.; Agalioti, T.; Vlahou, A.; Zoumpourlis, V.; Sotiropoulou, G. A tumor-protective role for human kallikrein-related peptidase 6 in breast cancer mediated by inhibition of epithelial-to-mesenchymal transition. Cancer Res. 2009, 69, 3779–3787. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.; Kolb, M.; Zaoui, K.; Flechtenmacher, C.; Grabe, N.; Weber, K.-J.; Plinkert, P.K.; Heß, J. 10 Kallikrein-related peptidase 6 regulates epithelial-to-mesenchymal transition and serves as prognostic biomarker for head and neck squamous cell carcinoma patients. Oral Oncol. 2015, 51, e30. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, N.; Berx, G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 2014, 4, 352. [Google Scholar] [CrossRef] [PubMed]
- Kuphal, S.; Bosserhoff, A.K. E-cadherin cell-cell communication in melanogenesis and during development of malignant melanoma. Arch. Biochem. Biophys. 2012, 524, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Dupin, E.; Le Douarin, N.M. Development of melanocyte precursors from the vertebrate neural crest. Oncogene 2003, 22, 3016–3023. [Google Scholar] [CrossRef] [PubMed]
- Thierauf, J.; Veit, J.A.; Affolter, A.; Bergmann, C.; Grünow, J.; Laban, S.; Lennerz, J.K.; Grünmüller, L.; Mauch, C.; Plinkert, P.K.; et al. Identification and clinical relevance of PD-L1 expression in primary mucosal malignant melanoma of the head and neck. Melanoma Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Büchsenschütz, K.; Veit, J.; Schuler, P.; Thierauf, J.; Laban, S.; Fahimi, F.; Bankfalvi, A.; Lang, S.; Sauerwein, W.; Hoffmann, T. Molekulare