MET and Small-Cell Lung Cancer



Abstract
:1. Introduction
2. Molecular Profiling of Small-Cell Lung Cancer (SCLC)

{kind=link}
{kind=link}
| Author | no. of Samples | Test(s) | no. (%) of Genomic Alterations | Type of Alterations |
|---|---|---|---|---|
| Shibata [9] | 15 tumors | Direct sequencing | 2 (13%) | PIK3CA mut |
| 13 cell lines | 3 (23%) | |||
| Yokomizo [10] | 10 tumors | DHPLC Direct sequencing | 1 (10%) | PTEN/MMAC1 mut |
| 34 cell lines | 6 (18%) | |||
| Tatematsu [11] | 122 tumors | Direct sequencing FISH | 5 (4%) | EGFR mut |
| 4 (3%) | EGFR ampl | |||
| Wakuda [12] | 60 tumors | Pyrosequencing | 13 in 9 cases (15%) | 4 PIK3CA ampl, 1 PIK3CA mut, 1 EGFR mut + PIK3CA mut, 1 KRAS mut, 1 AKT1 mut + PIK3CA ampl, 1 PIK3CA mut + MET ampl + PIK3CA ampl |
| Umemura [13] | 51 tumors | Whole-exome sequencing, copy-number analysis | 18 (36%) | PIK3CA pathway |
| 3 (6%) | PIK3CA mut | |||
| Peifer [14] | 97 tumors 2 cell lines | SNP; exome-, transcriptome- and genome-sequencing | 29 (100%) | TP53/Rb1 mut and loss |
| 18 (18%) | CREBBP/EP300 mut | |||
| 10 (16%) | MYC ampl | |||
| 9 (10%) | SLIT2 mut | |||
| 3 (10%) | PTEN mut | |||
| 3 (10%) | EPHA7 mut | |||
| 3 (10%) | MLL mut | |||
| 3 (6%) | FGFR1 ampl | |||
| Rudin [17] | 40 tumors 40 cell lines | Exome sequencing, RNA-sequencing, whole-genome sequencing, RT-PCR, FISH and IHC | 33 (78%) | TP53 mut |
| 14 (33%) | Rb1 mut | |||
| 15 (27%) | SOX2 ampl | |||
| 5 (9%) | RLF-MYCL1 fusion gene | |||
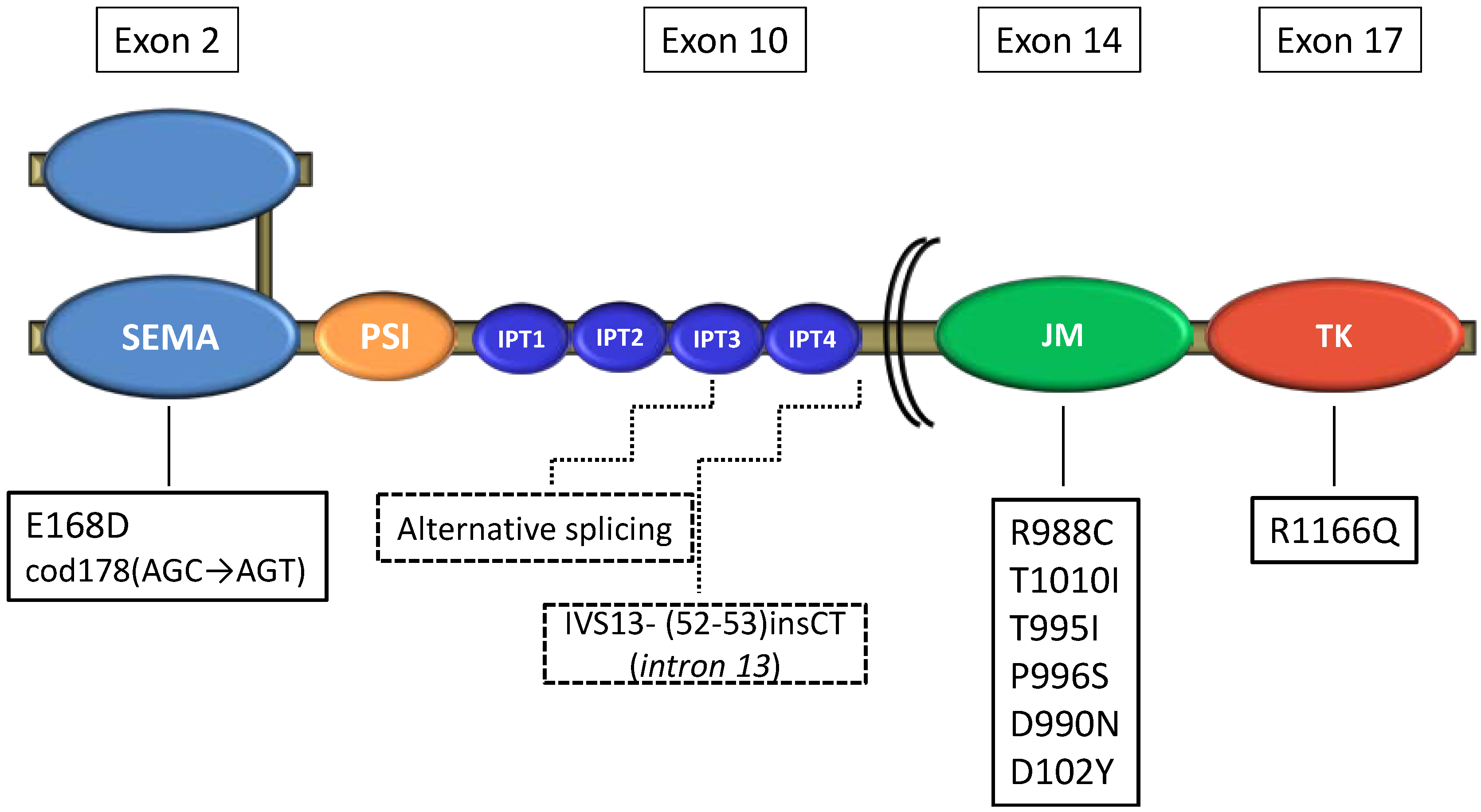
| Ma [18] | 32 tumors | Sequencing | 4 (12%) | JM mut, Sema mut, pre-JM intron 13 mut |
| 10 cell lines | 3 (30%) | JM mut, alternative transcript involving exon 10 | ||
| de Aguirre [19] | 44 tumors | Direct sequencing | 3 (8%) | JM mut, Sema mut |
| Voortman [20] | 46 tumors | Sequencing | 3 (6.5%) | JM mut |
| 13 cell lines | 3 (25%) | |||
| Bordi [21] | 113 tumors | Direct sequencing | 5 (4.4%) | JM mut, TK mut |
3. MET: Structure, Function and Aberrant Signaling in Tumor
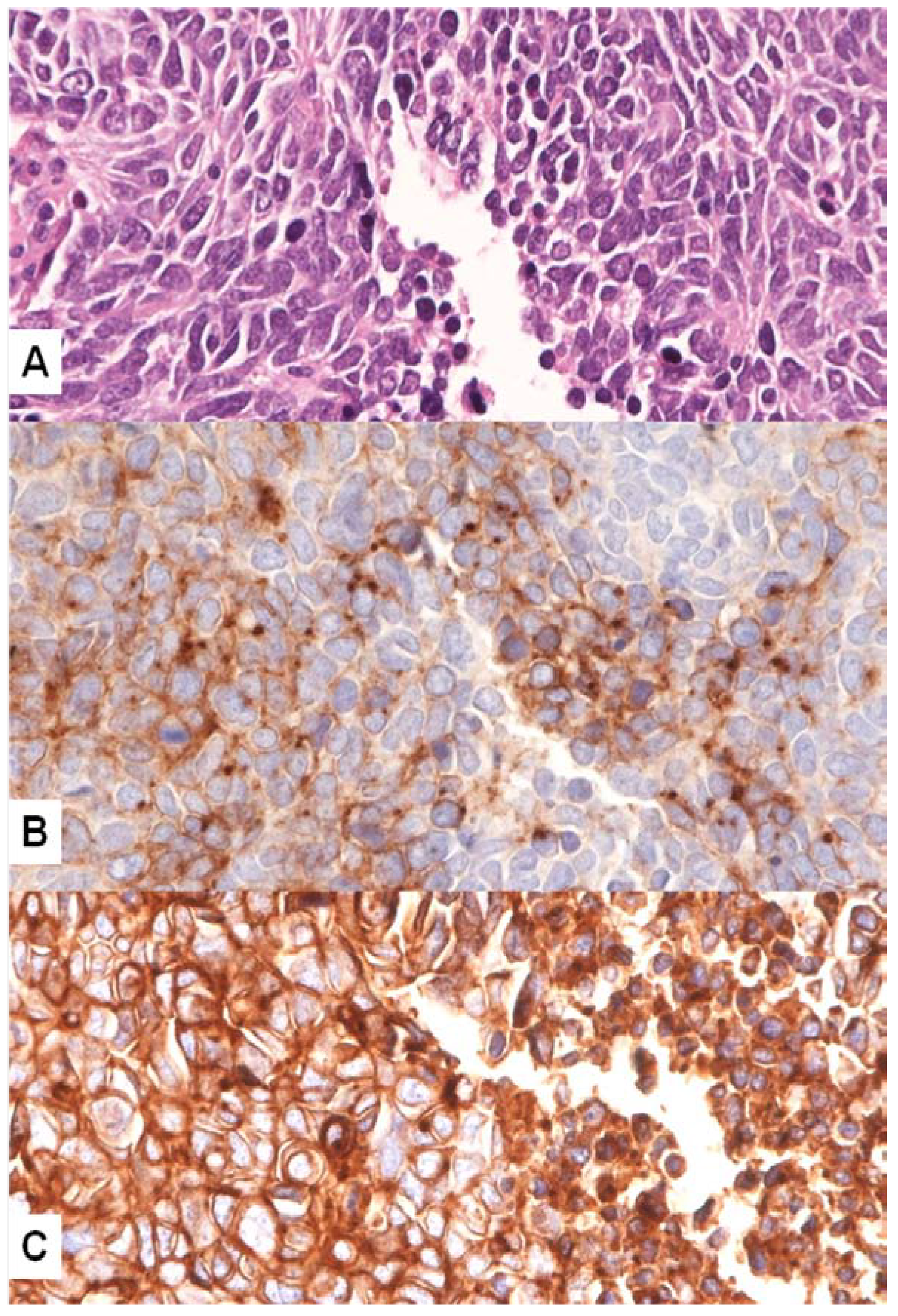
4. MET Pathway and SCLC
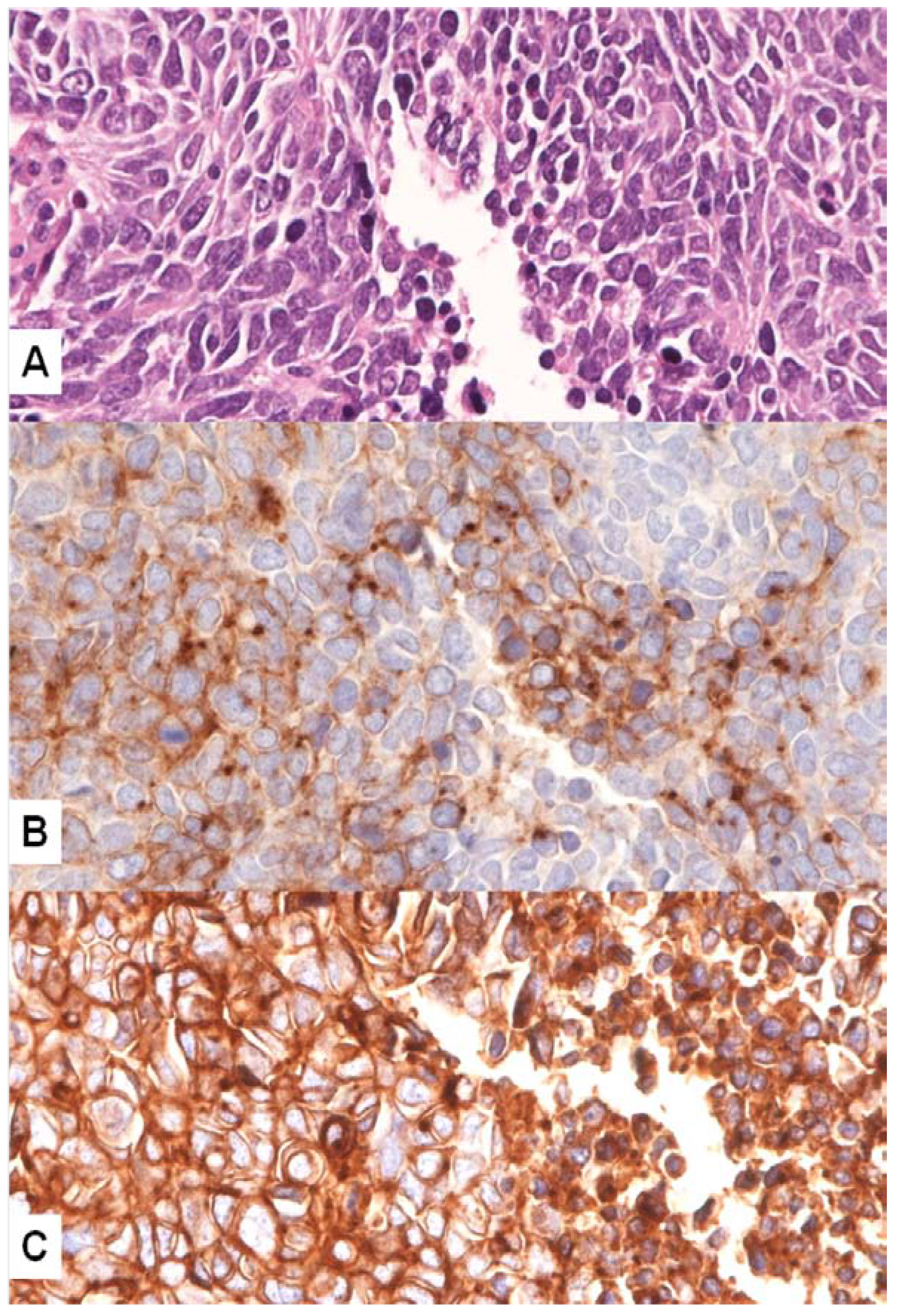
MET Mutations in SCLC
5. Therapeutic Implications
5.1. MET Inhibitors and Their Potential Activity in Restoring Chemo-Sensitivity: Data from Preclinical Studies
