
Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions


{kind=link}
Abstract
:1. Introduction
2. Hedgehog Signaling Modes of Action in Cancer
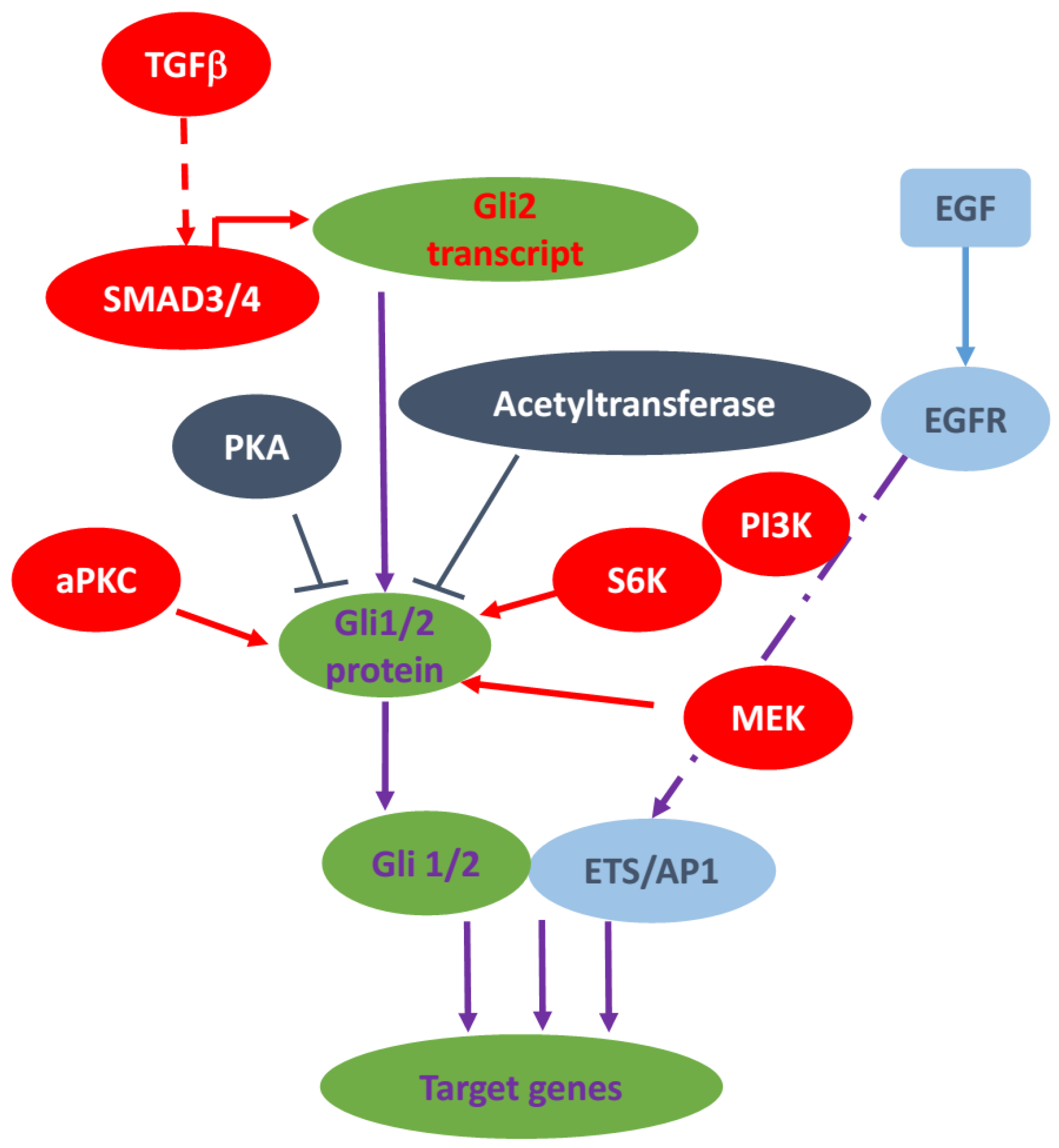
3. Non-Canonical Hedgehog Signaling in Cancer
3.1. The RAS-RAF-MEK Signaling Axis
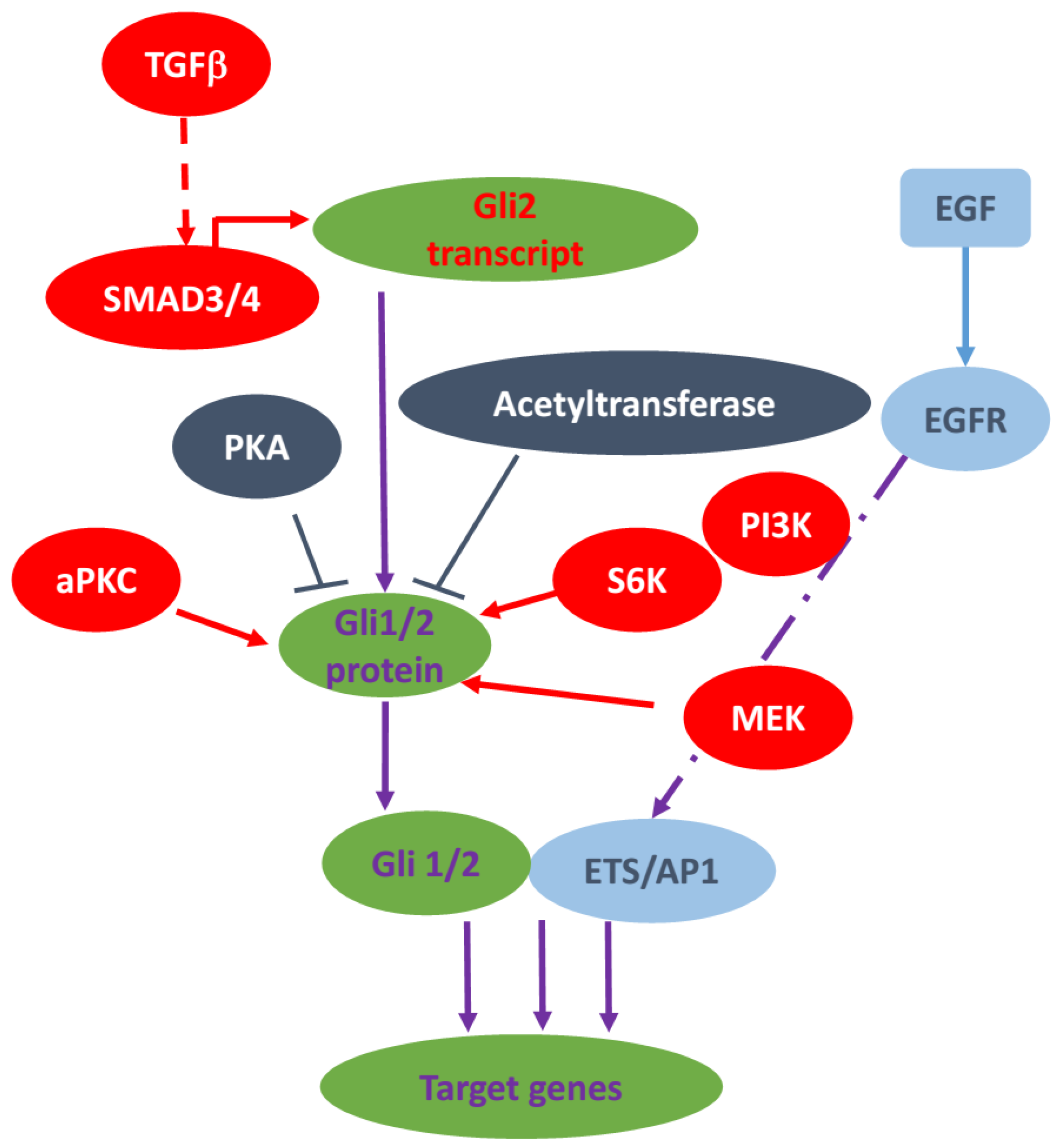
3.2. TGFβ Signaling Pathway
3.3. PKC Signaling
3.4. AKT/PI3K Signaling
3.5. TNFα/mTOR Pathway
3.6. Epigenetic Regulation of GLI
3.7. Other Non-Canonical Regulations of Hedgehog Signaling
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E., Jr. Genetic determinants of basal cell carcinoma risk. Med. Pediatr. Oncol. 2001, 36, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgard, R.; Unden, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the ptch and sufu genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Anderson, J.R.; Crossman, D.K.; Lynch, J.C.; Barr, F.G. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: A report from the children’s oncology group. Pediatr. Blood Cancer 2011, 57, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Aavikko, M.; Li, S.P.; Saarinen, S.; Alhopuro, P.; Kaasinen, E.; Morgunova, E.; Li, Y.; Vesanen, K.; Smith, M.J.; Evans, D.G.; et al. Loss of SUFU function in familial multiple meningioma. Am. J. Hum. Genet. 2012, 91, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Kijima, C.; Miyashita, T.; Suzuki, M.; Oka, H.; Fujii, K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Familial Cancer 2012, 11, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.E.; Tang, W.; Garcia, M.R.; Finkelstein, D.; Millay, D.P.; Liu, N.; Graff, J.; Galindo, R.L.; Olson, E.N. A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell 2012, 22, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.S.; Chen, E.; Elpek, N.M.; Fuller, A.Z.; Tenente, I.M.; Clagg, R.; Liu, S.; Blackburn, J.S.; Linardic, C.M.; Rosenberg, A.E.; et al. In vivo imaging of tumor-propagating cells, regional tumor heterogeneity, and dynamic cell movements in embryonal rhabdomyosarcoma. Cancer Cell 2012, 21, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Nitzki, F.; Zibat, A.; Frommhold, A.; Schneider, A.; Schulz-Schaeffer, W.; Braun, T.; Hahn, H. Uncommitted precursor cells might contribute to increased incidence of embryonal rhabdomyosarcoma in heterozygous Patched1-mutant mice. Oncogene 2011, 30, 4428–4436. [Google Scholar] [CrossRef] [PubMed]