Ansatzpunkte für systemische therapien adenoidzystischer karzinome im Kopf-Hals-Bereich. Laryngorhinootologie 2014, 93, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Phuchareon, J.; Ohta, Y.; Woo, J.M.; Eisele, D.W.; Tetsu, O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS ONE 2009, 4, e6040. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Feng, Y.; Lin, T.; Huang, X.-Y.; Gan, R.-H.; Zhao, Y.; Su, B.-H.; Ding, L.-C.; She, L.; Chen, J.; et al. CDH4 suppresses the progression of salivary adenoid cystic carcinoma via E-cadherin co-expression. Oncotarget 2016, 7, 82961–82971. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Li, B.-B.; Zhou, C.-X. Bmi-1 expression predicts prognosis in salivary adenoid cystic carcinoma and correlates with epithelial-mesenchymal transition-related factors. Ann. Diagn. Pathol. 2016, 22, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Wei, J.; Hou, R.; Wu, B.; Yang, Z.; Wang, L.; Lei, D.; Yang, X. Schwann cells promote EMT and the Schwann-like differentiation of salivary adenoid cystic carcinoma cells via the BDNF/TrkB axis. Oncol. Rep. 2015, 35, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Tan, X.; Sun, C.; Zhou, Q. High expression of SOX2 is associated with poor prognosis in patients with salivary gland adenoid cystic carcinoma. Int. J. Mol. Sci. 2014, 15, 8393–8406. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Zheng, H.; Bagnoli, M.; Guo, Y.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Ji, P.; Chen, K.; et al. MiR-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J. Pathol. 