
Targeting RTK Signaling Pathways in Cancer

Abstract
:1. Introduction
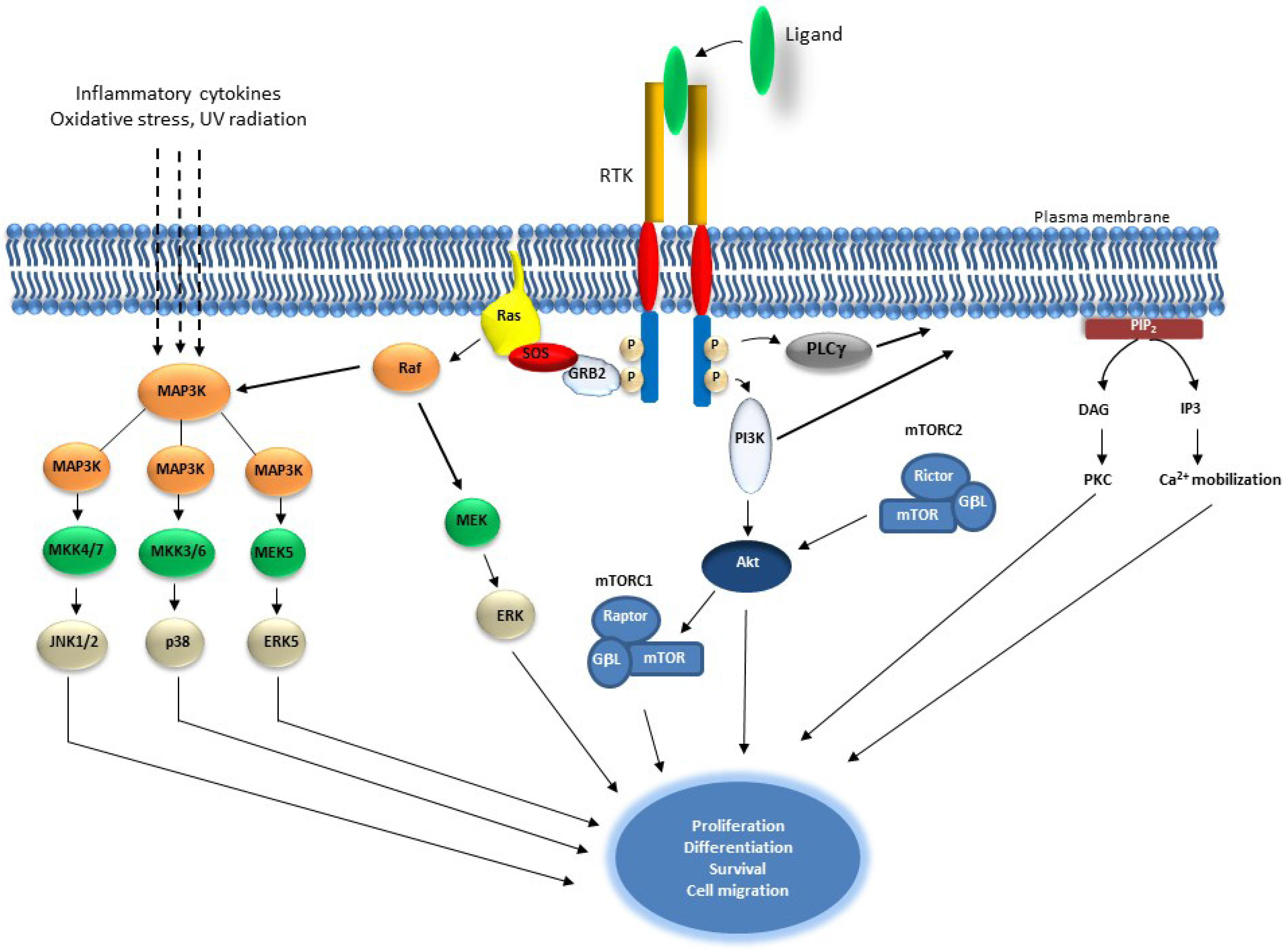
2. Receptor Tyrosine Kinase Signaling and Cancer
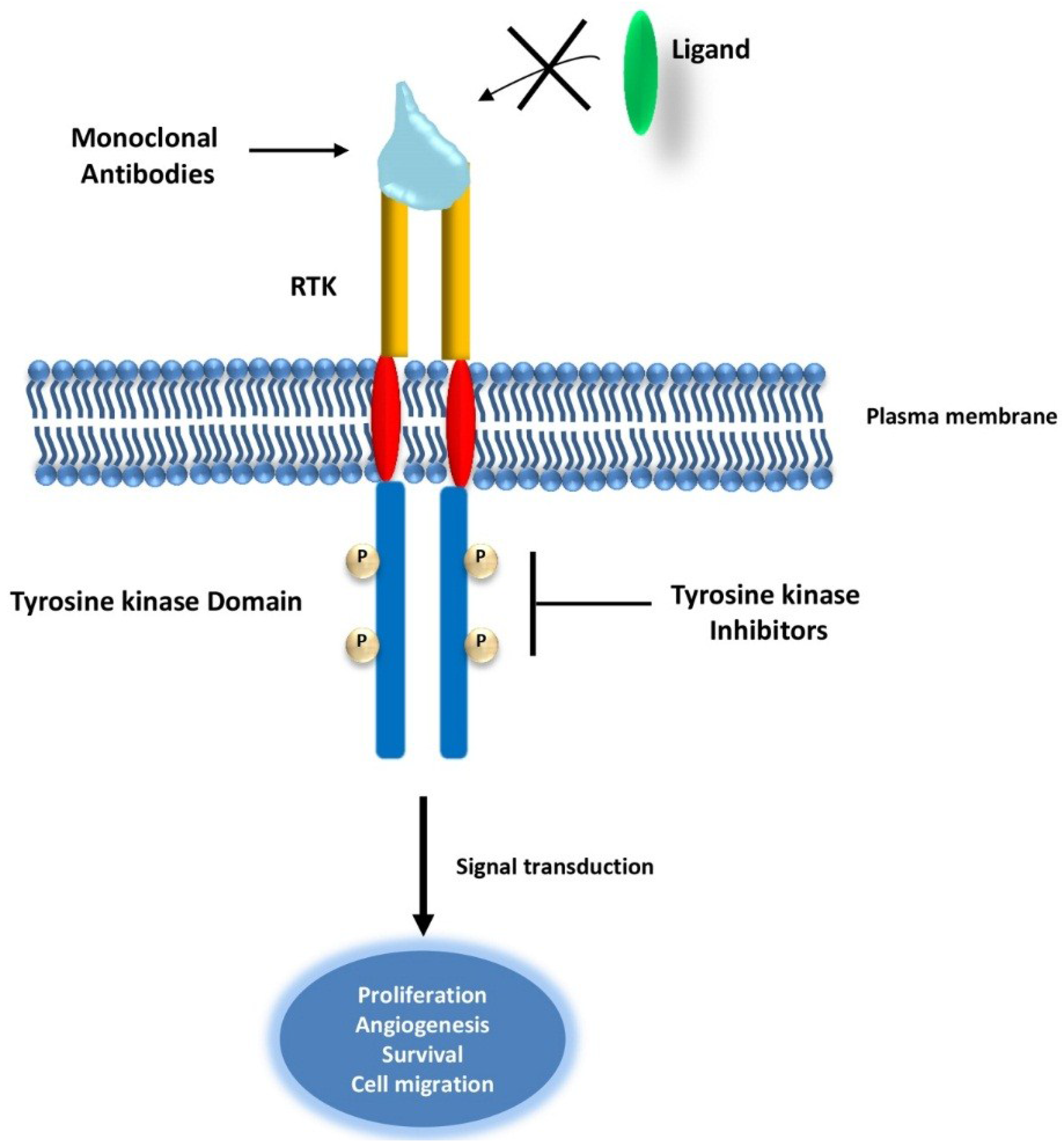
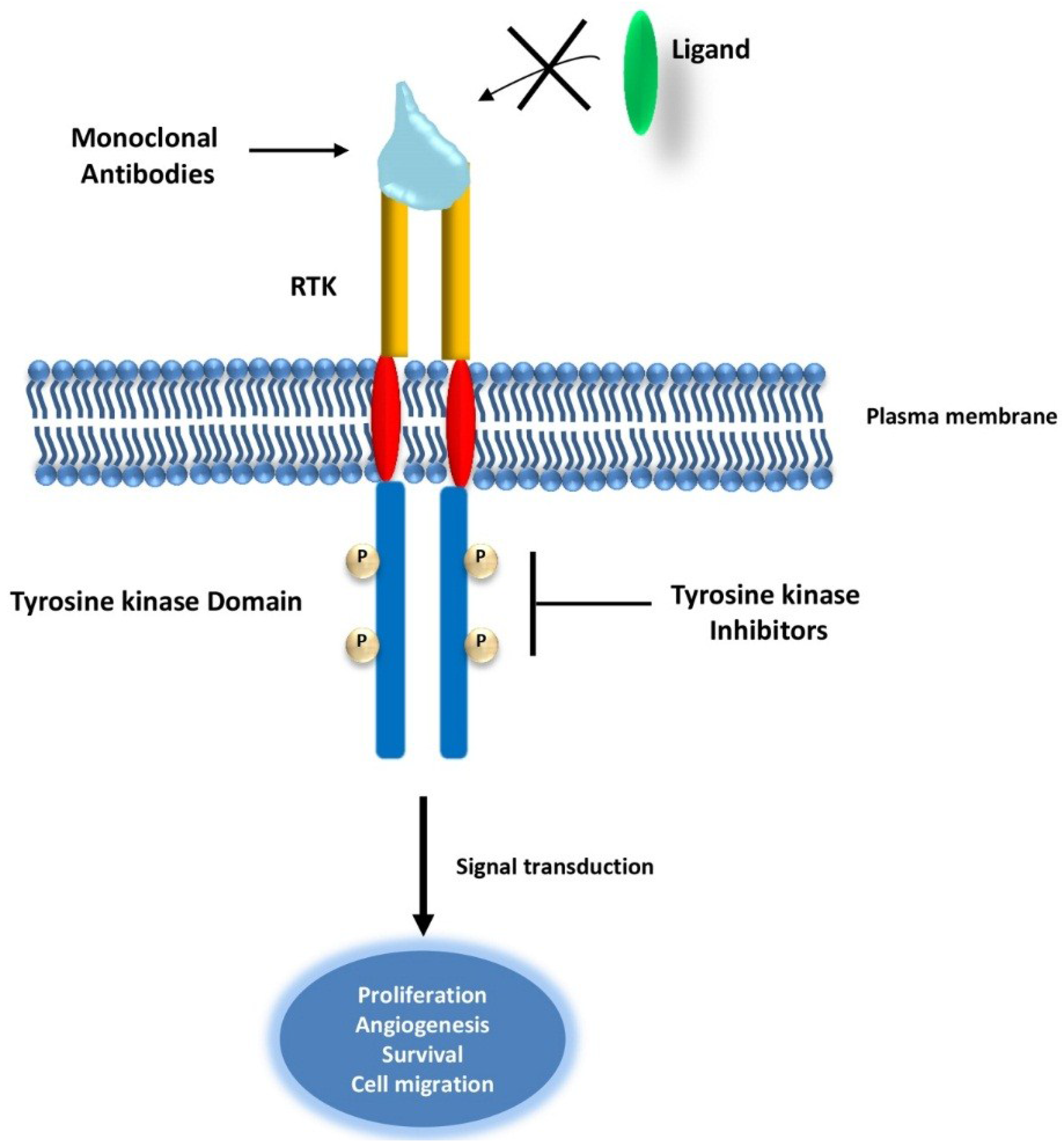
2.1. Targeting Receptor Tyrosine Kinases (RTKs) in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Compound | Cancer | References |
|---|---|---|---|
| EGFR family | |||
| HER2 | Trastuzumab (Herceptin) | HER2-positive breast cancer | [7,8,9] |
| EGFR | Cetuximab (Erbitux) | Metastatic colorectal cancer (RAS wild type) Metastatic non-small-cell lung cancer | [10] |
| Panitumumab (Vectibix) | [10,11,12] | ||
| Gefitinib (Iressa) | [13,14,15] | ||
| Erlotinib (Tarceva) | [13,16] | ||
| EGFR and HER2 | Lapatinib (Tykerb | HER2-positive breast cancer (Trastuzumab-resistant) | [14,17] |
| Afatinib | NSCLC HER2-positive breast cancer | [18,19,20,21] | |
| VEGFR | Sorafenib (Nexavar) | Renal, liver and thyroid cancer | [22,23,24] |
| Sunitinib (Sutent) | Renal cell cancer Gastrointestinal stromal tumor (GIST) | [25,26] | |
| Bevacizumab (Avastin) | Metastatic colorectal carcinoma | [27] | |
| PDGFR | Imatinib (Gleevec) | GIST (KIT+) | [28] |
| PDGFR and VEGFR | Sunitinib | Angiogenesis | [29,30,31,32] |
| Soratinib | |||
| Pazopanib | |||
| Nilotinib | |||
| FGFR and VEGFR | Brivanib (BMS-540215) | Human hepatocellular carcinoma model | [33] |
| VEGFR, PDGFR, FLT-3, c-KIT and FGFR | CHIR-258 (TKI-258) | Multiple myelomas | [34,35] |
| MET | SGX523 | MDCK and A549 cells and GTL16 xenograft models | [36] |
| C-KIT | Imatinib (Gleevec) | GIST | [37,38,39] |
2.1.1. EGFR-Targeted Therapy
2.1.2. VEGFR-Targeted Therapy
2.1.3. PDGFR-Targeted Therapy
2.1.4. FGFR-Targeted Therapy
2.1.5. MET-Targeted Therapy
2.1.6. c-KIT-Targeted Therapy
2.2. The RAS/MAP Kinase Pathway
Targeting the RAS/MAP Kinase Pathway in Cancer
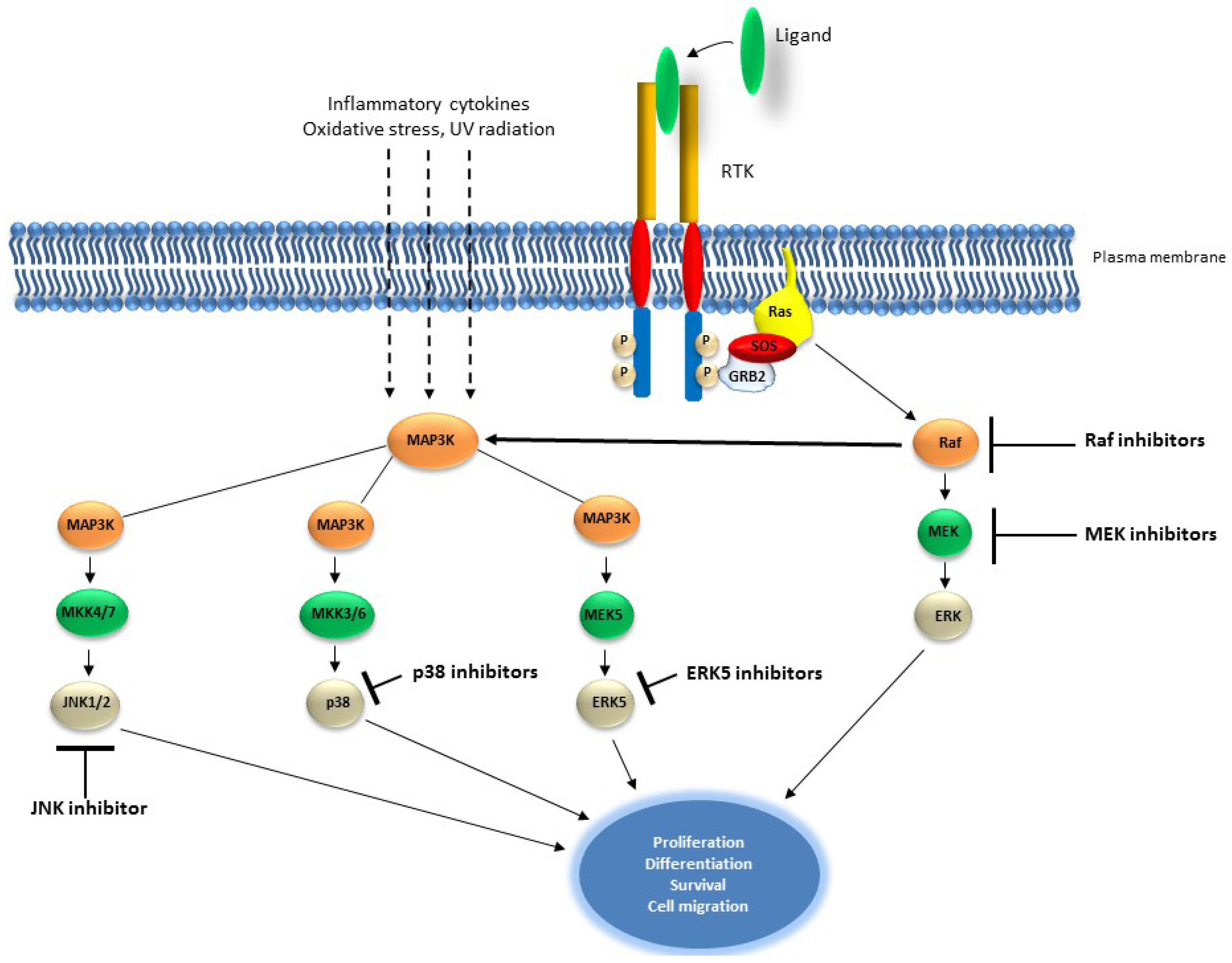
| Target | Compound | Cancer | References |
|---|---|---|---|
| MAP Kinase pathway | |||
| BRAFV600E | Sorafenib | Malignant melanoma | [143,144,145,146] |
| PLX4720 | |||
| PLX4032 | |||
| GSK2118436 | |||
| C-RAF | LErafAON (NeoPharm) | Ovarian and Breast cancer | [143,144,145,146] |
| ISIS 5132 | |||
| MEK | CI-1040 | Various cancers | [143,144,145,146] |
| PD-0325901 | |||
| AZD6244 | |||
| RDEA119/BAY 86-9766 | |||
| GDC-0973/XL581 | |||
| AZD8330/ARRY-424704 | |||
| MAP Kinase pathway | |||
| C-RAF, MEK | GSK1120212 | Various cancers | [143,144,145,146] |
| B-RAFV600E | |||
| BRAF wild type | |||
| PI3K/AKT pathway | |||
| PI3K/mTOR | NVP-BEZ235 | Various cancers | [150,151,152,153,154,155,156,157,158,159,160,161,162,163,164] |
| BGT226 | |||
| XL765/SAR245409 | |||
| SF1126 | |||
| GDC-0980 | |||
| PI-103 | |||
| PF-04691502 | |||
| PKI-587 | |||
| GSK2126458 | |||
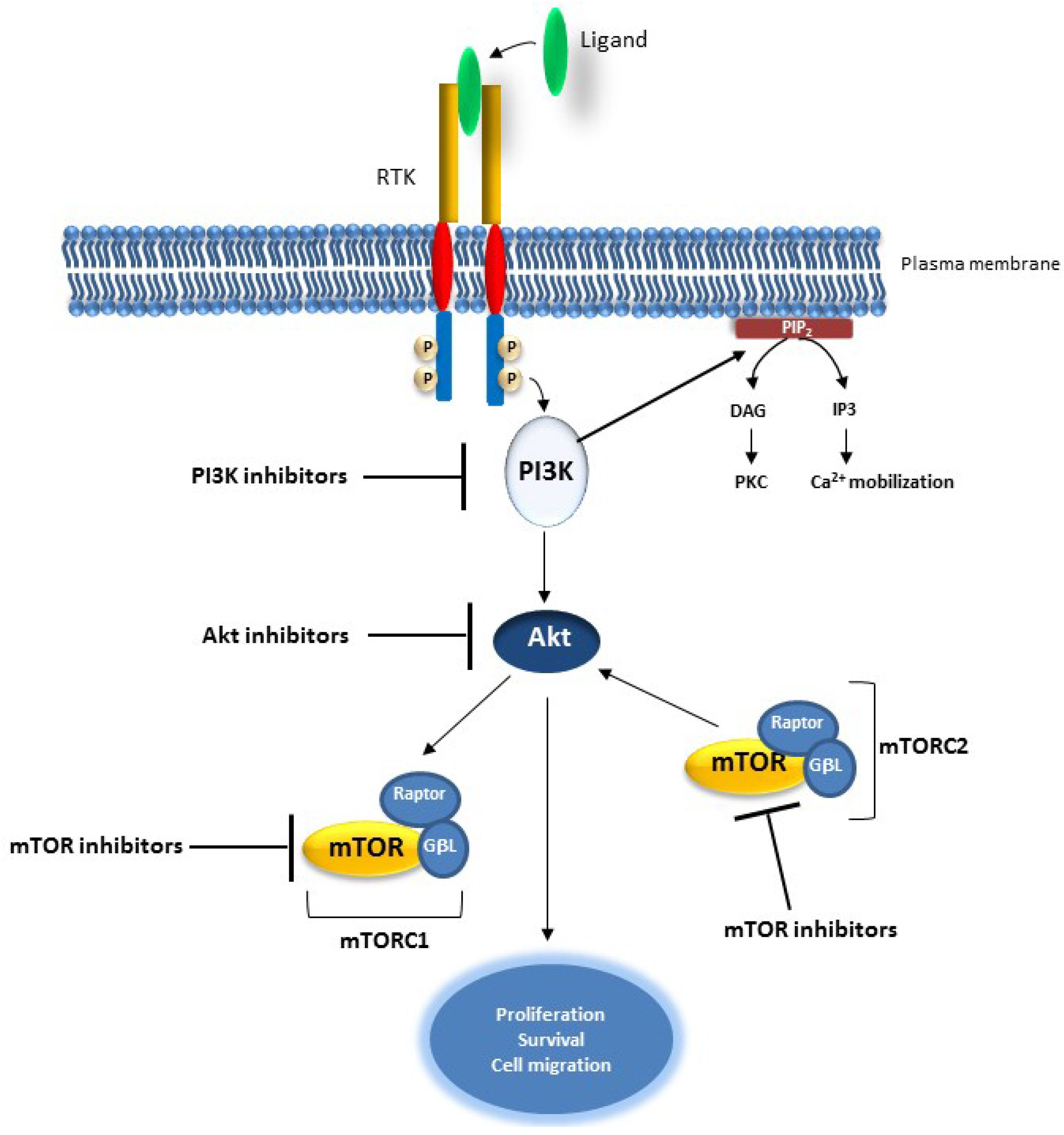
2.3. The PI3K/AKT Pathway
Targeting the PI3K/AKT Pathway in Cancer
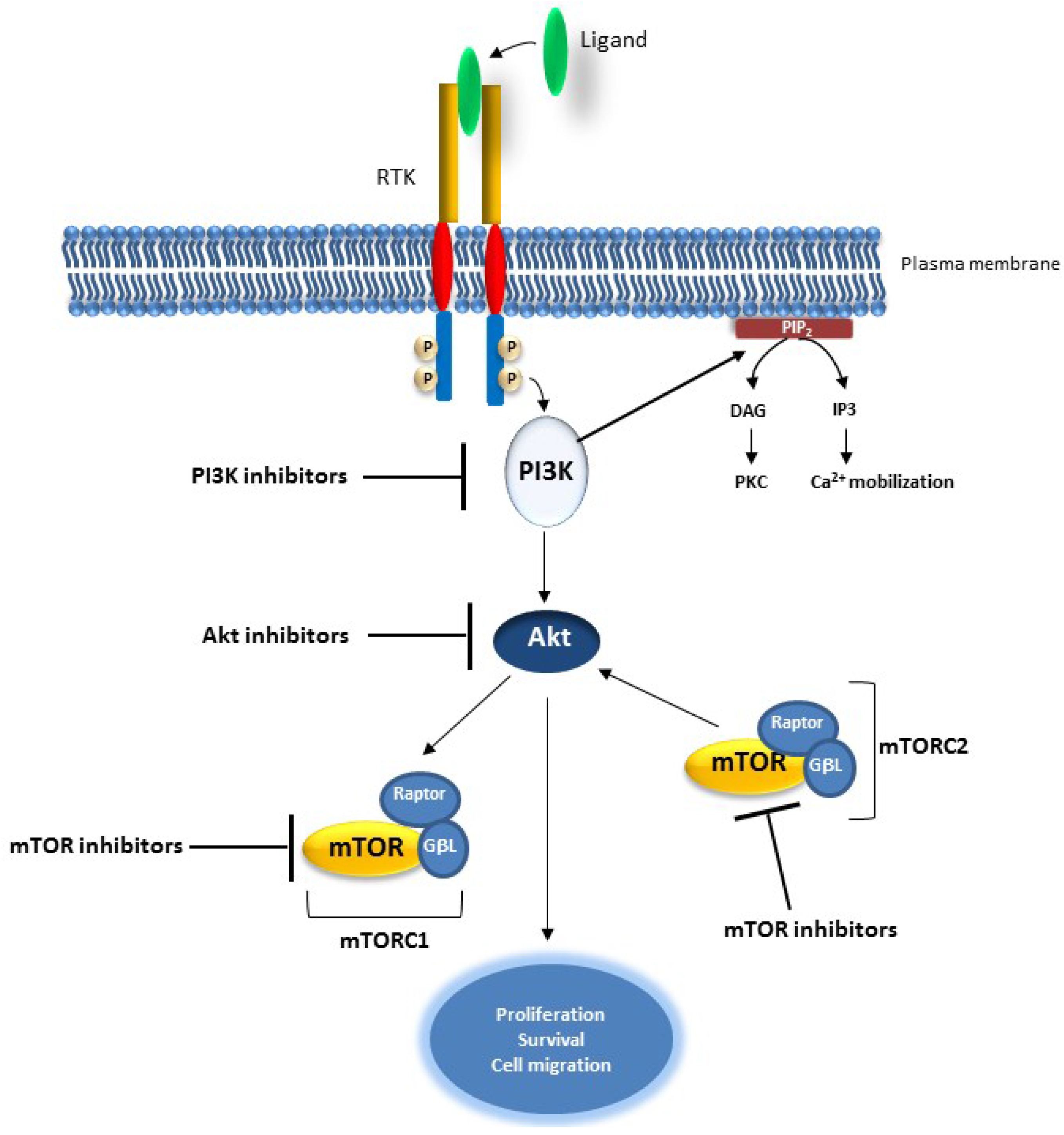
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 2007, 19, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Hubbard, S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Slamon, D.J.; Cobleigh, M.; Arnold, A.; Saleh, M.; Mortimer, J.E.; Murphy, M.; Stewart, S.J. Safety of treatment of metastatic breast cancer with trastuzumab beyond disease progression. J. Clin. Oncol. 2004, 22, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Donadio, M.; Clavarezza, M.; Redana, S.; Jacomuzzi, M.E.; Valabrega, G.; Danese, S.; Vietti-Ramus, G.; Durando, A.; Venturini, M.; et al. Outcome of patients with HER2-positive advanced breast cancer progressing during trastuzumab-based therapy. Oncologist 2006, 11, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008, 359, 1834–1836. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Pao, W.; Pham, D.; Li, A.R.; Rizvi, N.; Venkatraman, E.S.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M.; Miller, V.A. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 2006, 12, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Pippen, J., Jr.; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, M.J.; et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2009, 20, 5538–5546. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, F.; Shapiro, G.I.; Baum, I.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.H.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Sequist, L.V.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P.; Wheatley, D.; Carey, L.A.; Houston, S.; Mendelson, D.; Munster, P.; Frakes, L.; Kelly, S.; Garcia, A.A.; et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res. Treat. 2012, 133, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; Cosme de Oliveira, A.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, N.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Sleijfer, S.; Wiemer, E.; Seynaeve, C.; Verweij, J. Improved insight into resistance mechanisms to imatinib in gastrointestinal stromal tumors: A basis for novel approaches and individualization of treatment. Oncologist 2007, 12, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Michaelson, M.D.; Redman, B.G.; Hudes, G.R.; Wilding, G.; Figlin, R.A.; Ginsberg, M.S.; Kim, S.T.; Baum, S.M.; DePrimo, S.E.; et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Adams, V.R.; Leggas, M. Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clin. Ther. 2007, 29, 1338–1353. [Google Scholar] [CrossRef] [PubMed]
- Cauchi, C.; Somaiah, N.; Engstrom, P.F.; Litwin, S.; Lopez, M.; Lee, J.; Davey, B.B.; von Mehren, M. Evaluation of nilotinib in advanced GIST previously treated with imatinib and sunitinib. Cancer Chemother. Pharmacol. 2012, 69, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ngo, V.C.; Fargnoli, J.; Ayers, M.; Soo, K.C.; Koong, H.N.; Thng, C.H.; Ong, H.S.; Chung, A.; Chow, P.; et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 6146–6153. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Molife, R.; Evans, T.J.; Hardie, M.; Marriott, C.; Butzberger-Zimmerli, P.; Morrison, R.; Fox, J.A.; Heise, C.; Louie, S.; et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Li, Z.H.; Wei, E.; Wiesmann, M.; Chang, H.; Chen, C.; Reece, D.; Heise, C.; Stewart, A.K. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t (4; 14) multiple myeloma. Blood 2008, 105, 2941–2948. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, S.G.; Hendle, J.; Lee, P.S.; Smith, C.R.; Bounaud, P.Y.; Jessen, K.A.; Tang, C.M.; Huser, N.H.; Felce, J.D.; Froning, K.J.; et al. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol. Cancer Ther. 2009, 8, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.J.; Ferrao, P.T.; Hughes, T.P.; Ashman, L.K. Juxtamembrane Mutant V560GKit Is More Sensitive to Imatinib (STI571) Compared with Wild-Type c-Kit Whereas the Kinase Domain Mutant D816VKit Is Resistant 1 Supported by a grant from the National Health and Medical Research Council of Australia (NHMRC). MF is recipient of an Australian Postgraduate Award. LKA is a NHMRC Principal Research Fellow. Imatinib was provided by Novartis. 1. Mol. Cancer Ther. 2002, 1, 1115–1124. [Google Scholar] [PubMed]