- Pelczar, P.; Zibat, A.; van Dop, W.A.; Heijmans, J.; Bleckmann, A.; Gruber, W.; Nitzki, F.; Uhmann, A.; Guijarro, M.V.; Hernando, E.; et al. Inactivation of Patched1 in mice leads to development of gastrointestinal stromal-like tumors that express Pdgfralpha but not kit. Gastroenterology 2013, 144, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran-Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant epithelial-mesenchymal hedgehog signaling characterizes barrett’s metaplasia. Gastroenterology 2010, 138, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, L.S.; Chen, X.L.; Gatalica, Z.; Qiu, S.; Liu, Z.; Stoner, G.; Zhang, H.; Weiss, H.; Xie, J. Hedgehog signaling activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. Int. J. Biochem. Mol. Biol. 2012, 3, 46–57. [Google Scholar] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Ligon, K.L.; Rakhlin, E.Y.; Thayer, S.P.; Bronson, R.T.; Rowitch, D.; McMahon, A.P. A novel somatic mouse model to survey tumorigenic potential applied to the hedgehog pathway. Cancer Res. 2006, 66, 10171–10178. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Epstein, J.; Oro, A.; Douglas, V.; LeBoit, P.E.; Scott, M.P.; Epstein, E.H., Jr. Ultraviolet and ionizing radiation enhance the growth of bccs and trichoblastomas in patched heterozygous knockout mice. Nat. Med. 1999, 5, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Foltz, W.D.; Chaudary, N.; Hill, R.P.; Hedley, D.W. Tumor-stroma interaction in orthotopic primary pancreatic cancer xenografts during hedgehog pathway inhibition. Int. J. Cancer 2013, 133, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. Cancer 2012, 131, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.B.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An orally bioavailable small-molecule inhibitor of hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Liu, H.; Su, G.H.; Zhang, X.; Chin-Sinex, H.; Hanenberg, H.; Mendonca, M.S.; Shannon, H.E.; Chiorean, E.G.; Xie, J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol. Cancer Ther. 2013, 12, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.L.; Salci, K.R.; Shapovalova, Z.; McIntyre, B.A.; Bhatia, M. Nonhematopoietic cells represent a more rational target of in vivo hedgehog signaling affecting normal or acute myeloid leukemia progenitors. Exp. Hematol. 