5.2. MET Inhibitors in Combination with Chemotherapy: Data from Clinical Studies
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Van Meerbeeck, J.P.; Fennel, D.A.; de Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar]
- Takada, M.; Fukuoka, M.; Kawahara, M.; Sugiura, T.; Yokoyama, A.; Yokota, S.; Nishiwaki, Y.; Watanabe, K.; Noda, K.; Tamura, T.; et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: Results of the Japan Clinical Oncology Group Study 9104. J. Clin. Oncol. 2002, 20, 3054–3060. [Google Scholar] [CrossRef]
- Turrisi, A.T., 3rd; Kim, K.; Blum, R.; Sause, W.T.; Livingston, R.B.; Komaki, R.; Wagner, H.; Aisner, S.; Johnson, D.H. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N. Engl. J. Med. 1999, 340, 265–271. [Google Scholar] [CrossRef]
- Auperin, A.; Arriagada, R.; Pignon, J.P.; le Péchoux, C.; Gregor, A.; Stephens, R.J.; Kristjansen, P.E.; Johnson, B.E.; Ueoka, H.; Wagner, H.; et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. N. Engl. J. Med. 1999, 341, 476–484. [Google Scholar] [CrossRef]
- Albain, K.S.; Crowley, J.J.; LeBlanc, M.; Livingston, R.B. Determinants of improved outcome in small-cell lung cancer: An analysis of the 2,580-patient Southwest Oncology Group data base. J. Clin. Oncol. 1990, 8, 1563–1574. [Google Scholar] [PubMed]
- Wistuba, I.I.; Gazdar, A.F.; Minna, J.D. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Arriola, E.; Canadas, I.; Arumi, M.; Rojo, F.; Rovira, A.; Albanell, J. Genetic changes in small cell lung carcinoma. Clin. Transl. Oncol. 2008, 10, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Kokubu, A.; Tsuta, K.; Hirohashi, S. Oncogenic mutation of PIK3CA in small cell lung carcinoma: A potential therapeutic target pathway for chemotherapy-resistant lung cancer. Cancer Lett. 2009, 283, 6092–6096. [Google Scholar] [CrossRef]
- Yokomizo, A.; Tindall, D.J.; Drabkin, H.; Gemmill, R.; Franklin, W.; Yang, P.; Sugio, K.; Smith, D.I.; Liu, W. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene 1998, 17, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, A.; Shimizu, J.; Murakami, Y.; Horio, Y.; Nakamura, S.; Hida, T.; Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor mutations in small cell lung cancer. Clin. Cancer Res. 2008, 14, 6092–6096. [Google Scholar] [CrossRef] [PubMed]
