Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma


Abstract
:1. Introduction
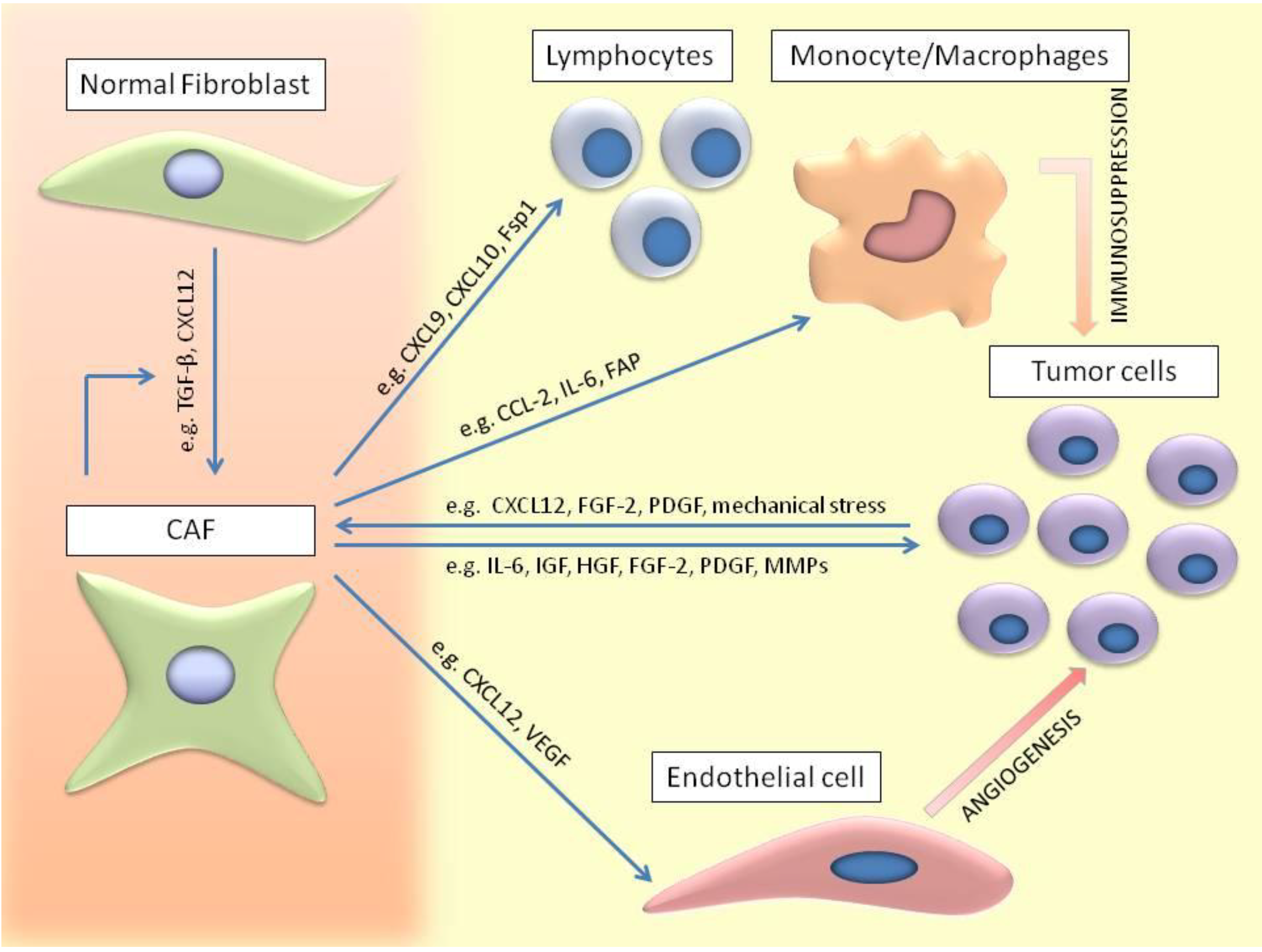
2. Characterization of CAFs

{kind=link}
{kind=link}
{kind=link}
| Fibroblast markers | Ref. | Activation (aggressive) markers | Ref. |
|---|---|---|---|
| Fibroblast specific protein 1 (FSP-1) | [10,15] | Alpha smooth muscle actin (αSma) | [23] |
| Vimentin | [10,16] | Podoplanin | [14] |
| Fibroblast activating protein (FAP) | [9,17] | Thrombospondin (Tsp-1) | [10] |
| Thy1 (CD90) | [13] | ||
| Tenascin C (TN-C) | [10] | ||
| Matrix metalloproteinase (MMP) | [19] | ||
| Neuron glial antigen 2 (NG2) | [12] | ||
| Periostin | [20] | ||
| Palladin | [8] | ||
| PDGFR (α/β) | [12,21,22] |
3. Origin and Generation of CAFs
4. Mechanism of CAFs Activation
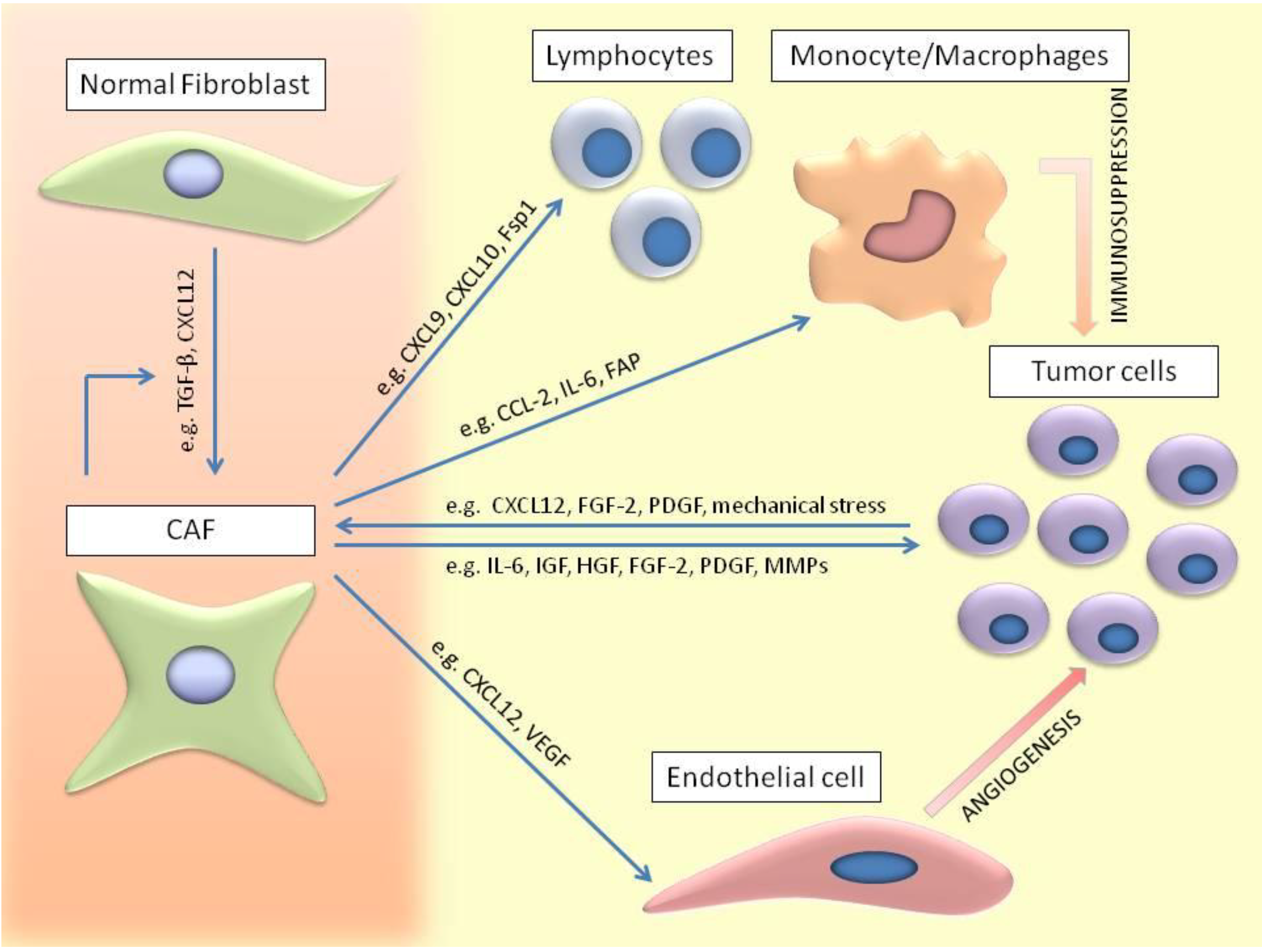
5. CAFs Involvement in Tumor Development
6. CAFs Involvement in Invasion and Metastasis
7. Interaction of CAFs with Immune Cells
8. Role of CAFs in Therapy Resistance
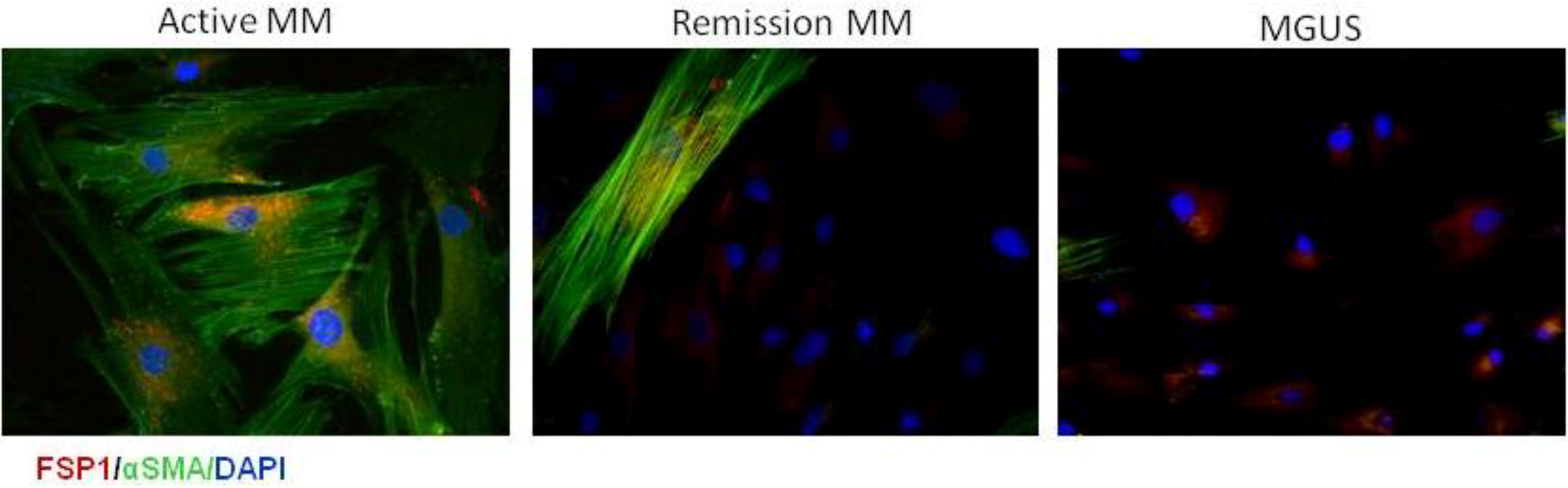
9. CAFs in Multiple Myeloma
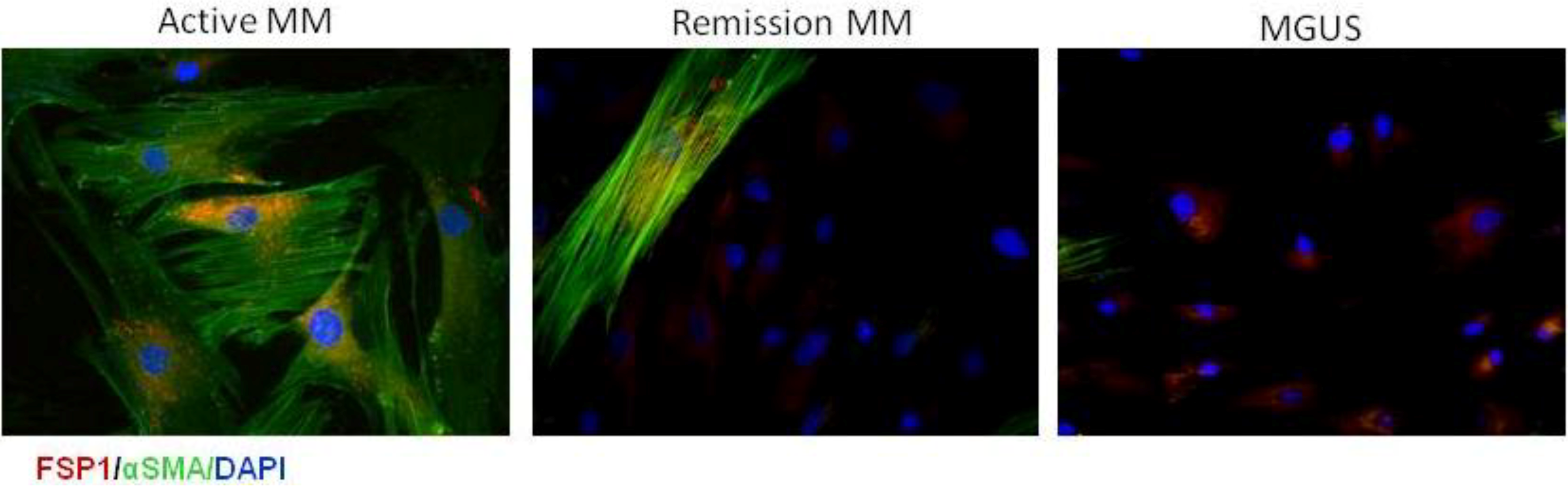
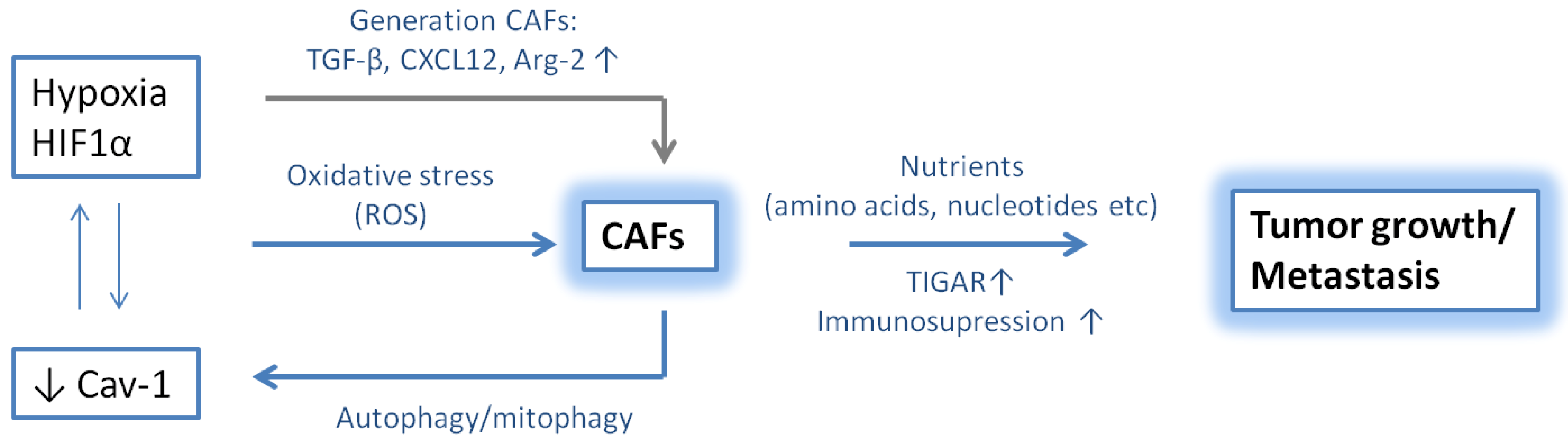
10. Hypoxic Niche in Multiple Myeloma
11. Role of CAFs and Hypoxia in Tumor Progression
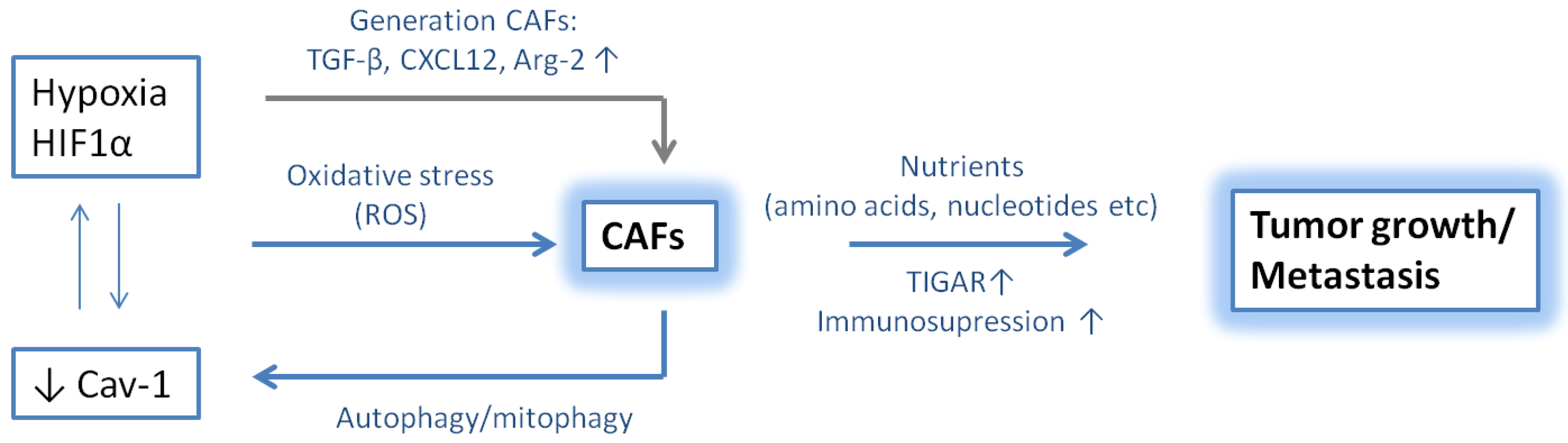
12. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Castells, M.; Thibault, B.; Delord, J.P.; Couderc, B. Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar]
- Madar, S.; Goldstein, I.; Rotter, V. Cancer associated fibroblasts—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol. Ther. 2007, 6, 618–619. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar] [CrossRef]
- Franco, O.E.; Shaw, A.K.; Strand, D.W.; Hayward, S.W. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 2010, 21, 33–39. [Google Scholar]
