
Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression


Abstract
:1. Introduction
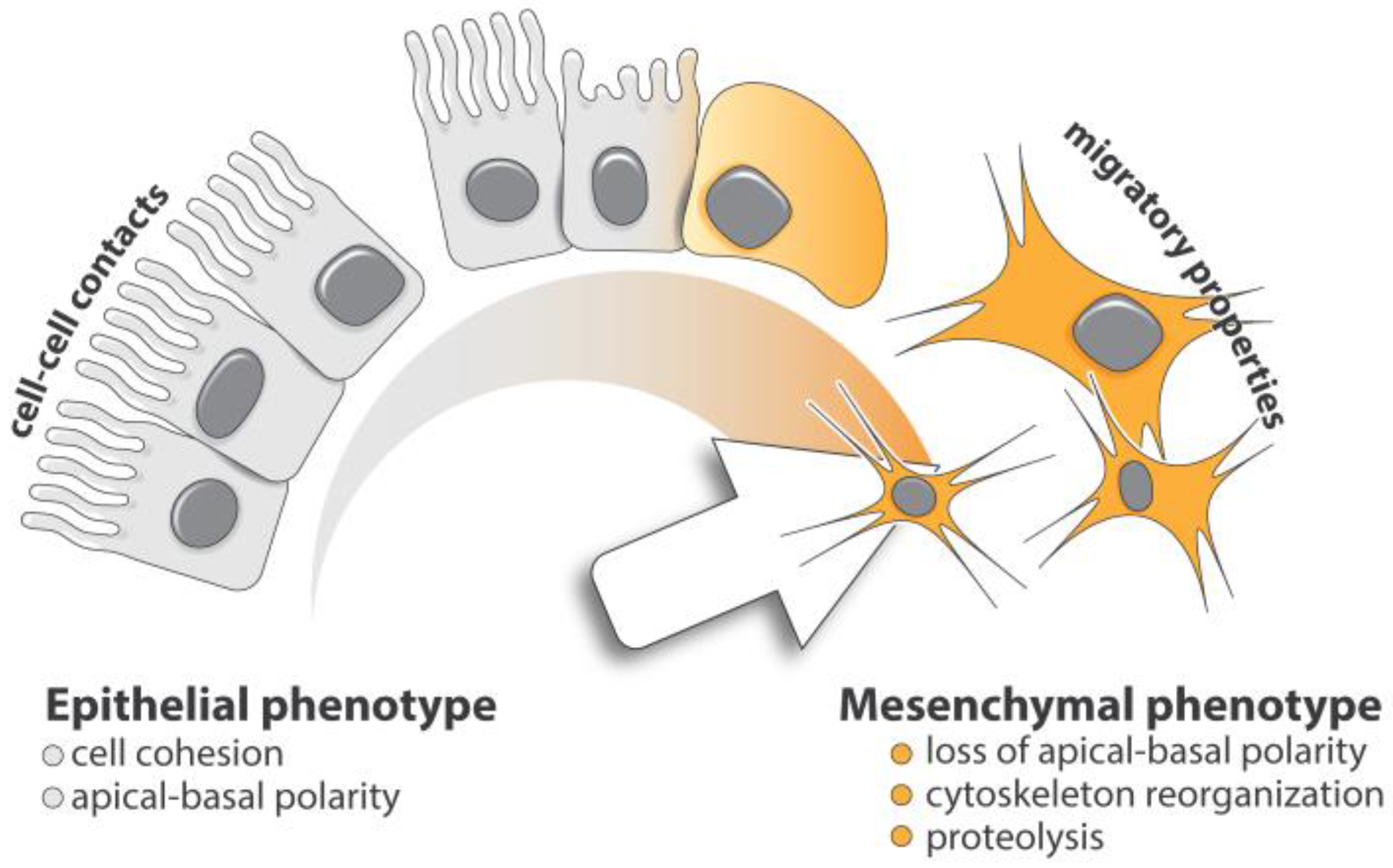
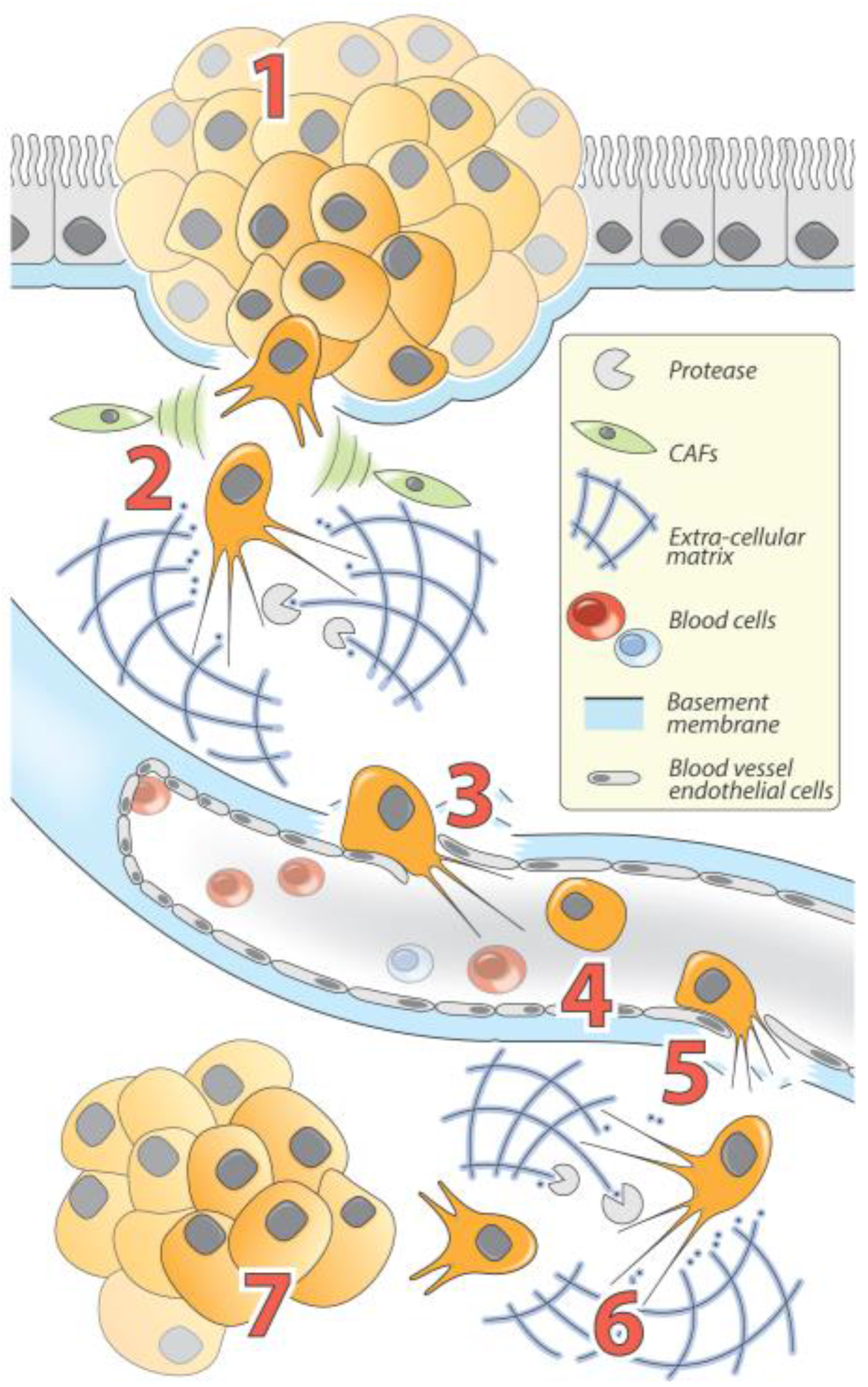
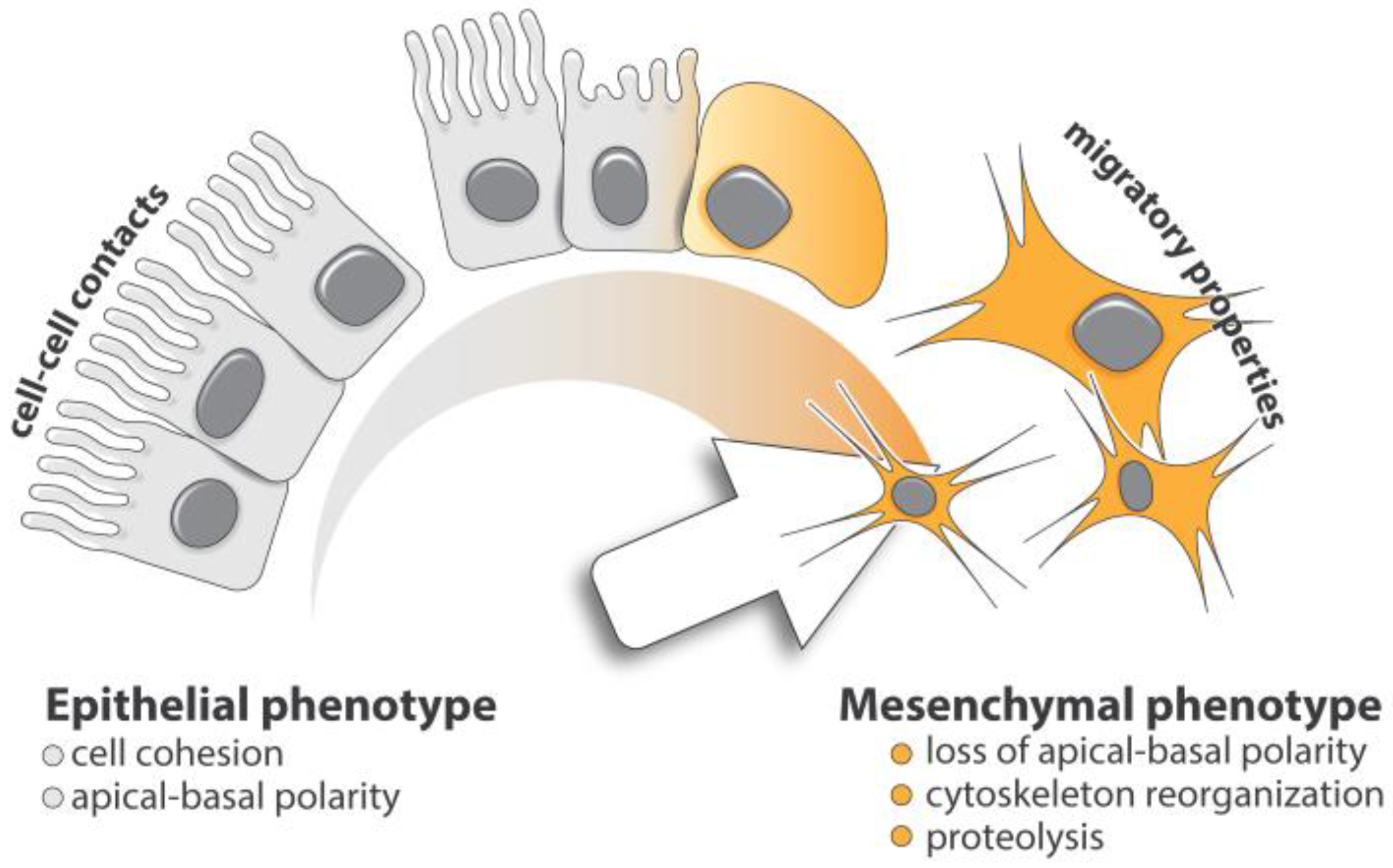
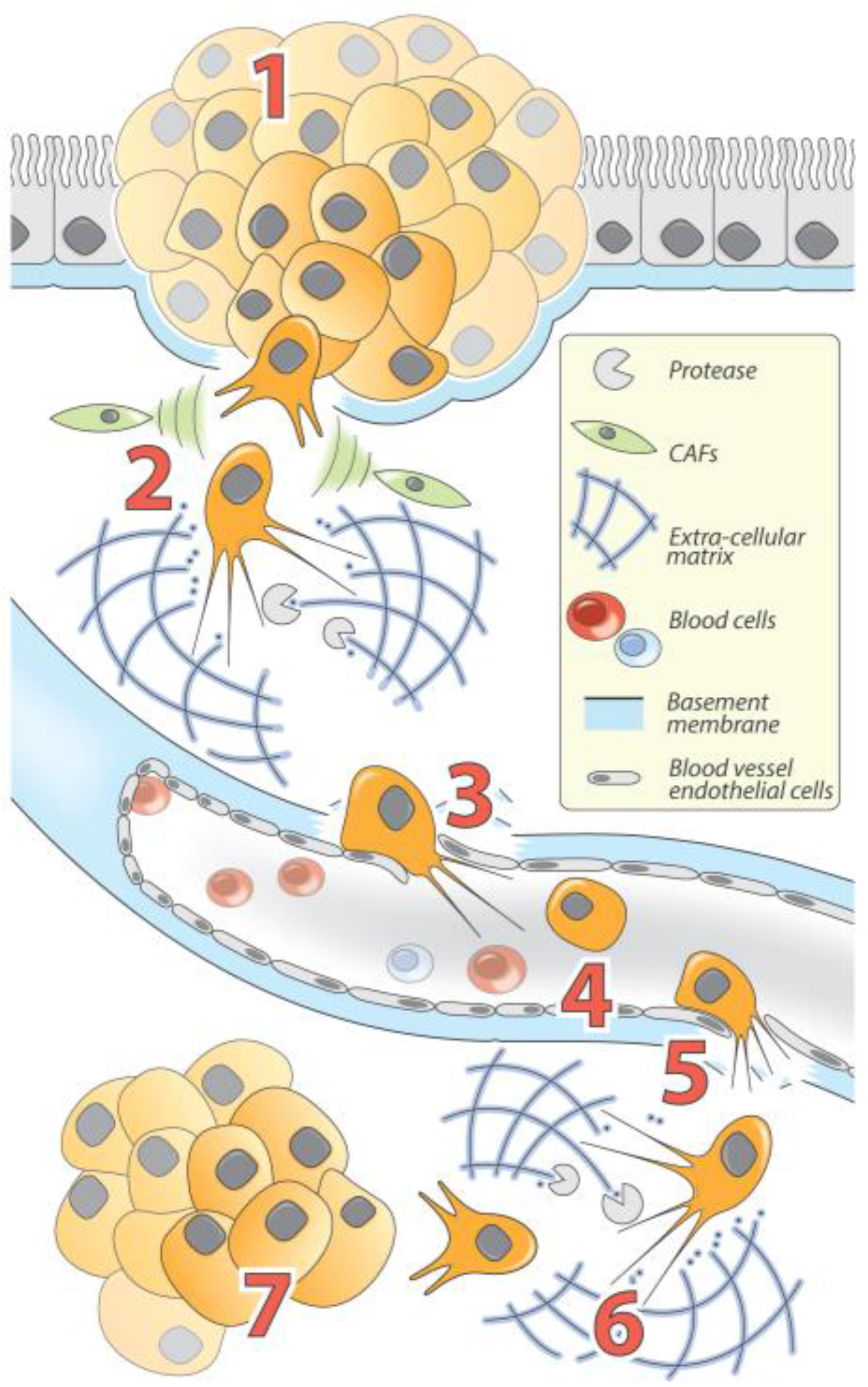
2. Cellular Basis of TGF-β-Induced EMT
3. Molecular Mechanisms of TGF-β-Induced EMT
3.1. Transcriptional Regulation of TGF-β-Induced EMT in Tumor Cells
3.2. Post-Transcriptional Regulation of TGF-β-Induced EMT in Tumor Cells
4. Role of Non-Coding RNAs in TGF-β-Induced EMT
4.1. miRNAs
4.2. Long Non-Coding RNAs
4.2.1. LncRNA-ATB
4.2.2. MALAT1
4.2.3. lncRNA-ZEB2NAT
4.2.4. HOTAIR
4.2.5. lncRNA-HIT
4.2.6. MEG3
4.3. Other Non-Coding RNA Species
4.3.1. Circular RNAs
4.3.2. PIWI-interacting RNAs
4.3.3. Small Nucleolar and Small Nuclear RNAs
4.3.4. Transfer RNAs
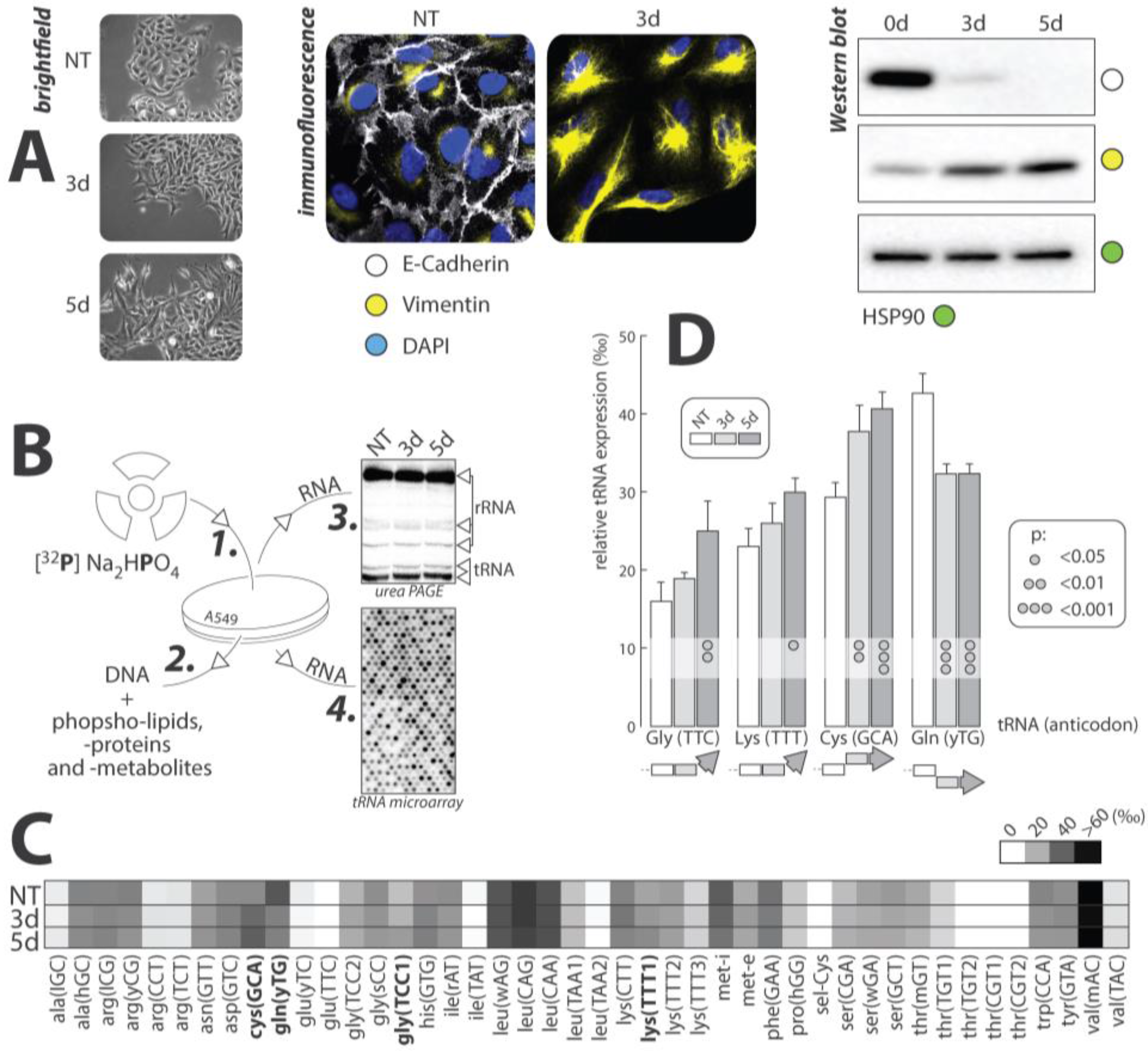
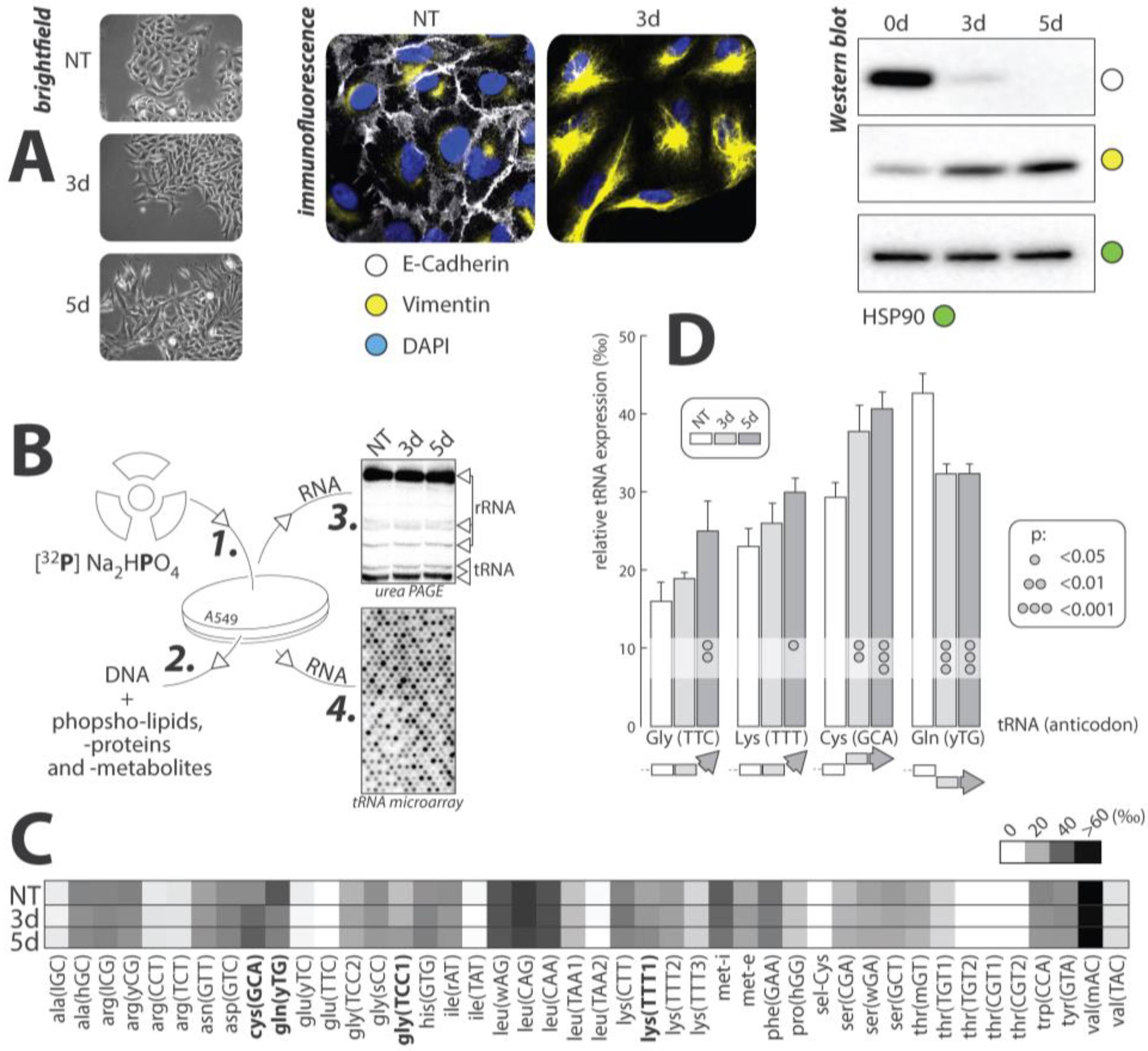
5. Evidence of Selective Regulation of tRNA Expression during TGF-β-Induced EMT
Acknowledgments
Conflicts of Interest
References
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Francart, M.-E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial-Mesenchymal Plasticity and Circulating Tumor Cells: Travel Companions to Metastases. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Schedin, P.; Borges, V. Breaking down barriers: the importance of the stromal microenvironment in acquiring invasiveness in young women’s breast cancer. Breast Cancer Res. BCR 2009, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Bourcy, M.; Suarez-Carmona, M.; Lambert, J.; Francart, M.-E.; Schroeder, H.; Delierneux, C.; Skrypek, N.; Thompson, E.W.; Jérusalem, G.; Berx, G.; et al. Tissue Factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State That Drives Metastasis of Circulating Tumor Cells. Cancer Res. 2016, 76, 4270–4282. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.