- Wakuda, K.; Kenmotsu, H.; Serizawa, M.; Koh, Y.; Isaka, M.; Takahashi, S.; Ono, A.; Taira, T.; Naito, T.; Murakami, H.; et al. Molecular profiling of small cell lung cancer in a Japanese cohort. Lung Cancer 2014, 84, 139–144. [Google Scholar] [CrossRef]
- Umemura, S.; Goto, K.; Mimaki, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; Nagai, K.; et al. Comprehensive genomic analysis of small cell lung cancer in Asian patients. J. Clin. Oncol. 2013, 31. Abstract 7512. [Google Scholar]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, LH.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analysis identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J.; et al. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef]
- Thomas, A.; Lee, J.-H.; Abdullaev, Z.; Park, K.S.; Pineda, M.; Saidkhodjaeva, L.; Miettinen, M.; Wang, Y.; Pack, S.D.; Giaccone, G. Characterization of fibroblast growth factor receptor 1 in small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. C-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar] [PubMed]
- De Aguirre, I.; Salvatierra, A.; Font, A.; Mate, J.L.; Perez, M.; Botia, M.; Taron, M.; Rosell, R. C-Met mutational analysis in the sema and juxtamembrane domains in small-cell-lung-cancer. Transl. Oncogenomics 2006, 1, 11–18. [Google Scholar] [PubMed]
- Voortman, J.; Harada, T.; Chang, R.P.; Killian, J.K.; Suuriniemi, M.; Smith, W.I.; Meltzer, P.S.; Lucchi, M.; Wang, Y.; Giaccone, G. Detection and therapeutic implications of c-Met mutations in small cell lung cancer and neuroendocrine tumors. Curr. Pharm. Des. 2013, 19, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Bordi, P.; Tiseo, M.; Barbieri, F.; Bavieri, M.; Sartori, G.; Marchetti, A.; Buttitta, F.; Bortesi, B.; Ambrosini-Spaltro, A.; Gnetti, L.; et al.; University Hospital of Parma, Parma, Italy Gene mutations in small-cell lung cancer (SCLC): Results of a panel of 6 genes in a cohort of Italian patients. Unpublished work. 2014. [Google Scholar]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ re generation and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Arnesano, A.; Galluzzo, M.; Comoglio, P.M.; Michieli, P. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of Met. J. Biol. Chem. 2008, 283, 21267–21277. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Furge, K.A.; Zhang, Y.W.; Vande Woude, G.F. Met receptor tyrosine kinase: Enhanced signaling through adapter proteins. Oncogene 2000, 19, 5582–5589. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Gao, M.; Meng, Q.; Laterra, J.J.; Symons, M.H.; Coniglio, S.; Pestell, R.G.; Goldberg, I.D.; Rosen, E.M. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene 2005, 24, 1749–1766. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Morotti, A.; Ponzetto, C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated cell proliferation and tubulogenesis. Mol. Cell. Biol. 2002, 22, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Maina, F.; Hilton, M.C.; Ponzetto, C.; Davies, A.M.; Klein, R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997, 11, 3341–3350. [Google Scholar] [CrossRef] [PubMed]
- Maina, F.; Hilton, M.C.; Andres, R.; Wyatt, S.; Klein, R.; Davies, A.M. Multiple roles for hepatocyte growth factor in sympathetic neuron development. Neuron 1998, 20, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Helmbacher, F.; Dessaud, E.; Arber, S.; de Lapeyrière, O.; Henderson, C.E.; Klein, R.; Maina, F. Met signaling is required for recruitment of motor neurons to PEA3-positive motor neurons. Neuron 2003, 39, 767–777. [Google Scholar] [CrossRef]
- Huh, C.G.; Factor, V.M.; Sanchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Garratt, A.N.; Wustefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dai, C.; Liu, Y. Hepatocyte growth factor suppresses renal interstitial myofibroblast activation and intercepts Smad signal transduction. Am. J. Pathol. 2003, 163, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Chmielowiec, J.; Borowiak, M.; Morkel, M.; Stradal, T.; Munz, B.; Werner, S.; Wehland, J.; Birchmeier, C.; Birchmeier, W. c-Met is essential for wound healing in the skin. J. Cell Biol. 2007, 177, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Danilkovitch-Miagkova, A.; Zbar, B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J. Clin. Investig. 