- Brentnall, T.A.; Lai, L.A.; Coleman, J.; Bronner, M.P.; Pan, S.; Chen, R. Arousal of cancer-associated stroma: Overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One 2012, 7, e30219. [Google Scholar]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One 2009, 4, e7965. [Google Scholar]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 2009, 4, e4992. [Google Scholar] [CrossRef]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- True, L.D.; Zhang, H.; Ye, M.; Huang, C.Y.; Nelson, P.S.; von Haller, P.D.; Tjoelker, L.W.; Kim, J.S.; Qian, W.J.; Smith, R.D.; et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod. Pathol. 2010, 23, 1346–1356. [Google Scholar] [CrossRef]
- Pula, B.; Jethon, A.; Piotrowska, A.; Gomulkiewicz, A.; Owczarek, T.; Calik, J.; Wojnar, A.; Witkiewicz, W.; Rys, J.; Ugorski, M.; et al. Podoplanin expression by cancer-associated fibroblasts predicts poor outcome in invasive ductal breast carcinoma. Histopathology 2011, 59, 1249–1260. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Schurch, W.; Seemayer, T.A.; Lagace, R.; Gabbiani, G. The intermediate filament cytoskeleton of myofibroblasts: An immunofluorescence and ultrastructural study. Virchows. Arch. A Pathol. Anat. Histopathol. 1984, 403, 323–336. [Google Scholar] [CrossRef]
- Wolf, B.B.; Quan, C.; Tran, T.; Wiesmann, C.; Sutherlin, D. On the edge of validation—Cancer protease fibroblast activation protein. Mini Rev. Med. Chem. 2008, 8, 719–727. [Google Scholar] [CrossRef]
- Nakagawa, H.; Liyanarachchi, S.; Davuluri, R.V.; Auer, H.; Martin, E.W., Jr.; de la Chapelle, A.; Frankel, W.L. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 2004, 23, 7366–7377. [Google Scholar] [CrossRef]
- Saad, S.; Gottlieb, D.J.; Bradstock, K.F.; Overall, C.M.; Bendall, L.J. Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Res. 2002, 62, 283–289. [Google Scholar]