-H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Wang, Y.; Zhao, G.; Xie, R.; Liu, C.; Xiao, X.; Wu, K.; Nie, Y.; Zhang, H.; et al. KLF8 involves in TGF-beta-induced EMT and promotes invasion and migration in gastric cancer cells. J. Cancer Res. Clin. Oncol. 2013, 139, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Cohen, J.R.; Fernando, R.I.; Huang, B.; Hamilton, D.H.; Palena, C. An autocrine loop between TGF-β1 and the transcription factor Brachyury controls the transition of human carcinoma cells into a mesenchymal phenotype. Mol. Cancer Ther. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Hardin, H.; Guo, Z.; Shan, W.; Montemayor-Garcia, C.; Asioli, S.; Yu, X.-M.; Harrison, A.D.; Chen, H.; Lloyd, R.V. The Roles of the Epithelial-Mesenchymal Transition Marker PRRX1 and miR-146b-5p in Papillary Thyroid Carcinoma Progression. Am. J. Pathol. 2014, 184, 2342–2354. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Fu, X.; Ran, W.; Wang, Z. Grhl2 reduces invasion and migration through inhibition of TGFβ-induced EMT in gastric cancer. Oncogenesis 2017, 6, e284. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhao, J.; Li, Y.; Li, H.; Hu, Z.; Pan, P.; Zhang, Y.; Du, E.; Liu, R.; Xu, Y. Elf5 inhibits TGF-β-driven epithelial-mesenchymal transition in prostate cancer by repressing SMAD3 activation. Prostate 2015, 75, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Nasarre, P.; Gemmill, R.; Baldys, A.; Pontis, J.; Korch, C.; Guilhot, J.; Ait-Si-Ali, S.; Drabkin, H. Global Decrease of Histone H3K27 Acetylation in ZEB1-Induced Epithelial to Mesenchymal Transition in Lung Cancer Cells. Cancers 2013, 5, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 2009, 15, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hussey, G.S.; Chaudhury, A.; Dawson, A.E.; Lindner, D.J.; Knudsen, C.R.; Wilce, M.C.J.; Merrick, W.C.; Howe, P.H. Identification of an mRNP Complex Regulating Tumorigenesis at the Translational Elongation Step. Mol. Cell 2011, 41, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Grelet, S.; Andries, V.; Polette, M.; Gilles, C.; Staes, K.; Martin, A.