2002, 109, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Farina, A.; Rubini, C.; Coccia, E.; Capogreco, M.; Colella, G.; Leonardi, R.; Campisi, G.; Carinci, F. Effect of c-Met expression on survival in head and neck squamous cell carcinoma. Tumor Biol. 2006, 27, 1670–1679. [Google Scholar] [CrossRef]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Radjabi, A.R.; Shinomiya, N.; Kistner, E.; Kenny, H.; Becker, A.R.; Turkyilmaz, M.A.; Salgia, R.; Yamada, S.D.; Vande Woude, G.F.; et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007, 67, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Arriola, E.; Canadas, I.; Arumi-Uria, M.; Dómine, M.; Lopez-Vilariño, J.A.; Arpí, O.; Salido, M.; Menéndez, S.; Grande, E.; Hirsch, F.R.; et al. MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines. Br. J. Cancer 2011, 105, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Weissfeld, L.A.; Singh-Kaw, P.; Weyant, R.J.; Testa, J.R.; Landreneau, R.J. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997, 57, 433–439. [Google Scholar] [PubMed]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, Y.L.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.J.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207. [Google Scholar] [PubMed]
- Okuda, K.; Sasaki, H.; Yukiue, H.; Yano, M.; Fujii, Y. MET gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008, 99, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Masuya, D.; Huang, C.; Liu, D.; Nakashima, T.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumor-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumor growth and prognosis in non-small cell lung cancer patients. Br. J. Cancer 2004, 90, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Maulik, G.; Kijima, T.; Ma, P.C.; Ghosh, S.K.; Lin, J.; Shapiro, G.I.; Schaefer, E.; Tibaldi, E.; Johnson, B.E.; Salgia, R. Modulation of the c-MET/hepatocyte growth factor pathway in small cell lung cancer. Clin. Cancer Res. 2002, 8, 620–627. [Google Scholar] [PubMed]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Thiagarajan, P.S.; Ma, P.C. MET signaling: Novel targeted inhibition and its clinical development in lung cancer. J. Thorac. Oncol. 