- Kikuchi, Y.; Kashima, T.G.; Nishiyama, T.; Shimazu, K.; Morishita, Y.; Shimazaki, M.; Kii, I.; Horie, H.; Nagai, H.; Kudo, A.; et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J. Histochem. Cytochem 2008, 56, 753–764. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κb-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Gantt, G.A.J.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; Leahy, P.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Y.; Qiu, S.; Chen, Z.N.; Fan, Z. A novel role of EMMPRIN/CD147 in transformation of quiescent fibroblasts to cancer-associated fibroblasts by breast cancer cells. Cancer Lett. 2013, 335, 380–386. [Google Scholar] [CrossRef]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. Micrornas reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef]
- Mi, Z.; Bhattacharya, S.D.; Kim, V.M.; Guo, H.; Talbot, L.J.; Kuo, P.C. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32, 477–487. [Google Scholar] [CrossRef]
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth—Bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S.; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Peng, Q.; Zhao, L.; Hou, Y.; Sun, Y.; Wang, L.; Luo, H.; Peng, H.; Liu, M. Biological characteristics and genetic heterogeneity between carcinoma-associated fibroblasts and their paired normal fibroblasts in human breast cancer. PLoS One 2013, 8, e60321. [Google Scholar]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.D.; van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar]
- Murata, T.; Mizushima, H.; Chinen, I.; Moribe, H.; Yagi, S.; Hoffman, R.M.; Kimura, T.; Yoshino, K.; Ueda, Y.; Enomoto, T.; et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011, 71, 6633–6642. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Ronnov-Jessen, L.; Petersen, O.W. Induction of α-smooth muscle actin by transforming growth factor-β 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 1993, 68, 696–707. [Google Scholar]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, D.; Wang, Y.; Sun, P.; Hou, X.; Larner, J.; Xiong, W.; Mi, J. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Neufert, C.; Becker, C.; Tureci, O.; Waldner, M.J.; Backert, I.; Floh, K.; Atreya, I.; Leppkes, M.; Jefremow, A.; Vieth, M.; et al. Tumor fibroblast-derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J. Clin. Invest. 2013, 123, 1428–1443. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Akashi, T.; Minami, J.; Ishige, Y.; Eishi, Y.; Takizawa, T.; Koike, M.; Yanagishita, M. Basement membrane matrix modifies cytokine interactions between lung cancer cells and fibroblasts. Pathobiology 2005, 72, 250–259. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, O. Tumour—Stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45, S163–S175. [Google Scholar] [CrossRef]
- Chiquet, M.; Gelman, L.; Lutz, R.; Maier, S. From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim. Biophys. Acta 2009, 1793, 911–920. [Google Scholar] [CrossRef]
- Gillette, M.; Bray, K.; Blumenthaler, A.; Vargo-Gogola, T. P190B RhoGAP overexpression in the developing mammary epithelium induces TGFβ-dependent fibroblast activation. PLoS One 2013, 8, e65105. [Google Scholar]
- Barcellos-Hoff, M.H.; Ravani, S.A. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000, 60, 1254–1260. [Google Scholar]
- Trimis, G.; Chatzistamou, I.; Politi, K.; Kiaris, H.; Papavassiliou, A.G. Expression of p21waf1/Cip1 in stromal fibroblasts of primary breast tumors. Hum. Mol. Genet. 