STAT3 Target Genes Relevant to Human Cancers

Abstract
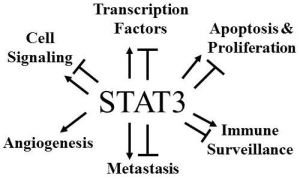
:
1. Introduction

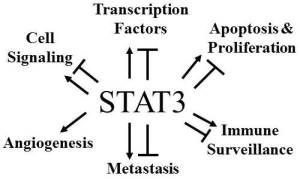{kind=link}
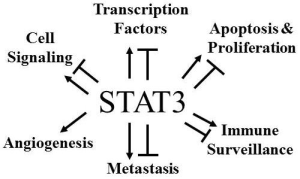{kind=link}
| STAT3-Regulated Genes | Direct Binding | STAT3 Binding Site(s) | Cell/Tissue Type(s) | Species | Reference | |
|---|---|---|---|---|---|---|
| Tumor Supporting Functions of STAT3 | ||||||
| Transcription Factors | ||||||
| ↑ | c-Fos | EMSA/ChIP | −348 to −339 bp | HepG2, A431 Cells | Human | [19,26,27] |
| ↑ | HIF-1α | EMSA/ChIP | −363 to −355 bp | A2058, v-Src-3T3 Cells, B16 Tumors | Human Murine | [28] |
| ↑ | c-Myc | EMSA/ChIP | +84 to +115 bp | HepG2, BAF-G277, KT-3, CCE ES Cells | Human Murine | [29] |
| ↑ | Sox2 | ChIP | −5.7 to −3.3 kb −528 to +238 bp | CCE ES Cells | Murine | [30] |
| ↑ | Nanog | ChIP | −871 to −585 bp | Mouse Embryonic Cells | Murine | [31] |
| ↑ | Twist | ChIP | −116 to −107 bp −103 to −96 bp | A431 Cells | Human | [19] |
| ↑ | Zeb1 | ChIP | −310 to −130 bp | SW1116, LoVo Cells | Human | [32] |
| ↓ | p53 | EMSA/ChIP | −128 bp | NIH-3T3, MEF Cells | Murine | [33] |
| ↑ | Oct-1 | ChIP | −3.5 to −2.5 kb | Eca-109 Cells | Human | [34] |
| Apoptosis and Proliferation | ||||||
| ↑ | Bcl-2 | ChIP | −1022 to −1002 bp | Hela Cells | Human | [35] |
| ↑ | Mcl-1 | EMSA | −94 to −86 bp | U266, v-Src-3T3 Cells | Human Murine | [36,37] |
| ↑ | Bcl-xL | ND | −600 to 0 bp | U266 Myeloma, NIH-3T3 Cells | Human Murine | [38] |
| ↑ | Survivin | EMSA/ChIP | −1174 to −1166 bp −1095 to −1087 bp | MDA-MB-453, NIH-3T3 Cells | Human Murine | [39] |
| ↓ | Fas | ChIP | −460 to −240 bp | Myeloma Cells | Human | [40] |
| ↑ | Hsp70 | EMSA/ChIP | −122 to −90 bp | VSM, HeLa Cells | Human | [41] |
| ↑ | Hsp90α | ChIP | −1642 to −1485 bp | Jurkat Cells | Human | [42] |
| ↑ | Hsp90β | EMSA | −643 to −623 bp | VSM Cells | Human | [41] |
| ↑ | Cyclin-D1 | EMSA/ChIP | −984 bp, −568 bp, −475 bp, −239 bp | 293T, 3YI, NIH-3T3, 2fTG Cells | Human Murine | [43,44,45] |
| Immune Suppression and Inflammation | ||||||
| ↑ | IL-10 | EMSA/ChIP | −120 to −111 bp | RPMI-8226 B Cells | Human | [46] |
| ↑ | IL-23 | ChIP | −1159 to +160 bp | B16 Tumors | Murine | [47] |
| ↑ | TGF-β | ChIP | −3155 to −2515 bp | CD4+ T Cells | Murine | [48] |
| ↑ | COX-2 | ChIP | −134 to −127 bp | U87MG Cells | Human | [49] |
| Metastasis | ||||||
| ↑ | MMP-1 | EMSA/ChIP | −79 to −42 bp | T24, HT-29 Cells | Human | [50] |
| ↑ | MMP-2 | EMSA | −1657 to −1620 bp −625 to −601 bp | C4 K1735 Cells | Murine | [51] |
| ↑ | MMP-3 | ChIP | −410 to −110 bp | HBVE Cells | Human | [52] |
| ↑ | MMP-9 | ChIP | −942 to −934 bp | MCF7 Cells | Human | [53] |
| ↑ | Fascin | ChIP | −1095 to −1067 bp −975 to −948 bp | 4T1, MDA-MB-231 Cells | Human Murine | [54] |
| ↑ | Vimentin | EMSA/ChIP | −757 to −749 bp | MDA-MB-231, C2C12 Cells | Human Murine | [55] |
| ↑ | RhoU | EMSA/ChIP | −1067 to −324 bp | MEF Cells | Murine | [56] |
| ↑ | ICAM-1 | EMSA/ChIP | −76 to −66 bp −175 to −97 bp | HepG2, BV2 Cells | Human Murine | [57] |
| ↑ | NGAL | ChIP | −170 bp | Primary Macrophages | Human | [58] |