- Wardelmann, E.; Thomas, N.; Merkelbach-Bruse, S.; Pauls, K.; Speidel, N.; Büttner, R.; Bihl, H.; Leutner, C.C.; Heinicke, T.; Hohenberger, P. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005, 6, 249–251. [Google Scholar] [CrossRef]
- Ashman, L.K.; Griffith, R. Therapeutic targeting of c-KIT in cancer. Expert Opin. Investig. Drugs 2013, 22, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ito, F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol. Pharm. Bull. 2011, 34, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Radinsky, R.; Risin, S.; Fan, D.; Dong, Z.; Bielenberg, D.; Bucana, C.D.; Fidler, I.J. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin. Cancer Res. 1995, 1, 19–31. [Google Scholar] [PubMed]
- Goldstein, N.S.; Armin, M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on cancer stage IV colon adenocarcinoma. Cancer 2001, 92, 1331–1346. [Google Scholar] [CrossRef]
- McKay, J.A.; Murray, L.J.; Curran, S.; Ross, V.G.; Clark, C.; Murray, G.I.; Cassidy, J.; McLeod, H.L. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur. J. Cancer 2002, 38, 2258–2264. [Google Scholar] [CrossRef]
- Gazdar, A. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harbor Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Petrova, T.V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2002, 19, 5598–5605. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Vasile, E.; Feng, D.; Sundberg, C.; Brown, L.F.; Detmar, M.J.; Lawitts, J.A.; Benjamin, L.; Tan, X.; Manseau, E.J.; et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 2002, 196, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Toffalini, F.; Demoulin, J.B. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 2010, 116, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Saxena, A.; Clark, W.C.; Robertson, J.T.; Oldfield, E.H.; Aaronson, S.A.; Ali, I.U. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992, 52, 4550–4553. [Google Scholar] [PubMed]
- Kumabe, T.; Sohma, Y.; Kayama, T.; Yoshimoto, T.; Yamamoto, T. Amplification of alpha-platelet-derived growth factor receptor gene lacking an exon coding for a portion of the extracellular region in a primary brain tumor of glial origin. Oncogene 1992, 7, 627–633. [Google Scholar] [PubMed]
- Puputti, M.; Tynninen, O.; Sihto, H.; Blom, T.; Mäenpää, H.; Isola, J.; Paetau, A.; Joensuu, H.; Nupponen, N.N. Amplification of KIT, PDGFRA, VEGFR2, and EGFR in gliomas. Mol. Cancer Res. 2006, 4, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Wang, X.Y.; Qian, J.; Hosek, S.M.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Amplification of the platelet-derived growth factor receptor-A (PDGFRA) gene occurs in oligodendrogliomas with grade IV anaplastic features. J. Neuropathol. Exp. Neurol. 2000, 59, 495–503. [Google Scholar] [PubMed]
- Arai, H.; Ueno, T.; Tangoku, A.; Yoshino, S.; Abe, T.; Kawauchi, S.; Oga, A.; Furuya, T.; Oka, M.; Sasaki, K. Detection of amplified oncogenes by genome DNA microarrays in human primary esophageal squamous cell carcinoma: Comparison with conventional comparative genomic hybridization analysis. Cancer Genet. Cytogenet. 2003, 146, 16–21. [Google Scholar] [CrossRef]
