
Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis

{kind=link}
Abstract
:1. Introduction
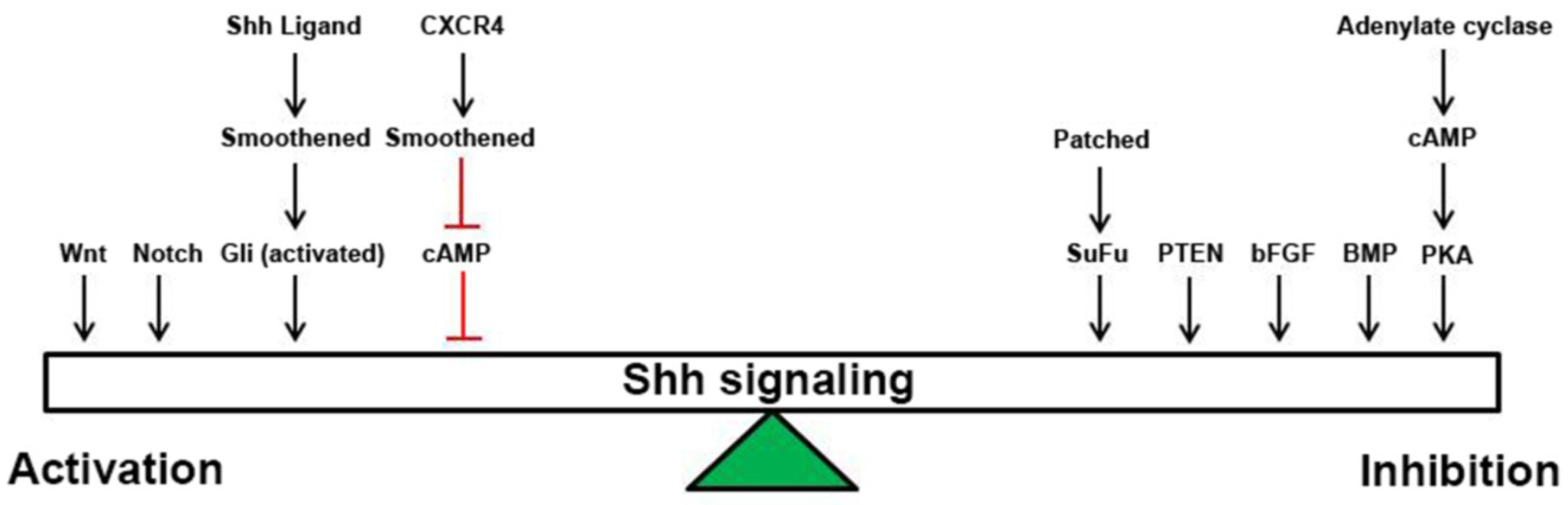
2. Shh Signaling
3. Wnt Signaling
4. Notch Signaling
5. BMP Signaling
6. bFGF Signaling
7. PTEN/Akt/PIK3/PKB Signaling
8. CXCR4 Signaling
9. cAMP Signaling
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Samkari, A.; White, J.C.; Packer, R.J. Medulloblastoma: Toward biologically based management. Semin. Pediatr. Neurol. 2015, 22, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, 4th ed.; IARC: Lyon, France, 2007. [Google Scholar]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-Van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, A.R.; Pritchard, J.I.; Hansen, S.; Benson, M.; Stoeck, J.; Hatton, B.A.; Russell, T.L.; Ellenbogen, R.G.; Bernstein, I.D.; Beachy, P.A.; et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004, 64, 7794–7800. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo model: Hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, C.; Monard, D. SHH pathway and cerebellar development. Cerebellum 2009, 8, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Rubin, J.B.; Gibson, H.D.; DeHaan, E.N.; Alvarez-Hernandez, X.; Segal, R.A.; Luster, A.D. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development 2001, 128, 1971–1981. [Google Scholar] [PubMed]
- Borghesani, P.R.; Peyrin, J.M.; Klein, R.; Rubin, J.; Carter, A.R.; Schwartz, P.M.; Luster, A.; Corfas, G.; Segal, R.A. BDNF stimulates migration of cerebellar granule cells. Development 2002, 129, 1435–1442. [Google Scholar] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M. Vismodegib. Clin. Cancer Res. 2012, 18, 3218–3222. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, Z.; Rikani, A.A.; Choudhry, A.M.; Tariq, S.; Zakaria, F.; Asghar, M.W.; Sarfraz, M.K.; Haider, K.; Shafiq, A.A.; Mobassarah, N.J. Sonic hedgehog signalling pathway: A complex network. Ann. Neurosci. 2014, 21, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Das, T.; Gulati, P.; Biswas, N.K.; Rote, S.; Chatterjee, U.; Ghosh, S.N.; Deb, S.; Saha, S.K.; Chowdhury, A.K.; et al. Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS ONE 2015, 10, e0116390. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.A.; Takada, R.; McMahon, A.P. Proteolytic processing yields two secreted forms of sonic hedgehog. Mol. Cell. Biol. 1995, 15, 2294–2303. [Google Scholar] [PubMed]
- Pepinsky, R.B.; Zeng, C.; Wen, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.; Goswami, D.; Manonmani, A.; Sharma, P.; Ranganath, H.A.; VijayRaghavan, K.; Shashidhara, L.S.; Sowdhamini, R.; Mayor, S. Nanoscale organization of hedgehog is essential for long-range signaling. Cell 2008, 133, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, P.; Exner, S.; Schurmann, S.; Pickhinke, U.; Bandari, S.; Ortmann, C.; Kupich, S.; Schulz, P.; Hansen, U.; Seidler, D.G.; et al. Scube2 enhances proteolytic Shh processing from the surface of Shh-producing cells. J. Cell. Sci. 2014, 127, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.K. Hedgehog pathway inhibitors: A patent review (2009–present). Expert Opin. Ther. Pat. 2013, 23, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Powers, G.L.; Hammer, K.D.; Domenech, M.; Frantskevich, K.; Malinowski, R.L.; Bushman, W.; Beebe, D.J.; Marker, P.C. Phosphodiesterase 4D inhibitors limit prostate cancer growth potential. Mol. Cancer Res. 2015, 13, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.P.; Kuhl, M. An updated overview on Wnt signaling pathways: A prelude for more. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt/beta-catenin signaling target genes. Available online: http://web.stanford.edu/~rnusse/pathways/targets.html (accessed on 27 June 2015).