2013, 41, 858–869.e4. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.K.; Fei, D.L.; Schilling, N.S.; Ahmed, Y.F.; Hwa, J.; Robbins, D.J. G protein galphai functions immediately downstream of smoothened in hedgehog signalling. Nature 2008, 456, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.P.; Mongeau, M.E.; Klimstra, D.S.; Morris, J.P.; Lee, Y.C.; Kawaguchi, Y.; Wright, C.V.; Hebrok, M.; Lewis, B.C. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinogen. 2009, 48, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Gotschel, F.; Berg, D.; Gruber, W.; Bender, C.; Eberl, M.; Friedel, M.; Sonntag, J.; Rungeler, E.; Hache, H.; Wierling, C.; et al. Synergism between Hedgehog-Gli and EGFR signaling in Hedgehog-responsive human medulloblastoma cells induces downregulation of canonical Hedgehog-target genes and stabilized expression of GLI1. PLoS ONE 2013, 8, e65403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhou, J.; Song, W.; Wu, D.; Dang, Q.; Zhang, L.; Li, L.; Wang, X.; He, D. Role of GLI-1 in epidermal growth factor-induced invasiveness of ARCaPE prostate cancer cells. Oncol. Rep. 2013, 30, 904–910. [Google Scholar] [PubMed]
- Fogarty, M.P.; Emmenegger, B.A.; Grasfeder, L.L.; Oliver, T.G.; Wechsler-Reya, R.J. Fibroblast growth factor blocks sonic hedgehog signaling in neuronal precursors and tumor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Tgfbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Mechlin, C.W.; Tanner, M.J.; Chen, M.; Buttyan, R.; Levin, R.M.; Mian, B.M. Gli2 expression and human bladder transitional carcinoma cell invasiveness. J. Urol. 2010, 184, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Verrecchia, F.; Mauviel, A. Cloning of the human GLI2 promoter: Transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H. Protein kinase c (PKC) isozymes and cancer. New J. Sci. 2014, 2014, 36. [Google Scholar] [CrossRef]
- Li, J.; Evers, B.M. Hedgehog and Protein Kinase C Signaling. In Hedgehog Signaling Activation in Human Cancer and Its Clinical Implications; Xie, J., Ed.; Springer: New York, NY, USA, 2011; pp. 77–83. [Google Scholar]
