
Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer

Abstract
:1. Introduction
2. Epigenetics
3. Epigenetics and Lung Cancer
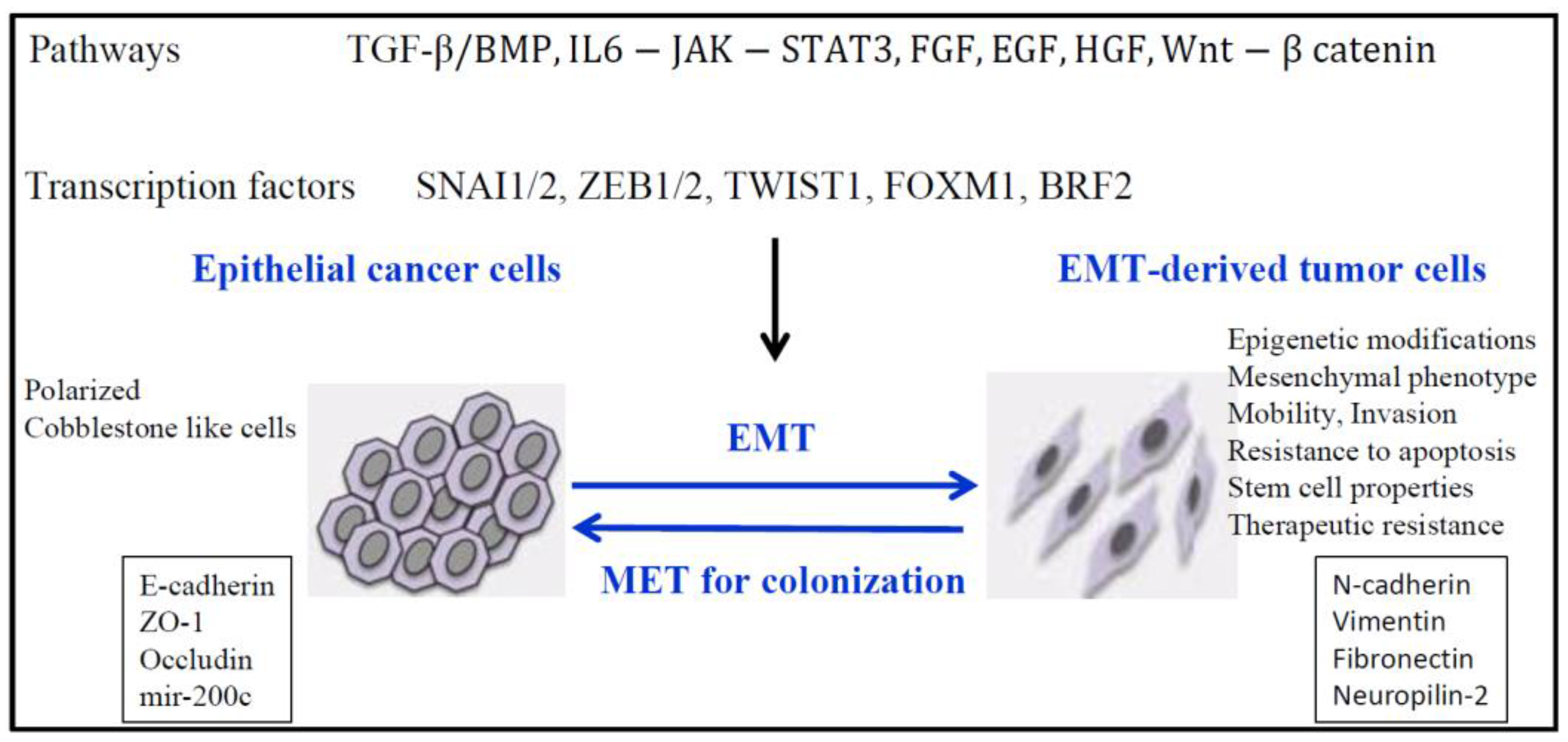
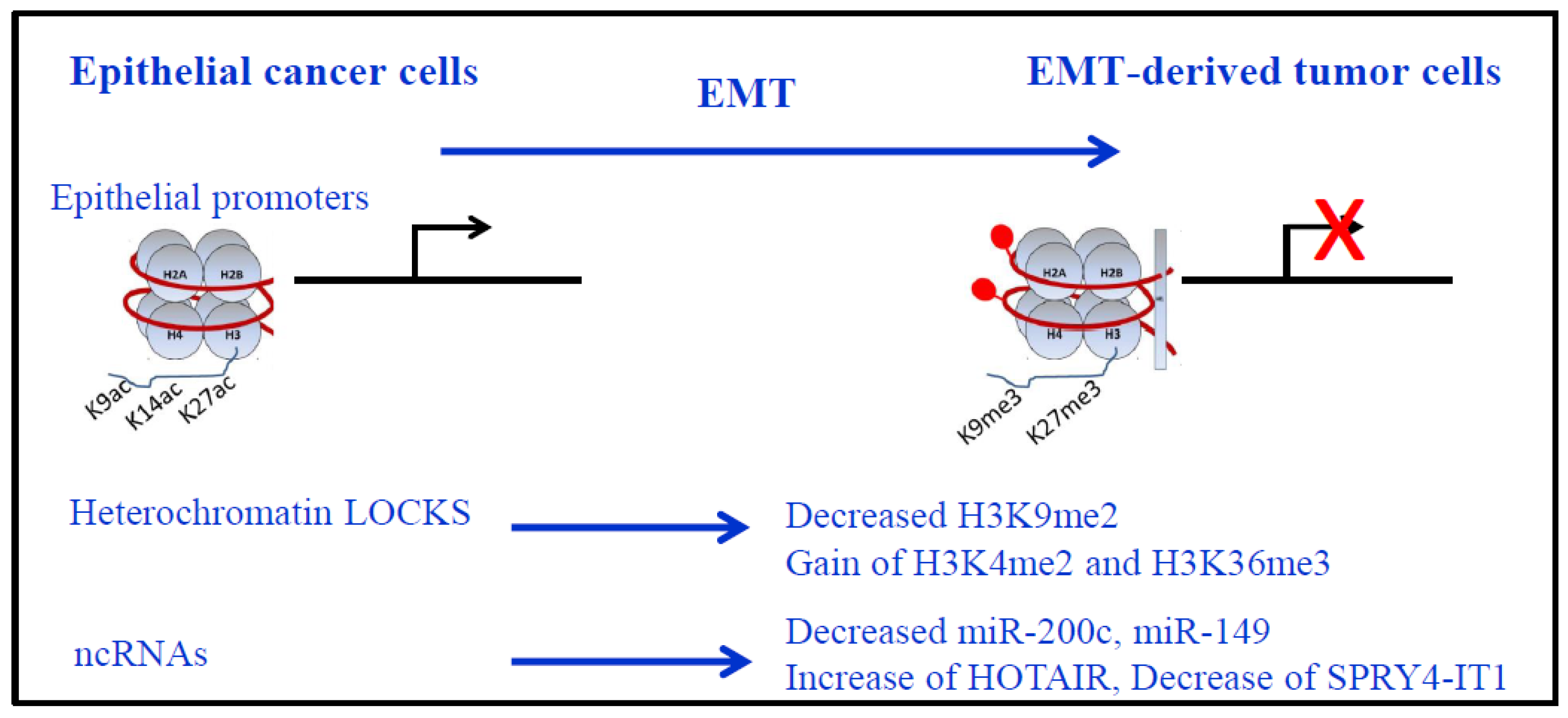
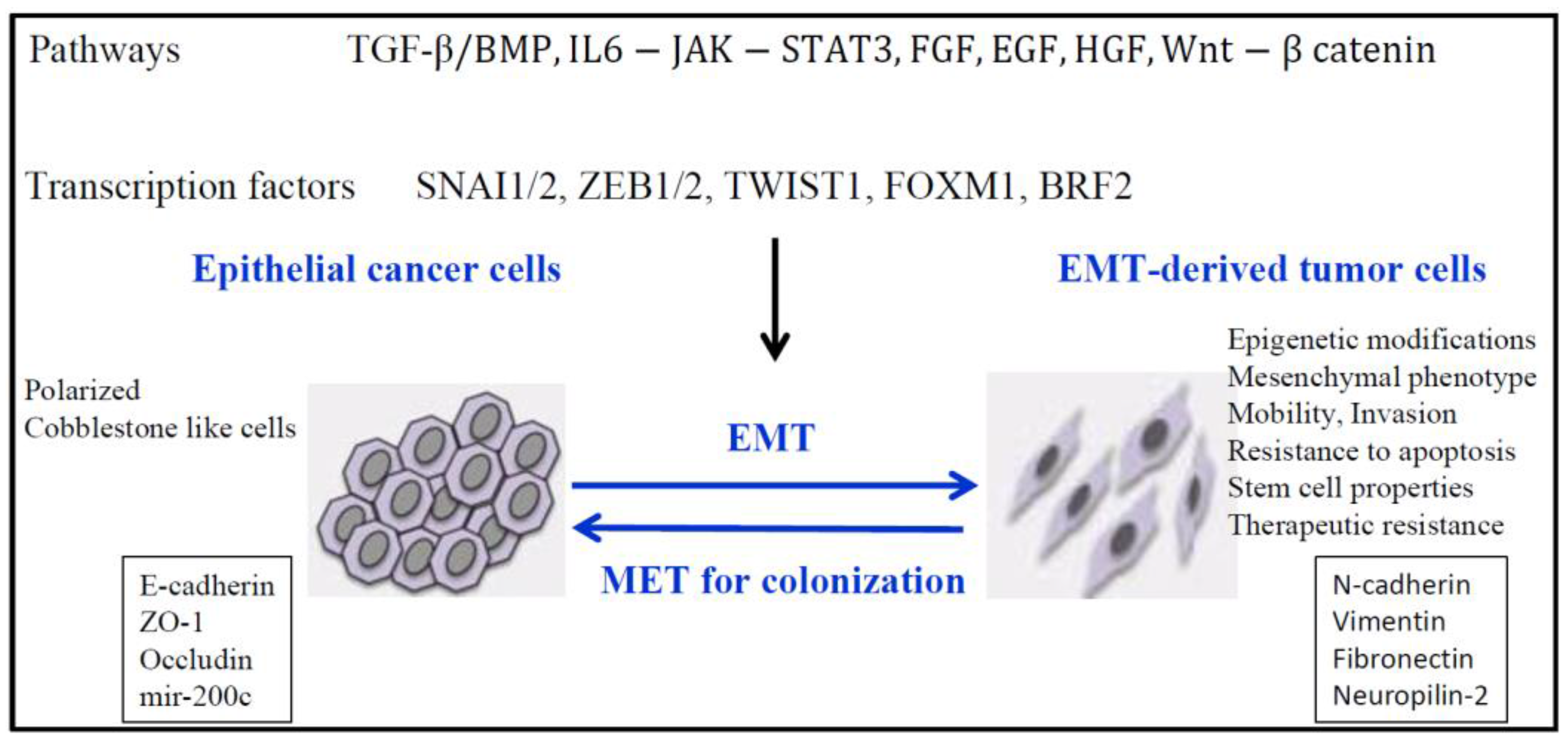
4. EMT and Epigenetics in Lung Cancer
5. Therapeutic Inhibition of EMT
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Adenocarcinoma |
| ceRNA | competing endogenous RNA |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Recptor |
| EMT | Epithelial to Mesenchymal transition |
| EZH2 | enhancer of zeste 2 polycomb repressive complex 2 subunit |
| HDAC | Histone deacetylase |
| HGF | Hepatocyte Growth Factor |
| lncRNA | long non-coding RNA |
| MET | Mesenchymal to Epithelial Transition |
| NSCLC | noRsmall cell lung cancer |
| PRC2 | polycomb repressive complex 2 |
| SC | squamous cell carcinoma |
| SCLC | small cell lung cancer |
| VEGF | Vascular Endothelial Growth Factor |
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Engelman, J.A. SnapShot: Non-small cell lung cancer. Cancer Cell 2012, 21, 448.e2. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, II; Gazdar, A.F.; Minna, J.D. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001, 28, 3–13. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Gemmill, R.M.; Potiron, V.A.; Roche, J.; Lu, X.; Baron, A.E.; Korch, C.; Garrett-Mayer, E.; Lagana, A.; Howe, P.H.; et al. Neuropilin-2 is upregulated in lung cancer cells during TGF-beta1-induced epithelial-mesenchymal transition. Cancer Res. 2013, 73, 7111–7121. [Google Scholar] [CrossRef] [PubMed]
- Gemmill, R.M.; Nasarre, P.; Nair-Menon, J.; Cappuzzo, F.; Landi, L.; D’Incecco, A.; Uramoto, H.; Yoshida, T.; Haura, E.B.; Armeson, K.; et al. The neuropilin 2 isoform NRP2b uniquely supports TGFbeta-mediated progression in lung cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Clarhaut, J.; Gemmill, R.M.; Potiron, V.A.; Ait-Si-Ali, S.; Imbert, J.; Drabkin, H.A.; Roche, J. ZEB-1, a repressor of the semaphorin 3F tumor suppressor gene in lung cancer cells. Neoplasia 2009, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Gemmill, R.M.; Drabkin, H.A. The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. Onco. Targets Ther. 2014, 7, 1663–1687. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, M.; Hoang, S.A.; Baranova, N.; Chodaparambil, S.; Kumar, M.; Allison, D.F.; Xu, X.; Wamsley, J.J.; Gray, L.; Jones, D.R.; et al. Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenet. Chromatin 2013, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Johnsen, S.A. Targeted therapy of epigenomic regulatory mechanisms controlling the epithelial to mesenchymal transition during tumor progression. Cell Tissue Res. 2014, 356, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011, 300, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Shi, L.; Wang, D.C.; Chen, C.; Wang, X. Role of epigenetics in lung cancer heterogeneity and clinical implication. Semin. Cell Dev. Biol. 2017, 64, 18–25. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, K.; Shia, A.; Schmid, P. Epigenetic Regulation of EMT in non-small cell lung cancer. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 128, 802. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, T. Tet family of 5-methylcytosine dioxygenases in mammalian development. J. Hum. Genet. 2013, 58, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Diagnostic and therapeutic potential of microRNAs in lung cancer. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.R.; Cooper, T.A. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhou, C.; Yang, J.; Li, J.; Gong, Z. Long and short noncoding RNAs in lung cancer precision medicine: Opportunities and challenges. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Paliogiannis, P.; Sini, M.C.; Manca, A.; Palomba, G.; Doneddu, V.; Tanda, F.; Pascale, M.R.; Cossu, A. Long non-coding RNA CASC2 in human cancer. Crit. Rev. Oncol. Hematol. 2017, 111, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yan, Y.; Qian, L.; Gong, Z. Long non-coding RNAs act as regulators of cell autophagy in diseases (Review). Oncol. Rep. 2017, 37, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Ollikainen, N.; Guttman, M. Long non-coding RNAs: Spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.M.; Zhou, G.B. Long Non-coding RNAs and their roles in non-small-cell lung cancer. Genom. Proteom. Bioinform. 