2008, 17, 3596–3600. [Google Scholar] [CrossRef]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. USA 2009, 106, 3372–3377. [Google Scholar]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Li, Q.; Wang, W.; Yamada, T.; Matsumoto, K.; Sakai, K.; Bando, Y.; Uehara, H.; Nishioka, Y.; Sone, S.; Iwakiri, S.; et al. Pleural mesothelioma instigates tumor-associated fibroblasts to promote progression via a malignant cytokine network. Am. J. Pathol. 2011, 179, 1483–1493. [Google Scholar] [CrossRef]
- Kinoshita, H.; Hirata, Y.; Nakagawa, H.; Sakamoto, K.; Hayakawa, Y.; Takahashi, R.; Nakata, W.; Sakitani, K.; Serizawa, T.; Hikiba, Y.; et al. Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS One 2013, 8, e60914. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1α co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef]
- Vermeulen, L.; de Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–477. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Maman, S.; Edry-Botzer, L.; Sagi-Assif, O.; Meshel, T.; Yuan, W.; Lu, W.; Witz, I.P. The metastatic microenvironment: Lung-derived factors control the viability of neuroblastoma lung metastasis. Int. J. Cancer 2013, 133, 2296–2306. [Google Scholar]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Goicoechea, S.M.; Garcia-Mata, R.; Staub, J.; Valdivia, A.; Sharek, L.; McCulloch, C.G.; Hwang, R.F.; Urrutia, R.; Yeh, J.J.; Kim, H.J.; et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene 2014, 33, 1265–1273. [Google Scholar] [CrossRef]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef]
- Pena, C.; Cespedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edqvist, P.H.; Hagglof, C.; Birgisson, H.; Bojmar, L.; Jirstrom, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2012, 73, 1287–1297. [Google Scholar]
- Raz, Y.; Erez, N. An inflammatory vicious cycle: Fibroblasts and immune cell recruitment in cancer. Exp. Cell Res. 2013, 319, 1596–1603. [Google Scholar] [CrossRef]