-P.; Kileztky, C.; Terryn, C.; Dalstein, V.; Cheng, C.-W.; et al. The human NANOS3 gene contributes to lung tumour invasion by inducing epithelial-mesenchymal transition. J. Pathol. 2015, 237, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Hussey, G.S.; Link, L.A.; Brown, A.S.; Howley, B.V.; Chaudhury, A.; Howe, P.H. Establishment of a TGFβ-Induced Post-Transcriptional EMT Gene Signature. PLOS ONE 2012, 7, e52624. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.-Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Park, S.I.; McCauley, L.K.; Wang, C.-Y. Signaling between Transforming Growth Factor β (TGF-β) and Transcription Factor SNAI2 Represses Expression of MicroRNA miR-203 to Promote Epithelial-Mesenchymal Transition and Tumor Metastasis. J. Biol. Chem. 2013, 288, 10241–10253. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Yin, J.J.; Abou-Kheir, W.; Hynes, P.G.; Casey, O.M.; Fang, L.; Yi, M.; Stephens, R.M.; Seng, V.; Sheppard-Tillman, H.; et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 2013, 32, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Drak Alsibai, K.; Nicolas, A.; Bieche, I.; Morillon, A. Long Noncoding RNAs as New Architects in Cancer Epigenetics, Prognostic Biomarkers, and Potential Therapeutic Targets. BioMed Res. Int. 2015, 2015, e320214. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yang, F.; Wang, F.; Ma, J.; Guo, Y.; Tao, Q.; Liu, F.; Pan, W.; Wang, T.; Zhou, C.; et al. A Long Noncoding RNA Activated by TGF-β Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yi, X.-M.; Tang, C.-P.; Ge, J.-P.; Zhang, Z.-Y.; Zhou, W.-Q. Long non-coding RNA ATB promotes growth and epithelial-mesenchymal transition and predicts poor prognosis in human prostate carcinoma. Oncol. Rep. 2016, 36, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, T.; Uchi, R.; Nambara, S.; Saito, T.; Komatsu, H.; Hirata, H.; Ueda, M.; Sakimura, S.; Takano, Y.; Kurashige, J.; et al. A long noncoding RNA, lncRNA-ATB, is involved in the progression and prognosis of colorectal cancer. Anticancer Res. 2015, 35, 1385–1388. [Google Scholar] [PubMed]