- Teodorczyk, M.; Schmidt, M.H. Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Gaiano, N.; Nye, J.S.; Fishell, G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 2000, 26, 395–404. [Google Scholar] [CrossRef]
- Chiba, S. Notch signaling in stem cell systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Solecki, D.J.; Liu, X.L.; Tomoda, T.; Fang, Y.; Hatten, M.E. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron 2001, 31, 557–568. [Google Scholar] [CrossRef]
- Fan, X.; Mikolaenko, I.; Elhassan, I.; Ni, X.; Wang, Y.; Ball, D.; Brat, D.J.; Perry, A.; Eberhart, C.G. Notch1 and Notch2 Have Opposite Effects on Embryonal Brain Tumor Growth. Cancer Res. 2004, 64, 7787–7793. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, B.M.; Oliveira, I.D.; Alves, M.T.; Saba-Silva, N.; Capellano, A.M.; Cavalheiro, S.; Dastoli, P.; Toledo, S.R. SHH, WNT, and NOTCH pathways in medulloblastoma: When cancer stem cells maintain self-renewal and differentiation properties. Childs Nerv. Syst. 2014, 30, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Fiaschetti, G.; Schroeder, C.; Castelletti, D.; Arcaro, A.; Westermann, F.; Baumgartner, M.; Shalaby, T.; Grotzer, M.A. NOTCH ligands JAG1 and JAG2 as critical pro-survival factors in childhood medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 39. [Google Scholar] [CrossRef] [PubMed]
- De Antonellis, P.; Medaglia, C.; Cusanelli, E.; Andolfo, I.; Liguori, L.; de Vita, G.; Carotenuto, M.; Bello, A.; Formiggini, F.; Galeone, A.; et al. MiR-34a targeting of Notch ligand delta-like 1 impairs CD15+/CD133+ tumor-propagating cells and supports neural differentiation in medulloblastoma. PLoS ONE 2011, 6, e24584. [Google Scholar] [CrossRef] [PubMed]
- Ingram, W.J.; McCue, K.I.; Tran, T.H.; Hallahan, A.R.; Wainwright, B.J. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene 2008, 27, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Dave, R.K.; Ellis, T.; Toumpas, M.C.; Robson, J.P.; Julian, E.; Adolphe, C.; Bartlett, P.F.; Cooper, H.M.; Reynolds, B.A.; Wainwright, B.J. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS ONE 2011, 6, e14680. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.; Li, Y.; Miller, E.E.; Shih, D.J.; Taylor, M.D.; Stearns, T.M.; Bronson, R.T.; Ackerman, S.L.; Yoon, J.K.; Yun, K. Notch1-Induced Brain Tumor Models the Sonic Hedgehog Subgroup of Human Medulloblastoma. Cancer Res. 2013, 73, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Pritchard, J.; LeBlanc, M.; Hansen, S.; Ulrich, M.; Ditzler, S.; Pullar, B.; Stroud, M.R.; Olson, J.M. Notch signaling is not essential in sonic hedgehog-activated medulloblastoma. Oncogene 2010, 29, 3865–3872. [Google Scholar] [CrossRef] [PubMed]
- Julian, E.; Dave, R.K.; Robson, J.P.; Hallahan, A.R.; Wainwright, B.J. Canonical Notch signaling is not required for the growth of Hedgehog pathway-induced medulloblastoma. Oncogene 2010, 29, 3465–3476. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, M.R.; Weiss, W.A. BMPs oppose Math1 in cerebellar development and in medulloblastoma. Genes Dev. 2008, 22, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Alder, J.; Lee, K.J.; Jessell, T.M.; Hatten, M.E. Generation of cerebellar granule neurons in vivo by transplantation of BMP-treated neural progenitor cells. Nat. Neurosci. 1999, 2, 535–540. [Google Scholar] [PubMed]