- Servais, C.; Erez, N. From sentinel cells to inflammatory culprits: Cancer-associated fibroblasts in tumour-related inflammation. J. Pathol. 2013, 229, 198–207. [Google Scholar] [CrossRef]
- Fu, Z.; Zuo, Y.; Li, D.; Xu, W.; Chen, H.; Zheng, S. The crosstalk: Tumor-infiltrating lymphocytes rich in regulatory T cells suppressed cancer-associated fibroblasts. Acta Oncol. 2013, 52, 1760–1770. [Google Scholar] [CrossRef]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70, 7073–7083. [Google Scholar]
- Müerköster, S.; Wegehenkel, K.; Arlt, A. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1β. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Lemaire, M.; Deleu, S.; De Bruyne, E.; van Valckenborgh, E.; Menu, E.; Vanderkerken, K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv. Cancer Res. 2011, 110, 19–42. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 1–5. [Google Scholar]
- Jobin, M.E.; Fahey, J.L.; Price, A.Z. Long-term establishment of a human plasmacyte cell line derived from a patient with IgD multiple myeloma. J. Exp. Med. 1974, 140, 494–507. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; di Marzo, L.; de Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2013. [Google Scholar] [CrossRef]
- Slany, A.; Haudek-Prinz, V.; Meshcheryakova, A.; Bileck, A.; Lamm, W. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. J. Proteome Res. 2014, 13, 844–854. [Google Scholar] [CrossRef]
- Fuhler, G.M.; Baanstra, M.; Chesik, D.; Somasundaram, R.; Seckinger, A.; Hose, D.; Peppelenbosch, M.P.; Bos, N.A. Bone marrow stromal cell interaction reduces syndecan-1 expression and induces kinomic changes in myeloma cells. Exp. Cell Res. 2010, 316, 1816–1828. [Google Scholar] [CrossRef]
- Van Valckenborgh, E.; Matsui, W.; Agarwal, P.; Lub, S.; Dehui, X.; De Bruyne, E.; Menu, E.; Empsen, C.; van Grunsven, L.; Agarwal, J.; et al. Tumor-initiating capacity of CD138− and CD138+ tumor cells in the 5T33 multiple myeloma model. Leukemia 2012, 26, 1436–1439. [Google Scholar] [CrossRef]