- Ke, L.; Xu, S.-B.; Wang, J.; Jiang, X.-L.; Xu, M.-Q. High expression of long non-coding RNA ATB indicates a poor prognosis and regulates cell proliferation and metastasis in non-small cell lung cancer. Clin. Transl. Oncol. 2017, 19, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-J.; Wang, L.-J.; Yu, B.; Li, Y.-H.; Jin, Y.; Bai, X.-Z. LncRNA-ATB promotes trastuzumab resistance and invasion-metastasis cascade in breast cancer. Oncotarget 2015, 6, 11652–11663. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Song, Y. Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Lett. 2013, 339, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Álvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial–mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous RNA binding by Polycomb Repressive Complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Endo, H.; Yokoyama, M.; Abe, J.; Tamai, K.; Tanaka, N.; Sato, I.; Takahashi, S.; Kondo, T.; Satoh, K. Large noncoding RNA HOTAIR enhances aggressive biological behavior and is associated with short disease-free survival in human non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2013, 436, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-H.; Wang, X.-L.; Tang, H.-M.; Jiang, T.; Chen, J.; Lu, S.; Qiu, G.-Q.; Peng, Z.-H.; Yan, D.-W. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol. Rep. 2014, 32, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Hajjari, M.; Khoshnevisan, A.; Shin, Y.K. Molecular function and regulation of long non-coding RNAs: paradigms with potential roles in cancer. Tumour Biol. 2014, 35, 10645–10663. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J.; Zhang, G.; Li, Z.-P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; Mao, W.