- Zhao, J.; Roth, J.; Bode-Lesniewska, B.; Pfaltz, M.; Heitz, P.U.; Komminoth, P. Combined comparative genomic hybridization and genomic microarray for detection of gene amplifications in pulmonary artery intimal sarcomas and adrenocortical tumors. Genes Chromosomes Cancer 2002, 34, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-mediated carcinogenesis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Jacquemier, J.; Adelaide, J.; Parc, P.; Penault-Llorca, F.; Planche, J.; Delapeyriere, O.; Birnbaum, D. Expression of the FGFR1 gene in human breast-carcinoma cells. Int. J. Cancer 1994, 59, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.B.; Maia, A.T.; O’Reilly, M.; Teschendorff, A.E.; Chin, S.F.; Caldas, C.; Ponder, B.A. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008, 6, e108. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Orsetti, B.; Cervera, N.; Finetti, P.; Sircoulomb, F.; Rougé, C.; Lasorsa, L.; Letessier, A.; Ginestier, C.; Monville, F.; et al. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol. Cancer Res. 2005, 3, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Sircoulomb, F.; Ginestier, C.; Cervera, N.; Monville, F.; Gelsi-Boyer, V.; Esterni, B.; Geneix, J.; Finetti, P.; Zemmour, C.; et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer 2006. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Pierrot, I.; Gruel, N.; Stransky, N.; Vincent-Salomon, A.; Reyal, F.; Raynal, V.; Vallot, C.; Pierron, G.; Radvanyi, F.; Delattre, O. Characterization of the recurrent 8p11-12 amplicon identifies PPAPDC1B, a phosphatase protein, as a new therapeutic target in breast cancer. Cancer Res. 2008, 68, 7165–7175. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.J.; Pole, J.C.; Chin, S.F.; Teschendorff, A.; Naderi, A.; Ozdag, H.; Vias, M.; Kranjac, T.; Subkhankulova, T.; Paish, C.; et al. A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene 2005, 24, 5235–5245. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Kraft, P.; Jacobs, K.B.; Cox, D.G.; Yeager, M.; Hankinson, S.E.; Wacholder, S.; Wang, Z.; Welch, R.; Hutchinson, A.; et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007, 39, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Shin, K.H.; Park, J.G. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001, 61, 3541–3543. [Google Scholar] [PubMed]
- Kunii, K.; Davis, L.; Gorenstein, J.; Hatch, H.; Yashiro, M.; di Bacco, A.; Elbi, C.; Lutterbach, B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008, 68, 2340–2348. [Google Scholar] [CrossRef] [PubMed]
- Nord, H.; Segersten, U.; Sandgren, J.; Wester, K.; Busch, C.; Menzel, U.; Komorowski, J.; Dumanski, J.P.; Malmström, P.U.; de Ståhl, T.D. Focal amplifications are associated with high grade and recurrences in stage Ta bladder carcinoma. Int. J. Cancer 2010, 126, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Freier, K.; Schwaenen, C.; Sticht, C.; Flechtenmacher, C.; Mühling, J.; Hofele, C.; Radlwimmer, B.; Lichter, P.; Joos, S. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC). Oral Oncol. 2007, 43, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Gorringe, K.L.; Jacobs, S.; Thompson, E.R.; Sridhar, A.; Qiu, W.; Choong, D.Y.; Campbell, I.G. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin. Cancer Res. 2007, 13, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Rand, V.; Huang, J.; Stockwell, T.; Ferriera, S.; Buzko, O.; Levy, S.; Busam, D.; Li, K.; Edwards, J.B.; Eberhart, C.; et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc. Natl. Acad. Sci. USA 2005, 102, 14344–14349. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, B.W.; van Tilborg, A.A.; Lurkin, I.; Bonaventure, J.; de Vries, A.; Thiery, J.P.; van der Kwast, T.H.; Zwarthoff, E.C.; Radvanyi, F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur. J. Hum. Genet. 