- Rios, I.; Alvarez-Rodriguez, R.; Marti, E.; Pons, S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development 2004, 131, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ayrault, O.; Zindy, F.; Kim, J.H.; Roussel, M.F. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 2008, 22, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, A.R.; Pritchard, J.I.; Chandraratna, R.A.; Ellenbogen, R.G.; Geyer, J.R.; Overland, R.P.; Strand, A.D.; Tapscott, S.J.; Olson, J.M. BMP-2 mediates retinoid-induced apoptosis in medulloblastoma cells through a paracrine effect. Nat. Med. 2003, 9, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Fogarty, M.P.; Emmenegger, B.A.; Grasfeder, L.L.; Oliver, T.G.; Wechsler-Reya, R.J. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Emmenegger, B.A.; Hwang, E.I.; Moore, C.; Markant, S.L.; Brun, S.N.; Dutton, J.W.; Read, T.A.; Fogarty, M.P.; Singh, A.R.; Durden, D.L.; et al. Distinct roles for fibroblast growth factor signaling in cerebellar development and medulloblastoma. Oncogene 2013, 32, 4181–4188. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Castellino, R.C.; Durden, D.L. Mechanisms of disease: the PI3K-Akt-PTEN signaling node—An intercept point for the control of angiogenesis in brain tumors. Nat. Clin. Pract. Neurol. 2007, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Packer, R.J.; Rorke, L.B.; Emanuel, B.S. Chromosome abnormalities in pediatric brain tumors. Cancer Res. 1988, 48, 175–180. [Google Scholar] [PubMed]
- Lastowska, M.; Al-Afghani, H.; Al-Balool, H.H.; Sheth, H.; Mercer, E.; Coxhead, J.M.; Redfern, C.P.; Peters, H.; Burt, A.D.; Santibanez-Koref, M.; et al. Identification of a neuronal transcription factor network involved in medulloblastoma development. Communication 2013, 1, 35. [Google Scholar] [CrossRef]
- Metcalfe, C.; Alicke, B.; Crow, A.; Lamoureux, M.; Dijkgraaf, G.J.; Peale, F.; Gould, S.E.; de Sauvage, F.J. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer Res. 2013, 73, 7034–7042. [Google Scholar] [CrossRef] [PubMed]
- Castellino, R.C.; Barwick, B.G.; Schniederjan, M.; Buss, M.C.; Becher, O.; Hambardzumyan, D.; Macdonald, T.J.; Brat, D.J.; Durden, D.L. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS ONE 2010, 5, e10849. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, W.; Digon-Sontgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sorensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Tautermann, C.S. GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett. 2014, 24, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, R.; Dubuc, A.; Ward, S.; Yang, L.; Northcott, P.; Woerner, B.M.; Kroll, K.; Luo, J.; Taylor, M.D.; Wechsler-Reya, R.J.; et al. CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 2012, 72, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2015. [Google Scholar] [CrossRef]
- Tissir, F.; Wang, C.E.; Goffinet, A.M. Expression of the chemokine receptor Cxcr4 mRNA during mouse brain development. Brain Res. Dev. Brain Res. 2004, 149, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, K.; Zhang, E.E.; Ke, Y.H.; Liu, G.; Liu, J.J.; Rao, Y.; Feng, G.S. Shp2 acts downstream of SDF-1alpha/CXCR4 in guiding granule cell migration during cerebellar development. Dev. Biol. 2009, 334, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Hurowitz, E.H.; Melnyk, J.M.; Chen, Y.J.; Kouros-Mehr, H.; Simon, M.I.; Shizuya, H. Genomic characterization of the human heterotrimeric G protein alpha, beta, and gamma subunit genes. DNA Res. 2000, 7, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Mukhopadhyay, S. Primary cilium and sonic hedgehog signaling during neural tube patterning: Role of GPCRs and second messengers. Dev. Neurobiol. 