2016, 14, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.G.; Kelley, D.R.; Tenen, D.; Bernstein, B.; Rinn, J.L. Widespread RNA binding by chromatin-associated proteins. Genome Biol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Heery, R.; Finn, S.P.; Cuffe, S.; Gray, S.G. Long non-coding RNAs: Key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.R. The RNA code comes into focus. Nature 2017, 542, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug. Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung cancer epigenetics: Emerging biomarkers. Biomark Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Liloglou, T.; Bediaga, N.G.; Brown, B.R.; Field, J.K.; Davies, M.P. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014, 342, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Van Den Broeck, A.; Ozenne, P.; Eymin, B.; Gazzeri, S. Lung cancer: A modified epigenome. Cell Adh. Migr. 2010, 4, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Diederichs, S. Long Noncoding RNAs in Lung Cancer. Curr. Top. Microbiol. Immunol. 2016, 394, 57–110. [Google Scholar] [CrossRef] [PubMed]
- Simo-Riudalbas, L.; Perez-Salvia, M.; Setien, F.; Villanueva, A.; Moutinho, C.; Martinez-Cardus, A.; Moran, S.; Berdasco, M.; Gomez, A.; Vidal, E.; et al. KAT6B is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Res. 2015, 75, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Augert, A.; Zhang, Q.; Bates, B.; Cui, M.; Wang, X.; Wildey, G.; Dowlati, A.; MacPherson, D. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J. Thorac. Oncol. 2017, 12, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Poirier, J.T.; Rudin, C.M. Histone code aberrancies in small cell lung cancer. J. Thorac. Oncol. 2017, 12, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Kinoshita, I.; Shimizu, Y.; Kikuchi, E.; Konishi, J.; Oizumi, S.; Kaga, K.; Matsuno, Y.; Nishimura, M.; Dosaka-Akita, H. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer 2010, 116, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Huqun, Ishikawa, R.; Zhang, J.; Miyazawa, H.; Shimizu, Y.; Hagiwara, K.; Koyama, N. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer 2012, 118, 1599–1606. [Google Scholar] [CrossRef]