2002, 10, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.; de Muga, S.; Agell, L.; Juanpere, N.; Esgueva, R.; Lorente, J.A.; Mojal, S.; Serrano, S.; Lloreta, J. FGFR3 mutations in prostate cancer: Association with low-grade tumors. Mod. Pathol. 2009, 22, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Salvesen, H.B.; Chen, T.H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Brents, L.A.; Ely, S.A.; Bais, C.; Robbiani, D.F.; Mesri, E.A.; Kuehl, W.M.; Bergsagel, P.L. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood 2001, 97, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Dekker, N.; Jordan, R.C. Identification of novel fibroblast growth factor receptor 3 gene mutations in actinic cheilitis and squamous cell carcinoma of the lip. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2009, 107, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Hunter, C.; Smith, R.; Stephens, P.; Greenman, C.; Bignell, G.; Teague, J.; Butler, A.; Edkins, S.; Stevens, C.; et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005, 65, 7591–7595. [Google Scholar] [PubMed]
- Pollock, P.M.; Gartside, M.G.; Dejeza, L.C.; Powell, M.A.; Mallon, M.A.; Davies, H.; Mohammadi, M.; Futreal, P.A.; Stratton, M.R.; Trent, J.M.; et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007, 26, 7158–7162. [Google Scholar] [CrossRef] [PubMed]
- Cappellen, D.; de Oliveira, C.; Ricol, D.; de Medina, S.; Bourdin, J.; Sastre-Garau, X.; Chopin, D.; Thiery, J.P.; Radvanyi, F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 1999, 23, 18–20. [Google Scholar] [PubMed]
- Cheng, L.; Zhang, S.; Davidson, D.D.; MacLennan, G.T.; Koch, M.O.; Montironi, R.; Lopez-Beltran, A. Molecular determinants of tumor recurrence in the urinary bladder. Future Oncol. 2009, 5, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.C.; Medeiros, L.J.; Miranda, R.N. 8p11 myeloproliferative syndrome: A review. Hum. Pathol. 2010, 41, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Yagasaki, F.; Wakao, D.; Yokoyama, Y.; Uchida, Y.; Murohashi, I.; Kayano, H.; Taniwaki, M.; Matsuda, A.; Bessho, M. Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with at (4; 12)(p16; p13) chromosomal translocation. Cancer Res. 2001, 61, 8371–8374. [Google Scholar] [PubMed]
- Roumiantsev, S.; Krause, D.S.; Neumann, C.A.; Dimitri, C.A.; Asiedu, F.; Cross, N.C.; van Etten, R.A. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 2004, 5, 287–298. [Google Scholar] [CrossRef]
- Xiao, S.; Nalabolu, S.R.; Aster, J.C.; Ma, J.; Abruzzo, L.; Jaffe, E.S.; Stone, R.; Weissman, S.M.; Hudson, T.J.; Fletcher, J.A. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t (8; 13) leukaemia/lymphoma syndrome. Nat. Genet. 1998, 18, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Demiroglu, A.; Steer, E.J.; Heath, C.; Taylor, K.; Bentley, M.; Allen, S.L.; Koduru, P.; Brody, J.P.; Hawson, G.; Rodwell, R.; et al. The t (8; 22) in chronic myeloid leukemia fuses BCR to FGFR1: Transforming activity and specific inhibition of FGFR1 fusion proteins. Blood 2001, 98, 3778–3783. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Li, X.; Cowell, J.K. Genetic fingerprinting of the development and progression of T-cell lymphoma in a murine model of atypical myeloproliferative disorder initiated by the ZNF198–fibroblast growth factor receptor-1 chimeric tyrosine kinase. Blood 2009, 114, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Knights, V.; Cook, S.J. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol. Ther. 2010, 125, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Hahn, K.; Oglivie, G.; Rusk, T.; Devauchelle, P.; Leblanc, A.; Legendre, A.; Powers, B.; Leventhal, P.S.; Kinet, J.P.; Palmerini, F.; et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J. Vet. Intern. Med. 2008, 22, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Manley, P.W. Su-6668. SUGEN. Curr. Opin. Investig. Drugs (Lond. Engl.) 2001, 2, 1142–1148. [Google Scholar]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Marek, L.; Ware, K.E.; Fritzsche, A.; Hercule, P.; Helton, W.R.; Smith, J.E.; McDermott, L.A.; Coldren, C.D.; Nemenoff, R.A.; Merrick, D.T.; et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol. Pharmacol. 2009, 75, 196–207. [Google Scholar] [CrossRef] [PubMed]
- McDermott, L.A.; Simcox, M.; Higgins, B.; Nevins, T.; Kolinsky, K.; Smith, M.; Yang, H.; Li, J.K.; Chen, Y.; Ke, J.; et al. RO4383596, an orally active KDR, FGFR, and PDGFR inhibitor: Synthesis and biological evaluation. Bioorg. Med. Chem. 2005, 13, 4835–4841. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 3657–3660. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Furge, K.A.; Zhang, Y.W.; vande Woude, W.G. Met receptor tyrosine kinase: Enhanced signaling through adapter proteins. Oncogene 2000, 19, 5582–5589. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Dong, S.M.; Kim, S.Y.; Na, E.Y.; Shin, M.S.; Pi, J.H.; Kim, B.J.; Bae, J.H.; Hong, Y.K.; Lee, K.S.; et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999, 59, 307–310. [Google Scholar] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1995, 1, 147–154. [Google Scholar] [PubMed]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [PubMed]
- Ferracini, R.; di Renzo, M.F.; Scotlandi, K.; Baldini, N.; Olivero, M.; Lollini, P.; Cremona, O.; Campanacci, M.; Comoglio, P.M. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995, 10, 739–749. [Google Scholar] [PubMed]
- Hecht, M.; Papoutsi, M.; Tran, H.D.; Wilting, J.; Schweigerer, L. Hepatocyte growth factor/c-Met signaling promotes the progression of experimental human neuroblastomas. Cancer Res. 2004, 64, 6109–6118. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Tsukasaki, K.; O’neill, M.C.; Yamada, Y.; Onimaru, Y.; Matsumoto, K.; Ohashi, J.; Yamashita, Y.; Tsutsumi, S.; Kaneda, R.; et al. A genomic analysis of adult T-cell leukemia. Oncogene 2007, 26, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Dolled-Filhart, M.; Ocal, I.T.; Singh, B.; Lin, C.Y.; Dickson, R.B.; Rimm, D.L.; Camp, R.L. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 2003, 63, 1101–1105. [Google Scholar] [PubMed]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; Vande Woude, G.F.; et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Schiering, N.; Knapp, S.; Marconi, M.; Flocco, M.M.; Cui, J.; Perego, R.; Rusconi, L.; Cristiani, C. Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc. Natl. Acad. Sci. USA 2003, 100, 12654–12659. [Google Scholar] [CrossRef] [PubMed]
- Underiner, T.L.; Herbertz, T.; Miknyoczki, S.J. Discovery of small molecule c-Met inhibitors: Evolution and profiles of clinical candidates. Anti-Cancer Agents Med. Chem. 2010, 10, 7–27. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Previdi, S.; Abbadessa, G.; Dalò, F.; France, D.S.; Broggini, M. Breast cancer-derived bone metastasis can be effectively reduced through specific c-MET inhibitor tivantinib (ARQ 197) and shRNA c-MET knockdown. Mol. Cancer Ther. 2012, 11, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Bagai, R.; Fan, W.; Ma, P.C. ARQ-197, an oral small-molecule inhibitor of c-Met for the treatment of solid tumors. IDrugs Investig. Drugs J. 2010, 13, 404–414. [Google Scholar]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Guan, K.L. Regulation of Raf through phosphorylation and N terminus-C terminus interaction. J. Biol. Chem. 2003, 278, 36269–36276. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.; Gilley, R.; Cook, S. ERK5 and its role in tumour development. Biochem. Soc. Trans. 2012, 40, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [PubMed]