2015, 75, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Makinodan, E.; Marneros, A.G. Protein kinase A activation inhibits oncogenic Sonic hedgehog signalling and suppresses basal cell carcinoma of the skin. Exp. Dermatol. 2012, 21, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Phan, T.; Storm, D.R. The type 3 adenylyl cyclase is required for novel object learning and extinction of contextual memory: Role of cAMP signaling in primary cilia. J. Neurosci. 2011, 31, 5557–5561. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Kasahara, K.; Miyazaki, I.; Asanuma, M. Lithium treatment elongates primary cilia in the mouse brain and in cultured cells. Biochem. Biophys. Res. Commun. 2009, 388, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Tuson, M.; He, M.; Anderson, K.V. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Nicot, A.; Lelievre, V.; Tam, J.; Waschek, J.A.; DiCicco-Bloom, E. Pituitary adenylate cyclase-activating polypeptide and sonic hedgehog interact to control cerebellar granule precursor cell proliferation. J. Neurosci. 2002, 22, 9244–9254. [Google Scholar] [PubMed]
- Niewiadomski, P.; Zhujiang, A.; Youssef, M.; Waschek, J.A. Interaction of PACAP with Sonic hedgehog reveals complex regulation of the hedgehog pathway by PKA. Cell. Signal. 2013, 25, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, L.; Chen, Y.; Remke, M.; Shih, D.; Lu, F.; Wang, H.; Deng, Y.; Yu, Y.; Xia, Y.; et al. The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat. Med. 2014, 20, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, R.; Sun, T.; Warrington, N.M.; Rubin, J.B. Treating brain tumors with PDE4 inhibitors. Trends Pharmacol. Sci. 2011, 32, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [PubMed]
- Goldhoff, P.; Warrington, N.M.; Limbrick, D.D., Jr.; Hope, A.; Woerner, B.M.; Jackson, E.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin. Cancer Res. 2008, 14, 7717–7725. [Google Scholar] [CrossRef] [PubMed]
- Bassilana, F.; Carlson, A.; DaSilva, J.A.; Grosshans, B.; Vidal, S.; Beck, V.; Wilmeringwetter, B.; Llamas, L.A.; Showalter, T.B.; Rigollier, P.; et al. Target identification for a Hedgehog pathway inhibitor reveals the receptor GPR39. Nat. Chem. Biol. 2014, 10, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.L.; de Farias, C.B.; Abujamra, A.L.; Kapczinski, F.; Schwartsmann, G.; Brunetto, A.L.; Roesler, R. BDNF and PDE4, but not the GRPR, regulate viability of human medulloblastoma cells. J. Mol. Neurosci. 2010, 40, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jackson, E.; Woerner, B.M.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007, 67, 651–658. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, S.A.; Rubin, J.B. Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis. Cancers 2015, 7, 1484-1498. https://doi.org/10.3390/cancers7030848
Ward SA, Rubin JB. Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis. Cancers. 2015; 7(3):1484-1498. https://doi.org/10.3390/cancers7030848
Chicago/Turabian StyleWard, Stacey A., and Joshua B. Rubin. 2015. "Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis" Cancers 7, no. 3: 1484-1498. https://doi.org/10.3390/cancers7030848
APA StyleWard, S. A., & Rubin, J. B. (2015). Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis. Cancers, 7(3), 1484-1498. https://doi.org/10.3390/cancers7030848