- Cao, W.; Ribeiro Rde, O.; Liu, D.; Saintigny, P.; Xia, R.; Xue, Y.; Lin, R.; Mao, L.; Ren, H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS ONE 2012, 7, e52984. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Schrump, D.S. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009, 69, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Thu, K.L.; Aviel-Ronen, S.; Vucic, E.A.; Gazdar, A.F.; Lam, S.; Tsao, M.S.; Lam, W.L. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS ONE 2013, 8, e71670. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Dang, Y.; Chen, G.; Mo, Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 2015, 8, 13405–13410. [Google Scholar] [PubMed]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.L.; Olsen, M.; Lucci, A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar] [CrossRef]
- Christofori, G. Snail1 links transcriptional control with epigenetic regulation. EMBO J. 2010, 29, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 2002, 9, 213–224. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.; Sui, G.; Affar el, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Nasarre, P.; Gemmill, R.; Baldys, A.; Pontis, J.; Korch, C.; Guilhot, J.; Ait-Si-Ali, S.; Drabkin, H. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers 2013, 5, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, U.; Nalapareddy, K.; Saxena, M.; O'Neill, N.K.; Pinello, L.; Yuan, G.C.; Orkin, S.H.; Shivdasani, R.A. Acquired tissue-specific promoter bivalency is a basis for prc2 necessity in adult cells. Cell 2016, 165, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Fiorentino, F.P.; Alibes, A.; Perucho, M.; Sanchez-Cespedes, M.; Kohno, T.; Yokota, J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 2016, 7, 77378–77388. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. miR-149 Inhibits non-small-cell lung cancer cells EMT by targeting FOXM1. Biochem. Res. Int. 2013, 2013, 506731. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Song, L.; Bai, Y.; Kinose, F.; Li, J.; Ohaegbulam, K.C.; Munoz-Antonia, T.; Qu, X.; Eschrich, S.; Uramoto, H.; et al. ZEB1 mediates acquired resistance to the epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. PLoS ONE 2016, 11, e0147344. [Google Scholar] [CrossRef] [PubMed]