2015, 235, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.-T.; Huang, Y.-F.; Tsai, H.-Y.; Chen, C.-C.; Chang, C.-H.; Huang, S.-C.; Hsu, K.-F.; Chou, C.-Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015, 6, 2349–2365. [Google Scholar] [CrossRef] [PubMed]
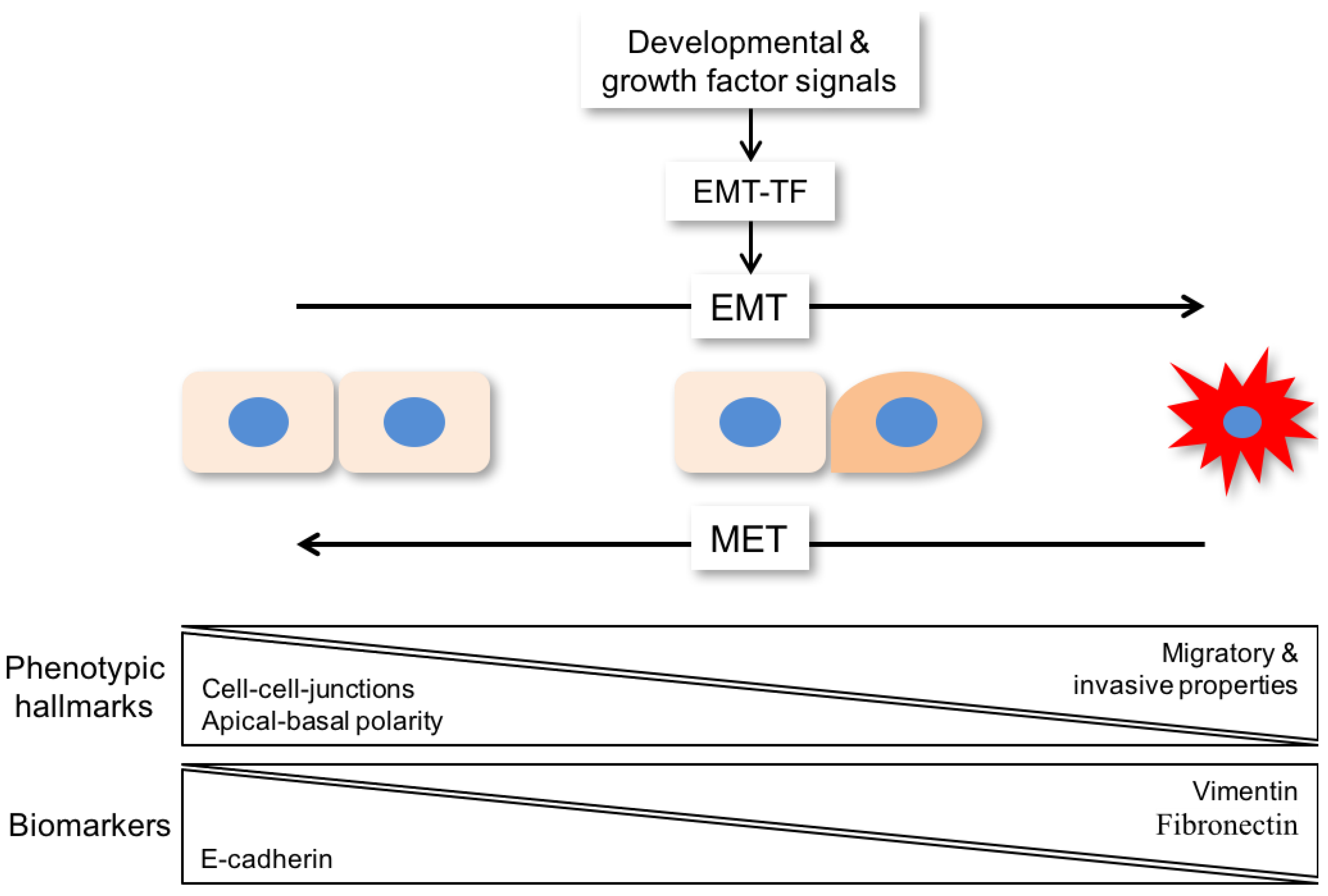

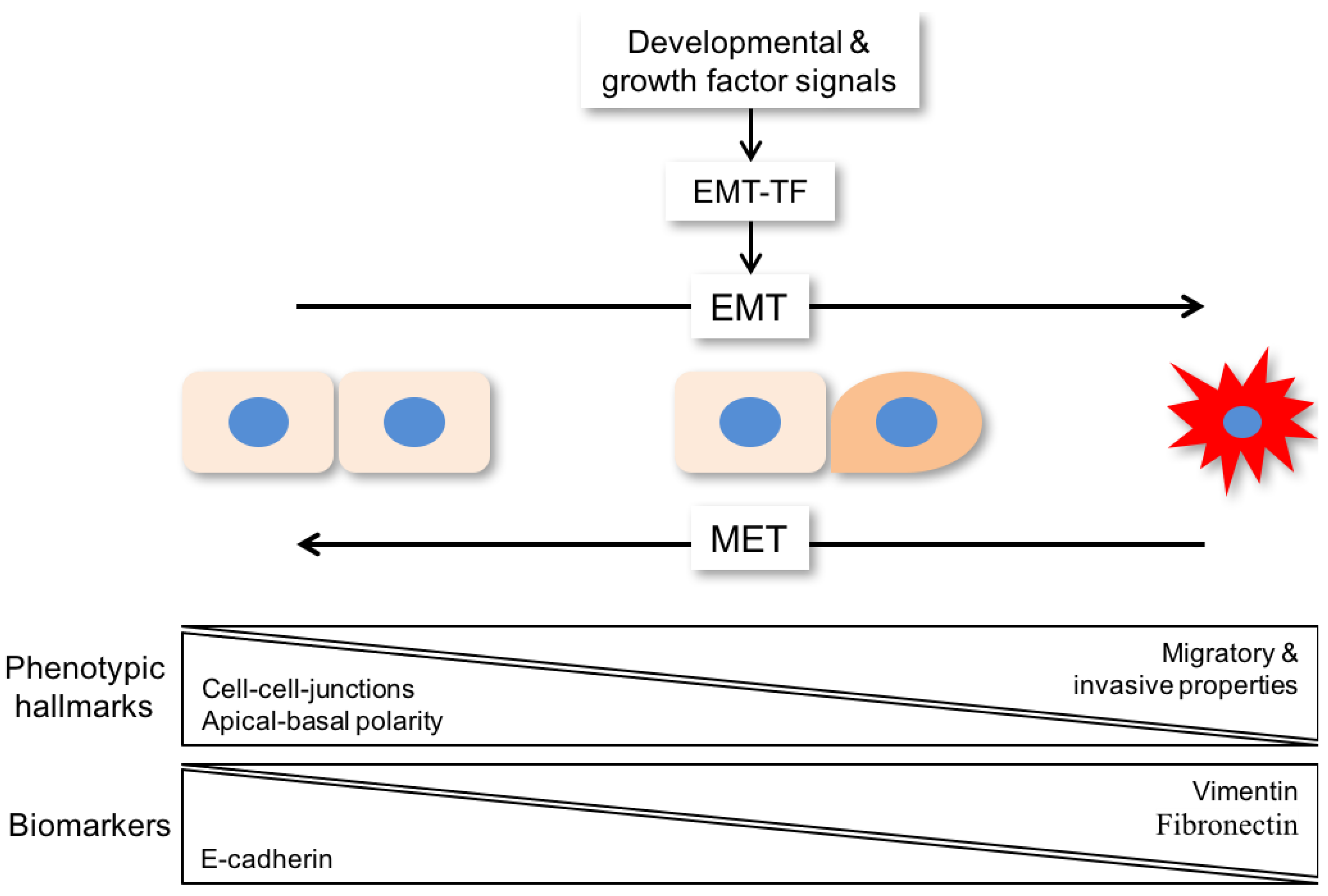{kind=link}
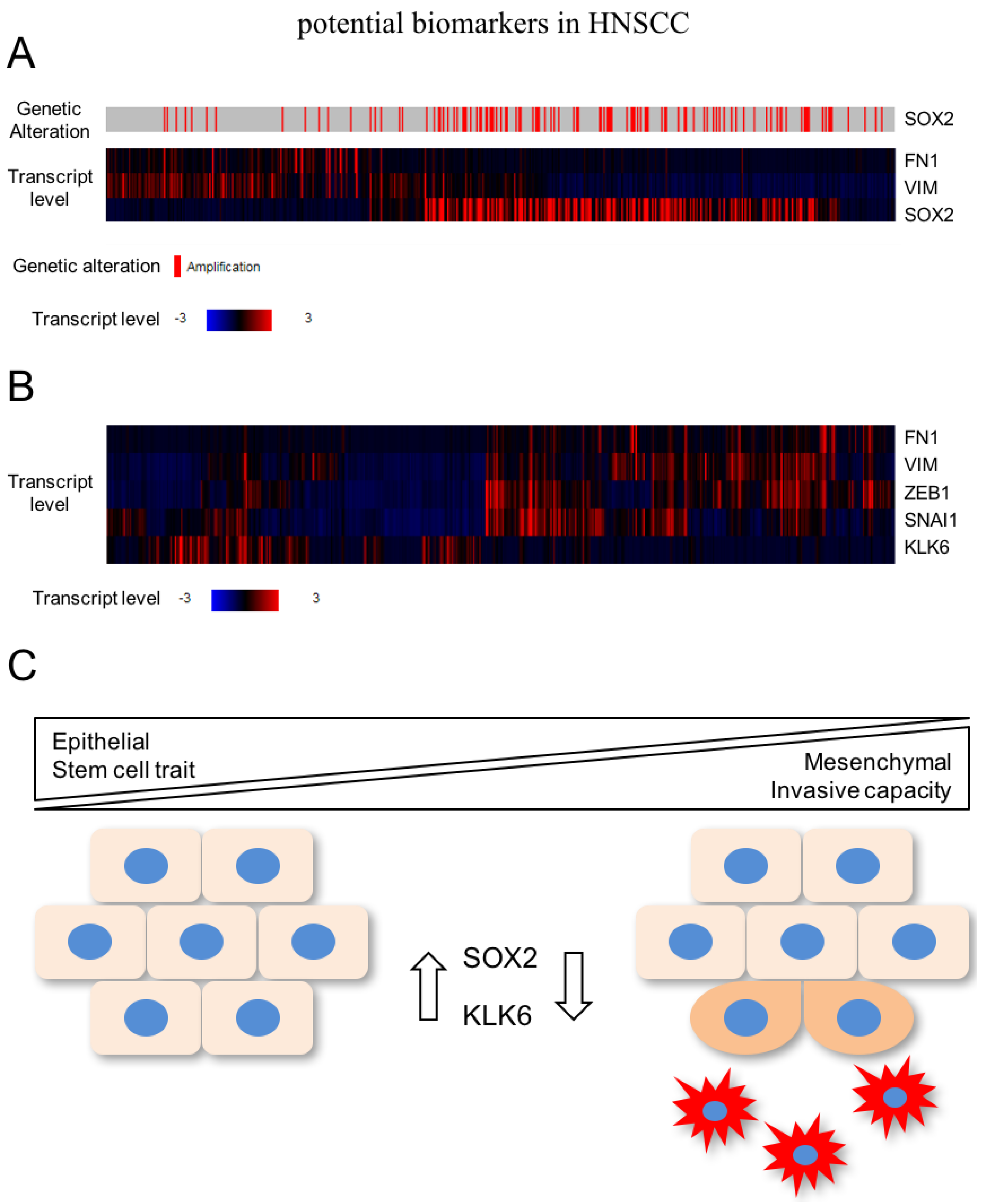
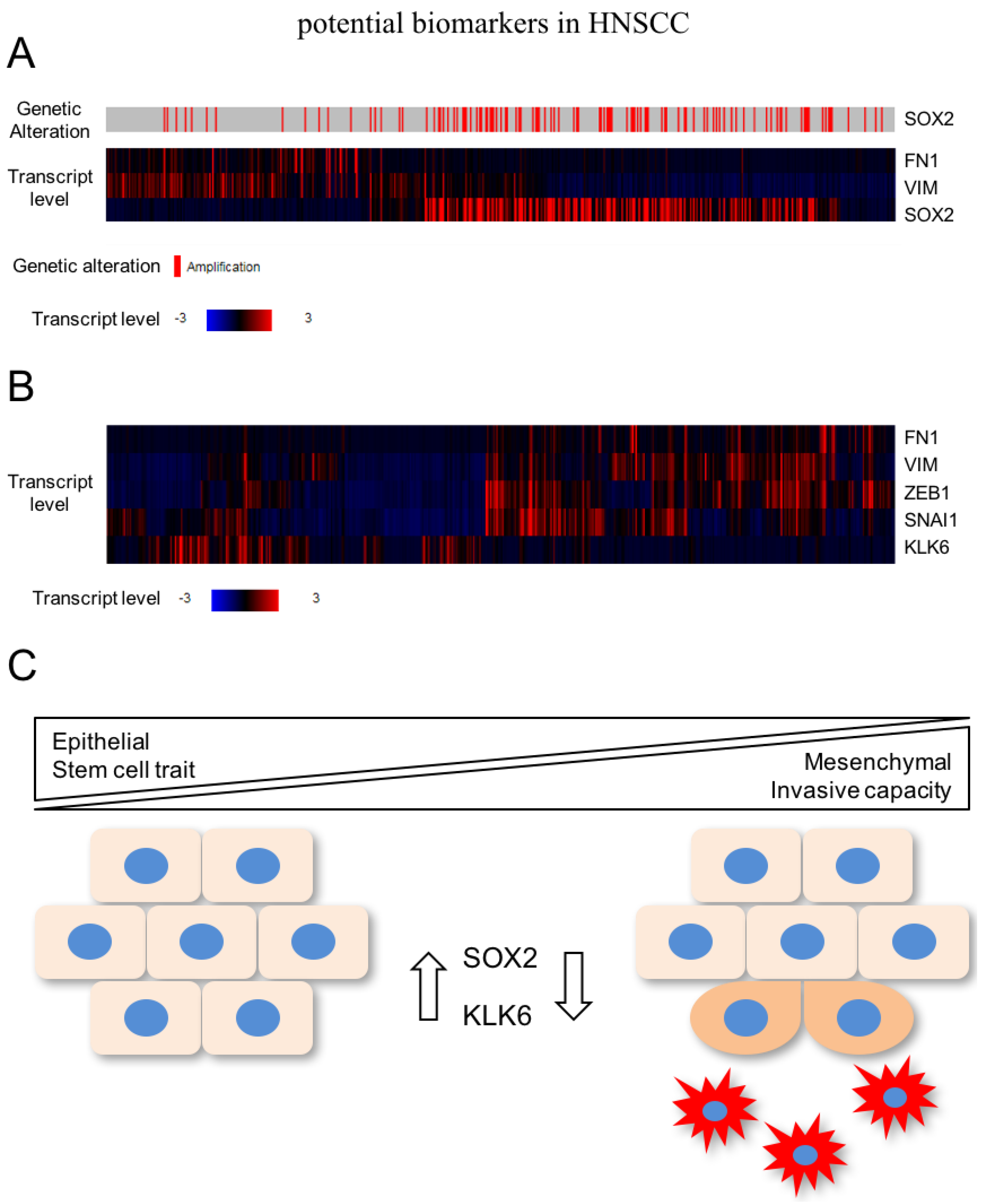{kind=link}
| Established EMT-Markers | Biological Behavior | Role in EMT/Tumorigenesis | Reference |
| Vimentin | Type III intermediate filament that is found in mesenchymal cells of various types | Marker of cells undergoing an epithelial-to-mesenchymal transition (EMT) during both normal development and metastatic progression | [16] |
| E-cadherin | Protein encoded by the CDH1 gene, also been designated as CD324, tumor suppressor gene | Loss of E-cadherin function/expression is implicated in cancer progression/metastasis, downregulation decreases the strength of cellular adhesion, resulting in an increase in cellular motility | [17] |
| N-cadherin | In embryogenesis, N-cadherin is the key molecule during gastrulation and neural crest development | Promotes tumor cell survival, migration, and invasion, and high levels are often associated with poor prognosis | [18] |
| Fibronectin | Many different cells are capable of incorporating plasma fibronectin into their extracellular matrix of any tissue | Cancer-associated fibroblasts (CAFs) are essential sources of increased extracellular matrix deposition and altered remodeling to pave the way for cancer cell invasion. | [19] |