2012, 7, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Rygaard, K.; Nakamura, T.; Spang-Thomsen, M. Expression of the proto-oncogenes c-met and c-kit and their ligands, hepatocyte growth factor/scatter factor and stem cell factor, in SCLC cell lines and xenografts. Br. J. Cancer 1993, 67, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Rygaard, K.; Klausen, B.; Nakamura, T.; Spangthomsen, M. Growth-inhibition and change in morphology and motility of SCLC cell-lines by hepatocyte growth-factor scatter factor. Int. J. Oncol. 1993, 2, 991–996. [Google Scholar] [PubMed]
- Maulik, G.; Madhiwala, P.; Brooks, S.; Ma, P.C.; Kijima, T.; Tibaldi, E.V.; Schaefer, E.; Parmar, K.; Salgia, R. Activated c-Met signals through PI3K with dramatic effects on cytoskeletal functions in small cell lung cancer. J. Cell. Mol. Med. 2002, 6, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Harshman, L.C.; Choueiri, T.K. Targeting the hepatocyte growth factor/c-Met signaling pathway in renal cell carcinoma. Cancer J. 2013, 19, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F.; Facchinetti, F.; Haspinger, E.R.; Garassino, M.C.; Trusolino, L.; de Braud, F.; Tiseo, M. Targeting the MET gene for the treatment of non-small-cell lung cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A.; et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005, 65, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Fletcher, L.B.; Wang, E.Q.; Yang, W.F.; Rutenberg-Schoenberg, M.L.; Beadling, C.; Mori, M.; Heinrich, M.C.; Deininger, M.W.; Druker, B.J.; et al. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010, 70, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Tretiakova, M.S.; Nallasura, V.; Jagadeeswaran, R.; Husain, A.N.; Salgia, R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: Implications for tumor invasion. Br. J. Cancer 2007, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Lu, B.-B.; Yang, J.-S.; Wang, K.M.; De, W. Adenovirus-mediated siRNA targeting c-Met inhibits proliferation and invasion of small-cell lung cancer (SCLC) cells. J. Surg. Res. 2011, 171, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Canadas, I.; Rojo, F.; Taus, A.; Arpí, O.; Arumí-Uría, M.; Pijuan, L.; Menéndez, S.; Zazo, S.; Dómine, M.; Salido, M.; et al. Targeting epithelial to mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin. Cancer Res. 2014, 20, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Cañadas, I.; Taus, A.; González, I.; Villanueva, X.; Gimeno, J.; Pijuan, L.; Dómine, M.; Sánchez-Font, A.; Vollmer, I.; Menéndez, S.; et al. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget 2014, 5, 5246–5256. [Google Scholar] [PubMed]