-M.; et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA-hit-mediated TGFβ-induced epithelial to mesenchymal transition in mammary epithelia. J. Biol. Chem. 2015, 290, 6857–6867. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Tange, S.; Ishimura, A.; Suzuki, T. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial-mesenchymal transition in lung cancer cell lines. J. Biol. Chem. 2016, 292, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; He, D.; Peng, Z.; Peng, W.; Shi, W.; Wang, J.; Li, B.; Zhang, C.; Duan, C. Circular RNAs in cancer: an emerging key player. J. Hematol. Oncol. 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation—Exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 2015, 5, 8057. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Kjems, J.; Damgaard, C.K. Circular RNA and miR-7 in cancer. Cancer Res. 2013, 73, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, J.; Zhou, P.; Le, Y.; Zhou, C.; Wang, S.; Xu, D.; Lin, H.-K.; Gong, Z. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am. J. Cancer Res. 2015, 5, 472–480. [Google Scholar] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Wei, Q.; Toden, S.; Yoshida, K.; Nagasaka, T.; Fujiwara, T.; Cai, S.; Qin, H.; Ma, Y.; Goel, A. Circular RNA ciRS-7—A promising prognostic biomarker and a potential therapeutic target in colorectal cancer. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Anderson, C.; Marshall, E.A.; Minatel, B.C.; Enfield, K.S.S.; Saprunoff, H.L.; Lam, W.L.; Martinez, V.D. Piwi-interacting RNAs in cancer: Emerging functions and clinical utility. Mol. Cancer 2016, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Hashim, A.; Rizzo, F.; Marchese, G.; Ravo, M.; Tarallo, R.; Nassa, G.; Giurato, G.; Santamaria, G.; Cordella, A.; Cantarella, C.; et al. RNA sequencing identifies specific PIWI-interacting small non-coding RNA expression patterns in breast cancer. Oncotarget 2014, 5, 9901–9910. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Hu, H.; Xue, X.; Shen, S.; Gao, E.; Guo, G.; Shen, X.; Zhang, X. Altered expression of piRNAs and their relation with clinicopathologic features of breast cancer. Clin. Transl. Oncol. 2013, 15, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ren, Y.; Xu, H.; Pang, D.; Duan, C.; Liu, C. The expression of stem cell protein Piwil2 and piR-932 in breast cancer. Surg. Oncol. 2013, 22, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Rapisuwon, S.; Vietsch, E.E.; Wellstein, A. Circulating biomarkers to monitor cancer progression and treatment. Comput. Struct. Biotechnol. J. 2016, 14, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Xu, T.; Ganapathy, S.; Shadfan, M.; Long, M.; Huang, T.H.-M.; Thompson, I.; Yuan, Z.-M. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 2014, 33, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Lu, H.; Zhou, Q.; Luo, K. LARP7 suppresses P-TEFb activity to inhibit breast cancer progression and metastasis. Elife 2014, 3, e02907. [Google Scholar] [CrossRef] [PubMed]
- Waldron, C.; Lacroute, F. Effect of growth rate on the amounts of ribosomal and transfer ribonucleic acids in yeast. J. Bacteriol. 1975, 122, 855–865. [Google Scholar] [PubMed]
- Goodenbour, J.M.; Pan, T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006, 34, 6137–6146. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Pavon-Eternod, M.; Gomes, S.; Geslain, R.; Dai, Q.; Rosner, M.R.; Pan, T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009, 37, 7268–7280. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.M.; Schmitt, B.M.; Villar, D.; White, R.J.; Marioni, J.C.; Kutter, C.; Odom, D.T. Codon-Driven Translational Efficiency Is Stable across Diverse Mammalian Cell States. PLOS Genet. 2016, 12, e1006024. [Google Scholar] [CrossRef] [PubMed]