- Wang, X.; Merritt, A.J.; Seyfried, J.; Guo, C.; Papadakis, E.S.; Finegan, K.G.; Kayahara, M.; Dixon, J.; Boot-Handford, R.P.; Cartwright, E.J.; et al. Targeted deletion of mek5causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell. Biol. 2005, 25, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Fearns, C.; Eliceiri, B.; Yang, Y.; Lee, J.D. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005, 65, 7699–7706. [Google Scholar] [PubMed]
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9expression and invasion. Oncogene 2003, 22, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pesakhov, S.; Harrison, J.S.; Kafka, M.; Danilenko, M.; Studzinski, G.P. The MAPK ERK5, but not ERK1/2, inhibits the progression of monocytic phenotype to the functioning macrophage. Exp. Cell Res. 2015, 330, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 2004, 15, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Toshiharu, S.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar]
- Davies, C.; Tournier, C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 2012, 40, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lijian, H.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar]
- Chen, N.; Nomura, M.; She, Q.B.; Ma, W.Y.; Bode, A.M.; Wang, L.; Flavell, R.A.; Dong, Z. Suppression of skin tumorigenesis in c-Jun NH2-terminal kinase-2-deficient mice. Cancer Res. 2001, 61, 3908–3912. [Google Scholar] [PubMed]
- She, Q.B.; Chen, N.; Bode, A.M.; Flavell, R.A.; Dong, Z. Deficiency of c-Jun-NH2-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002, 62, 1343–1348. [Google Scholar] [PubMed]
- Qingshan, C.; Zhang, Y.; Beezhold, K.J.; Bhatia, D.; Zhao, H.; Chen, J.; Castranova, V.; Shi, X.; Chen, F. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J. Hepatol. 2009, 50, 323–333. [Google Scholar]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin β1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.L.; Symons, M.; Jou, T.S. Regulation of anoikis by Cdc42 and Rac1. Exp. Cell Res. 2004, 295, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Rosen, N. Mutant BRAF melanomas—Dependence and resistance. Cancer Cell 2011, 19, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 2007, 28, 742–762. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; de Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-pkb/akt pathway. Cold Spring Harbor Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- GarciaEcheverria, C. Blocking the mTOR pathway: A drug discovery perspective. Biochem. Soc. Trans. 2011, 39, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Howes, A.L.; Chiang, G.G.; Lang, E.S.; Ho, C.B.; Powis, G.; Vuori, K.; Abraham, R.T. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol. Cancer Ther. 2007, 6, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Rodon, J.; Bedell, C.; Kwak, E.L.; Baselga, J.; Braña, I.; Pandya, S.S.; Scheffold, C.; Laird, A.D.; Nguyen, L.T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.W.; Ma, W.W.; Senzer, N.; Brahmer, J.R.; Adjei, A.A.; Davies, M.; Lazar, A.J.; Vo, A.; Peterson, S.; Walker, L.; et al. A multicenter phase 1 study of PX-866 in combination with docetaxel in patients with advanced solid tumours. Br. J. Cancer 2013, 109, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Munugalavadla, V.; Mariathasan, S.; Slaga, D.; Du, C.; Berry, L.; del Rosario, G.; Yan, Y.; Boe, M.; Sun, L.; Friedman, L.S.; et al. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene 2014, 33, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; de Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2011, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Hoellenriegel, J.; Meadows, S.A.; Sivina, M.; Wierda, W.G.; Kantarjian, H.; Keating, M.J.; Giese, N.; O’Brien, S.; Yu, A.; Miller, L.L.; et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011, 118, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, E.; Chaudhuri, A.; Leiphrakpam, P.D.; Haferbier, K.L.; Brattain, M.G.; Chowdhury, S. Akt inhibitor MK-2206 promotes anti-tumor activity and cell death by modulation of AIF and Ezrin in colorectal cancer. BMC Cancer 2014, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeong, E.G.; Yoo, N.J.; Lee, S.H. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br. J. Cancer 2008, 98, 1533–1535. [Google Scholar] [CrossRef] [PubMed]
- Malanga, D.; Scrima, M.; de Marco, C.; Fabiani, F.; de Rosa, N.; de Gisi, S.; Malara, N.; Savino, R.; Rocco, G.; Chiappetta, G.; et al. Activating E17K mutation in the gene encoding the protein kinase AKT in a subset of squamous cell carcinoma of the lung. Cell Cycle 2008, 7, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Stemke-Hale, K.; Tellez, C.; Calderone, T.L.; Deng, W.; Prieto, V.G.; Lazar, A.J.; Gershenwald, J.E.; Mills, G.B. A novel AKT3 mutation in melanoma tumours and cell lines. Br. J. Cancer 2008, 99, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3′-kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar] [PubMed]
- Pedrero, J.M.G.; Carracedo, D.G.; Pinto, C.M.; Zapatero, A.H.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer 2005, 114, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Woenckhaus, J.; Steger, K.; Werner, E.; Fenic, I.; Gamerdinger, U.; Dreyer, T.; Stahl, U. Genomic gain of PIK3CA and increased expression of p110alpha are associated with progression of dysplasia into invasive squamous cell carcinoma. J. Pathol. 2002, 198, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.P.; Kuo, W.L.; Stokoe, D.; Olshen, A.B.; Treseler, P.A.; Chin, K.; Chen, C.; Polikoff, D.; Jain, A.N.; Pinkel, D.; et al. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization implications of the phosphatidylinositol 3-kinase pathway. Cancer Res. 2002, 62, 3636–3640. [Google Scholar] [PubMed]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, B.A.; Huang, L.; Wood, M.; Cheng, J.Q.; Testa, J.R. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 1998, 21, 81–86. [Google Scholar] [CrossRef]
- Bellacosa, A.; de Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V.; et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Molecular and cellular pathogenesis of melanoma initiation and progression. Cell. Mol. Life Sci. 2013, 70, 4055–4065. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758-1784. https://doi.org/10.3390/cancers7030860
Regad T. Targeting RTK Signaling Pathways in Cancer. Cancers. 2015; 7(3):1758-1784. https://doi.org/10.3390/cancers7030860
Chicago/Turabian StyleRegad, Tarik. 2015. "Targeting RTK Signaling Pathways in Cancer" Cancers 7, no. 3: 1758-1784. https://doi.org/10.3390/cancers7030860
APA StyleRegad, T. (2015). Targeting RTK Signaling Pathways in Cancer. Cancers, 7(3), 1758-1784. https://doi.org/10.3390/cancers7030860