- Portoso, M.; Ragazzini, R.; Brencic, Z.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017, 36, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.R.; Guttman, M. Re-evaluating the foundations of lncRNA-Polycomb function. EMBO J. 2017, 36, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Padua Alves, C.; Fonseca, A.S.; Muys, B.R.; de Barros, E.L.B.R.; Burger, M.C.; de Souza, J.E.; Valente, V.; Zago, M.A.; Silva, W.A., Jr. Brief report: The lincRNA hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 2013, 31, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, X.H.; Lu, K.H.; Nie, F.Q.; Xia, R.; Kong, R.; Yang, J.S.; Xu, T.P.; Liu, Y.W.; Zou, Y.F.; et al. EZH2-mediated epigenetic suppression of long noncoding RNA SPRY4-IT1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial-mesenchymal transition. Cell Death Dis. 2014, 5, e1298. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Ott, C.J.; Johnstone, R.W.; Bradner, J.E. A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 2017, 17, 160–183. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Adeegbe, D.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, M.; Ohira, T.; Chan, D.; Webster, R.B.; Kato, H.; Drabkin, H.A.; Gemmill, R.M. Induction of E-cadherin in lung cancer and interaction with growth suppression by histone deacetylase inhibition. J. Thorac. Oncol. 2009, 4, 1455–1465. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Epigenetic Modification | Function | Writer | Eraser | Reader | ||
|---|---|---|---|---|---|---|
| DNA | CpG Methylation | Transcriptional repression | DNMT1/3A/3B | TET1/2/3 | MeCP, MBD1-4, UHRF1 | |
| RNA | m5C | tRNA stabilization, translation, immune response | DNMT2 (=TRDMT1) NSUN family | TET | not identified | |
| m6A | RNA splicing, export, stability, immune tolerance | METTL3/4, WTAP | FTO, ALKBH5 | YTHD family, HuR HNRNPA2B1 | ||
| Histones | Lysine Acetylation | Transcriptional activation | HAT | HDAC1-11, SIRT1-7 | BRD bromodomain | |
| Lysine Methylation | HH3K4 | Transcriptional activation | MLL1-5, SET1A/B, SET7/9, ASH1L | LSD1, JARID1a/b | Chromodomain, Tudor, MBT repeat, PHD finger | |