- Celegato, M.; Borghese, C.; Casagrande, N.; Carbone, A.; Colombatti, A.; Aldinucci, D. Bortezomib down-modulates the survival factor interferon regulatory factor 4 in hodgkin lymphoma cell lines and decreases the protective activity of hodgkin lymphoma-associated fibroblasts. Leukemia Lymphoma 2014, 55, 149–159. [Google Scholar] [CrossRef]
- Hu, J.; van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Vanderkerken, K. Understanding the hypoxic niche of multiple myeloma: Therapeutic implications and contributions of mouse models. Dis. Model. Mech. 2012, 5, 763–771. [Google Scholar] [CrossRef]
- Cosse, J.P.; Michiels, C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med. Chem. 2008, 8, 790–797. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Schipani, E. Role of carcinoma-associated fibroblasts and hypoxia in tumor progression. Curr. Top. Microbiol. Immunol. 2010, 345, 31–45. [Google Scholar]
- Colla, S.; Storti, P.; Donofrio, G.; Todoerti, K.; Bolzoni, M.; Lazzaretti, M.; Abeltino, M.; Ippolito, L.; Neri, A.; Ribatti, D.; et al. Low bone marrow oxygen tension and hypoxia-inducible factor-1α overexpression characterize patients with multiple myeloma: Role on the transcriptional and proangiogenic profiles of CD138+ cells. Leukemia 2010, 24, 1967–1970. [Google Scholar] [CrossRef]
- Hu, J.; Handisides, D.R.; van Valckenborgh, E.; De Raeve, H.; Menu, E.; Vande Broek, I.; Liu, Q.; Sun, J.D.; van Camp, B.; Hart, C.P.; et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood 2010, 116, 1524–1527. [Google Scholar] [CrossRef]
- Storti, P.; Bolzoni, M.; Donofrio, G.; Airoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013, 27, 1697–1706. [Google Scholar] [CrossRef]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NF-κb activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef]