| ↑ | POMC | EMSA | −399 to −374 bp | AtT20 Cells | Murine | [59] |
| ↑ | SAA1 | EMSA/ChIP | −226 to +24 bp | HepG2 Cells | Human | [60] |
| Angiogenesis | ||||||
| ↑ | VEGF-A | EMSA/ChIP | −848 bp | v-Src-3T3 Cells | Murine | [61] |
| ↑ | bFGF | ChIP | −997 to −989 bp | HUVEC | Human | [62] |
| ↑ | HGF | EMSA/ChIP | −149 bp, −110 bp | SP1, RINm5F Cells | Murine | [63,64] |
| Cell Signaling | ||||||
| ↑ | AKT | ChIP | Proximal −2.2 kb | 293 Cells | Human | [65] |
| ↑ | PIM-1 | ChIP | −934 to −905 bp | Microglial Cells | Murine | [66] |
| ↑ | TNF-R2 | ChIP | −1578 bp, −364 bp | SW480 Cells | Human | [67] |
| ↑ | S1P-R1 | EMSA/ChIP | −588 bp | MB49 Cells, B16 Tumors | Murine | [68] |
| ↑ | MUC-1 | EMSA/ChIP | −503 to −495 bp | T74D, ZR-75-1 Cells | Human | [69,70] |
| Transcription Factors | ||||||
| ↑ | FOXO1 | ChIP | −515 bp | CD4+ T Cells | Murine | [71] |
| ↑ | FOXO3A | ChIP | +196 bp | CD4+ T Cells | Murine | [71] |
| ↑ | Foxp3 | ND | Intron 1 | 293 Cells | Human | [72] |
| ↓ | Necdin | EMSA/ChIP | −588 bp | v-Src-3T3 | Murine | [73] |
| Survival and Metastasis | ||||||
| ↑ | p21CIP1/WAF1 | EMSA/ChIP | −4183 bp −2540 bp −640 bp | MG63, A431, HT-29, WiDr, HepG2 Cells | Human | [74,75,76] |
| ↑ | PI3K p50α | ChIP | −276 bp | Mammary Gland | Murine | [77] |
| ↑ | PI3K p55α | ChIP | −624 bp | Mammary Gland | Murine | [77] |
| Tumor Immune Surveillance | ||||||
| ↑↓ | IL-6 | ChIP | −73 to −54 bp | NIH-3T3, CT26 | Murine | [78,79,80] |
| ↑↓ | TNF-α | ND | −1452 bp | Macrophages, SCK1 | Murine | [80,81] |
| ↑↓ | IFN-γ | ChIP | 105 to 542 bp | T Cells | Human Murine | [78,80,82] |
| ↑↓ | RANTES | EMSA/ChIP | −120 to −1 bp | PC3, NIH-3T3 Cells | Human Murine | [78,80,83] |
| ↑ | CRP | EMSA | −112 to −105 bp | Hep3B Cells | Human | [84] |
| ↑ | STAT1 | ChIP | −604 to −596 bp −444 to −435 bp −363 to −356 bp −246 to −239 bp | MDA-MB-468 Cells | Human | [85] |
| ↑ | RORγt | ChIP | 1st Intron | TH17 Cells | Murine | [86] |
| ↑ | RORα | ChIP | 1st Intron | TH17 Cells | Murine | [86] |
| ↑ | BATF | ChIP | 2nd Intron | TH17 Cells | Murine | [86] |
| ↑ | IRF4 | ChIP | Proximal Promoter | TH17 Cells | Murine | [86] |
| ↑ | IL-6Rα | ChIP | 1st Intron | TH17 Cells | Murine | [86] |
| ↑ | IL-23R | ChIP | UD | TH17 Cells | Murine | [86] |
| ↑ | IL-17A | ChIP | −144 bp | TH17 Cells | Murine | [86,87] |
| ↑ | IL-17F | ChIP | −309 bp −326 bp | TH17 Cells | Murine | [86,87] |
| Other | ||||||
| ↑ | TIMP-1 | EMSA/ChIP | −49 to −41 bp | HepG2, WI38, CD4+ T Cells | Human Murine | [88,89] |
| ↑ | JunB | EMSA | −196 to −91 bp | HepG2 Cells | Human | [26,90] |
| ↑ | iNOS | EMSA/ChIP | −142 to −130 bp −84 to −60 bp | A431 Cells | Human | [18] |
| ↑↓ | CDC25A | ChIP | −222 to +58 bp | HepG2, Saos Cells | Human | [91] |
2. Tumor Supporting Functions of STAT3
2.1. Transcription Factors
2.2. Apoptosis and Proliferation
2.3. Immune Suppression and Inflammation
2.4. Metastasis
2.5. Angiogenesis
2.6. Cell Signaling
3. Tumor Suppressing Functions of STAT3
3.1. Transcription Factors
3.2. Survival and Metastasis
4. Dual Functions by STAT3 in Tumor Growth
4.1. Tumor Immune Function