| HH3K9 | Transcriptional repression | G9a(EHMT2) SUV39H1/2 | LSD1, GASC1 | |||
| HH3K27 | Transcriptional repression | EZH1/2, G9a ** | UTX, JMJD3 | |||
| HH3K36 | Transcriptional activation | SETD2, ASH1L, ASF1A, NSD1-3, SMYD2 | Rph1/KDM4 Jhdm1b/Kdm2b | |||
| HH3K79 | Transcriptional regulation | DOT1L, RE-IIBP | not known | |||
| Lung Cancer | Gene | Function | Mutation | References |
|---|---|---|---|---|
| SCLC | KAT3A/CREBBP | histone acetytransferase | inactivating mutation | [7] |
| KAT3B/EP300 | histone acetytransferase | inactivating mutation | [7] | |
| KAT6B | H3K23 histone acetytransferase | genomic loss | [61] | |
| KMT2D/MLL2 | H3K4me1/2 histone methyltransferase | frequent inactivation | [7,62] | |
| KDM6A/UTX | H3K27 histone demethylase | truncating mutation in a small number of SCLC patients | [62,63] | |
| PBRM1 | chromatin remodeling factor | mutation | [62] | |
| ARID1A | mutation | [62] | ||
| ARID1B | mutation | [62] | ||
| NSCLC | KMT2D/MLL2 | H3K4me1/2 histone methyltransferase | mutation in 20% SC | [3] |
| SETD2 | H3K36 histone methyltransferase | 9% ADC | [3] | |
| DOT1L | H3K79 histone methyltransferase | 3% ADC | [64] | |
| ARID1A | chromatin remodeling factor | 7% ADC | [3] | |
| ARID1B | 6% ADC | |||
| ARID2 | 7% ADC | |||
| SMARCA4/BRG1 | 6% ADC | |||
| BRD3 | Bromodomain, binds hyperacetylated chromatin | [65] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roche, J.; Gemmill, R.M.; Drabkin, H.A. Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer. Cancers 2017, 9, 72. https://doi.org/10.3390/cancers9070072
Roche J, Gemmill RM, Drabkin HA. Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer. Cancers. 2017; 9(7):72. https://doi.org/10.3390/cancers9070072
Chicago/Turabian StyleRoche, Joëlle, Robert M. Gemmill, and Harry A. Drabkin. 2017. "Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer" Cancers 9, no. 7: 72. https://doi.org/10.3390/cancers9070072
APA StyleRoche, J., Gemmill, R. M., & Drabkin, H. A. (2017). Epigenetic Regulation of the Epithelial to Mesenchymal Transition in Lung Cancer. Cancers, 9(7), 72. https://doi.org/10.3390/cancers9070072