| SNAI1/2 | Snail superfamily of zinc-finger transcription factors, involved in cell differentiation and survival. Snail1: essential for gastrulation. Snail2: embryonic development | Snail1: Common sign of poor prognosis in metastatic cancer, and tumors with elevated Snail1 expression show high rates of treatment failure | [20] |
| Snail2: Tumor metastasis promotes EMT through activation of SNAIL2 in HNSCC | |||
| TWIST1 | Helix-loop-helix transcription factor, plays an essential and pivotal role in multiple stages of embryonic development | Promotes the formation of cancer stem cells and EMT, targeting TWIST1-related molecules significantly inhibits tumor growth and thus improves the survival of cancer patients | [21] |
| ZEB1/2 | Zinc finger E-box binding homeobox 1/2, acts as transcriptional repressor | Dual role: (1) repressor for epithelial genes. (2) a transcriptional activator when associated with YAP (Hippo Pathway); also known to induce EMT in various cancers, but has also been linked to promote treatment failure in an EMT-independent manner | [22] |
| Markers Associated with EMT | Biological Behavior | Role in EMT/Tumorigenesis | Reference |
| PD-L1 and PD-1 | PD-L1: cluster of differentiation 274(CD274) or B7 homolog 1 (B7-H1), 40kDa type 1 transmembrane protein, plays a role in suppressing the immune system during pregnancy, tissue allografts, autoimmune disease, and others, ligand of programmed cell death protein-1 (PD-1). PD-1: Cell surface receptor that plays a role in promoting self-tolerance by suppressing T cell inflammatory activity | Many tumor cells express PD-L; inhibition of the interaction between PD-1 and PD-L1 can enhance T-cell responses in vitro and mediate preclinical antitumor activity. This is known as immune checkpoint blockade | [23] |
| SOX2 | Pluripotency-associated transcription factor SOX2 (sex determining region Y-box 2), essential during mammalian embryogenesis, adult tissue regeneration, and homeostasis | Identified as a lineage-survival oncogene in lung and esophageal SCC and recurrent copy number gain of chromosome 3q26, the gene locus encoding SOX2 represents a frequent alteration in HNSCC | [24] |
| KLK6 | Kallikrein-related peptidase 6 (KLK6), Family of 15 secreted serine proteases with trypsin or chymotrypsin-like activity, encoded by a cluster of genes located on chromosome 19q13.3–13.4 | Common feature for many human cancers, promising biomarker for early diagnosis or unfavorable prognosis. KLK6 can degrade components of the extracellular matrix and is implicated in tissue remodeling and induction of tumor-relevant processes such as proliferation, migration, and invasion | [25] |
| BMI1 | BMI1 (B lymphoma Mo-MLV insertion region 1 homolog) has been reported as an oncogene by regulating p16 and p19 | BMI1 deregulation is associated with enhanced migration, invasion, and poor prognosis in salivary adenoid cystic carcinoma | [26] |