- Geslain, R.; Eriani, G. Regulation of translation dynamic and neoplastic conversion by tRNA and their pieces. Transl. Austin. 2014, 2, e28586. [Google Scholar] [CrossRef] [PubMed]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef] [PubMed]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sørensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; Gimotty, P.A.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Klochendler, A.; Weinberg-Corem, N.; Moran, M.; Swisa, A.; Pochet, N.; Savova, V.; Vikeså, J.; Van de Peer, Y.; Brandeis, M.; Regev, A.; et al. A Transgenic Mouse Marking Live Replicating Cells Reveals In Vivo Transcriptional Program of Proliferation. Dev. Cell 2012, 23, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.S.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA NY 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Jobava, R.; Parisien, M.; Putnam, A.; Krokowski, D.; Gao, X.-H.; Guan, B.-J.; Yuan, Y.; Jankowsky, E.; Feng, Z.; et al. Angiogenin-cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol. Cell. Biol. 2014, 34, 2450–2463. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Feng, J.; Liu, Q.; Sun, F.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009, 583, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kuscu, C.; Dutta, A. Biogenesis and Function of Transfer RNA-Related Fragments (tRFs). Trends Biochem. Sci. 2016, 41, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lin, Y.; Li, C.; Hu, X.; Liu, Y.; He, M.; Luo, J.; Sun, G.; Wang, T.; Li, W.; et al. MicroRNA-720 promotes in vitro cell migration by targeting Rab35 expression in cervical cancer cells. Cell Biosci. 2015, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Choi, P.-W.; Trachtenberg, A.J.; Ng, A.C.; Kuo, W.P.; Ng, S.-K.; Dinulescu, D.M.; Matzuk, M.M.; Berkowitz, R.S.; Ng, S.-W. Epithelialization of mouse ovarian tumor cells originating in the fallopian tube stroma. Oncotarget 2016, 7, 66077–66086. [Google Scholar] [CrossRef] [PubMed]