- Chiavarina, B.; Whitaker-Menezes, D.; Migneco, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Tanowitz, H.B.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. HIF1-α functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle 2010, 9, 3534–3551. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Iwano, M.; Haase, V.H. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 2008, 7, 1128–1132. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One 2013, 8, e55146. [Google Scholar]
- Kim, J.W.; Evans, C.; Weidemann, A.; Takeda, N.; Lee, Y.S.; Stockmann, C.; Branco-Price, C.; Brandberg, F.; Leone, G.; Ostrowski, M.C.; et al. Loss of fibroblast hif-1alpha accelerates tumorigenesis. Cancer Res. 2012, 72, 3187–3195. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma. Cancers 2014, 6, 1363-1381. https://doi.org/10.3390/cancers6031363
De Veirman K, Rao L, De Bruyne E, Menu E, Van Valckenborgh E, Van Riet I, Frassanito MA, Di Marzo L, Vacca A, Vanderkerken K. Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma. Cancers. 2014; 6(3):1363-1381. https://doi.org/10.3390/cancers6031363
Chicago/Turabian StyleDe Veirman, Kim, Luigia Rao, Elke De Bruyne, Eline Menu, Els Van Valckenborgh, Ivan Van Riet, Maria Antonia Frassanito, Lucia Di Marzo, Angelo Vacca, and Karin Vanderkerken. 2014. "Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma" Cancers 6, no. 3: 1363-1381. https://doi.org/10.3390/cancers6031363
APA StyleDe Veirman, K., Rao, L., De Bruyne, E., Menu, E., Van Valckenborgh, E., Van Riet, I., Frassanito, M. A., Di Marzo, L., Vacca, A., & Vanderkerken, K. (2014). Cancer Associated Fibroblasts and Tumor Growth: Focus on Multiple Myeloma. Cancers, 6(3), 1363-1381. https://doi.org/10.3390/cancers6031363