- Rolle, C.E.; Kanteti, R.; Surati, M.; Nandi, S.; Dhanasingh, I.; Yala, S.; Tretiakova, M.; Arif, Q.; Hembrough, T.; Brand, T.M.; et al. Combined MET inhibition and topoisomerase I inhibition block cell growth of small cell lung cancer. Mol. Cancer Ther. 2014, 13, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, H.; Oguri, T.; Maeno, K.; Takakuwa, O.; Kunii, E.; Yagi, Y.; Uemura, T.; Kasai, D.; Miyazaki, M.; Niimi, A. Significance of c-MET overexpression in cytotoxic anticancer drug-resistant small-cell lung cancer cells. Cancer Sci. 2014, 105, 1032–1039. [Google Scholar] [PubMed]
- Lorigan, P.; Soria, J.; Stephenson, J.; Maru, A.; Gervais, R.; Zhu, M.; McCaffery, I.; Jiang, Y.; McGreivy, J.; Glisson, B. Safety and pharmacokinetics of first-line AMG 479 (mAb to IGF1R) or AMG 102 (mAb to HGF/SF) with platinum-based chemotherapy in extensive-stage small cell lung cancer (SCLC). ESMO 2010. [Google Scholar] [CrossRef]
- Glisson, B.; Kazarnowicz, A.; Nackaerts, K.; Orlov, S.; Ramlau, R.; Besse, B.; Cobo Dols, M.; Menon, H.; Paz-Ares Rodriguez, L.; Zhang, Y.; et al. A multicenter, double-blind, placebo-controlled, randomized phase 2 study of ganitumab or rilotumumab with platinum-based chemotherapy as first-line treatment for extensive-stage small-cell lung cancer (SCLC). J. Thorac. Oncol. 2013, 8, S220–S221. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelsomino, F.; Rossi, G.; Tiseo, M. MET and Small-Cell Lung Cancer. Cancers 2014, 6, 2100-2115. https://doi.org/10.3390/cancers6042100
Gelsomino F, Rossi G, Tiseo M. MET and Small-Cell Lung Cancer. Cancers. 2014; 6(4):2100-2115. https://doi.org/10.3390/cancers6042100
Chicago/Turabian StyleGelsomino, Francesco, Giulio Rossi, and Marcello Tiseo. 2014. "MET and Small-Cell Lung Cancer" Cancers 6, no. 4: 2100-2115. https://doi.org/10.3390/cancers6042100
APA StyleGelsomino, F., Rossi, G., & Tiseo, M. (2014). MET and Small-Cell Lung Cancer. Cancers, 6(4), 2100-2115. https://doi.org/10.3390/cancers6042100