- Grelet, S.; McShane, A.; Hok, E.; Tomberlin, J.; Howe, P.H.; Geslain, R. SPOt: A novel and streamlined microarray platform for observing cellular tRNA levels. PLOS ONE 2017, 12, e0177939. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Non-Coding RNA | Relevant Examples | Specific Function | Most Described Targets | Related Cancers | References |
|---|---|---|---|---|---|
| miRNAs | miR-1 * miR-200 family * miR-205 * miR-203 * | Epithelial maintenance | ZEB1/2↓ Slug↓ Bmi1↓ | Breast Lung Prostate | [5,35,37,39,40] |
| LncRNAs | LncRNA-ATB † MALAT1 † lncRNA-ZEB2NAT † HOTAIR † lncRNA-HIT | Tumor cell invasion; Organ colonization; Proliferation; Cancer Stem Cells | ZEB1/2↑ IL-11↑ miR-200↓ miR-205↓ E-cadherin↓ | Prostate Lung Breast Kidney Pancreas Liver Colon Uterus | [44,45,46,47,49,51,52,53,54,55,57,58,59,61] |
| MEG3 * | TGF-β pathway regulation | TGFBR1↑ TGFB2↑ SMAD2↑ | Breast | [62,63] | |
| circRNAs | CDR1as/ciRS-7 * | miRNA sponge | miRNA-7↓ | Colon | [66,67,69] |
| piRNAs | Pir-932 † | Stemness properties | Latexin↓ | Breast | [74] |
| snoRNAs snRNAs | 7SK snRNA * | Tumor cell invasion | Slug↓ FOXC2↓ ZEB2↓ Twist1↓ | Breast | [77] |
| tRNAs | tRNAGluUUC † | Tumor progression | EXOSC2↓ GRIPAP1↓ | Breast | [80] |
| MicroRNA-720 † | Tumor cell motility | Rab35↓ | Uterus | [94] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grelet, S.; McShane, A.; Geslain, R.; Howe, P.H. Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression. Cancers 2017, 9, 75. https://doi.org/10.3390/cancers9070075
Grelet S, McShane A, Geslain R, Howe PH. Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression. Cancers. 2017; 9(7):75. https://doi.org/10.3390/cancers9070075
Chicago/Turabian StyleGrelet, Simon, Ariel McShane, Renaud Geslain, and Philip H. Howe. 2017. "Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression" Cancers 9, no. 7: 75. https://doi.org/10.3390/cancers9070075
APA StyleGrelet, S., McShane, A., Geslain, R., & Howe, P. H. (2017). Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression. Cancers, 9(7), 75. https://doi.org/10.3390/cancers9070075