| BDNF | Brain-derived neurotrophic factor (BDNF) acts on certain neurons of the central and the peripheral nervous system, helping to support the survival of existing neurons and encourage the growth and differentiation of new neurons and synapses | Elevated expression of the brain-derived neurotrophic factor (BDNF) and its receptor NTRK2 together with reduced E-cadherin expression is a common feature of salivary adenoid cystic carcinoma (ACC) and significantly correlated with invasion, metastasis, and poor prognosis of ACC | [27] |
| NTRK2 | Receptor tyrosine kinase involved in the development and maturation of the central and the peripheral nervous systems through regulation of neuron survival, proliferation, migration, differentiation, and synapse formation and plasticity | NTRK2 levels are positively correlated with expression of the EMT-related protein S100A4 but negatively associated with E-cadherin levels | [27] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thierauf, J.; Veit, J.A.; Hess, J. Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer. Cancers 2017, 9, 76. https://doi.org/10.3390/cancers9070076
Thierauf J, Veit JA, Hess J. Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer. Cancers. 2017; 9(7):76. https://doi.org/10.3390/cancers9070076
Chicago/Turabian StyleThierauf, Julia, Johannes Adrian Veit, and Jochen Hess. 2017. "Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer" Cancers 9, no. 7: 76. https://doi.org/10.3390/cancers9070076
APA StyleThierauf, J., Veit, J. A., & Hess, J. (2017). Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer. Cancers, 9(7), 76. https://doi.org/10.3390/cancers9070076