- Neill, G.W.; Ghali, L.R.; Green, J.L.; Ikram, M.S.; Philpott, M.P.; Quinn, A.G. Loss of protein kinase Calpha expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 2003, 63, 4692–4697. [Google Scholar] [PubMed]
- Cai, Q.; Li, J.; Gao, T.; Xie, J.; Evers, B.M. Protein kinase Cdelta negatively regulates hedgehog signaling by inhibition of Gli1 activity. J. Biol. Chem. 2009, 284, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. Gli activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require Hedgehog-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Gurung, B.; Feng, Z.; Hua, X. Menin directly represses Gli1 expression independent of canonical hedgehog signaling. Mol. Cancer Res. 2013, 11, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Tu, K.; Li, C.; Lu, Z.; Roberts, L.R.; Zheng, X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1712. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; de Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.H.; Ayad, N.G.; et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Wang, J.; Liu, Y.; Peng, Y.; Tan, W. GPCR-like signaling mediated by smoothened contributes to acquired chemoresistance through activating Gli. Mol. Cancer 2014, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-activated protein kinase directly phosphorylates and destabilizes hedgehog pathway transcription factor GLI1 in medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Toftgard, R.; Zaphiropoulos, P.G.; Shimokawa, T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer 2010, 1, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevag, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer. 2013, 132, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers 2015, 7, 1684-1698. https://doi.org/10.3390/cancers7030857
Gu D, Xie J. Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers. 2015; 7(3):1684-1698. https://doi.org/10.3390/cancers7030857
Chicago/Turabian StyleGu, Dongsheng, and Jingwu Xie. 2015. "Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions" Cancers 7, no. 3: 1684-1698. https://doi.org/10.3390/cancers7030857
APA StyleGu, D., & Xie, J. (2015). Non-Canonical Hh Signaling in Cancer—Current Understanding and Future Directions. Cancers, 7(3), 1684-1698. https://doi.org/10.3390/cancers7030857