4.2. Other
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Wegenka, U.M.; Buschmann, J.; Lutticken, C.; Heinrich, P.C.; Horn, F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol. Cell. Biol. 1993, 13, 276–288. [Google Scholar]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. JAK-STAT pathways and transcriptional activation in response to ifns and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar]
- Fu, X.Y. From PTK-STAT signaling to caspase expression and apoptosis induction. Cell Death Differ. 1999, 6, 1201–1208. [Google Scholar]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Matikainen, S.; Sareneva, T.; Ronni, T.; Lehtonen, A.; Koskinen, P.J.; Julkunen, I. Interferon-alpha activates multiple stat proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood 1999, 93, 1980–1991. [Google Scholar]
- Puthier, D.; Bataille, R.; Amiot, M. IL-6 up-regulates Mcl-1 in human myeloma cells through JAK/STAT rather than Ras/MAP kinase pathway. Eur. J. Immunol. 1999, 29, 3945–3950. [Google Scholar] [CrossRef]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a STAT3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar]
- Wen, Z.; Darnell, J.E., Jr. Mapping of STAT3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of stat1 and stat3. Nucleic Acids Res. 1997, 25, 2062–2067. [Google Scholar]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Serrat, M.A.D.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef]
- Bousquet, C.; Susini, C.; Melmed, S. Inhibitory roles for SHP-1 and SOCS-3 following pituitary proopiomelanocortin induction by leukemia inhibitory factor. J. Clin. Investig. 1999, 104, 1277–1285. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of STAT3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef]
- Abou-Ghazal, M.; Yang, D.S.; Qiao, W.; Reina-Ortiz, C.; Wei, J.; Kong, L.Y.; Fuller, G.N.; Hiraoka, N.; Priebe, W.; Sawaya, R.; et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 2008, 14, 8228–8235. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Mukherjee, N.; Chakravarti, A.; Robe, P.; Zhai, G.; Chakladar, A.; Loeffler, J.; Black, P.; Frank, D.A. A STAT3 gene expression signature in gliomas is associated with a poor prognosis. Transl. Oncogenomics 2007, 2, 99–105. [Google Scholar]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated Stat3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of twist gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef]
- Ecker, A.; Simma, O.; Hoelbl, A.; Kenner, L.; Beug, H.; Moriggl, R.; Sexl, V. The dark and the bright side of STAT3: Proto-oncogene and tumor-suppressor. Front. Biosci. 2009, 14, 2944–2958. [Google Scholar]
- Clevenger, C.V. Roles and regulation of stat family transcription factors in human breast cancer. Am. J. Pathol. 2004, 165, 1449–1460. [Google Scholar] [CrossRef]
- Ling, X.; Arlinghaus, R.B. Knockdown of STAT3 expression by rna interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005, 65, 2532–2536. [Google Scholar] [CrossRef]
- Chan, K.S.; Sano, S.; Kiguchi, K.; Anders, J.; Komazawa, N.; Takeda, J.; DiGiovanni, J. Disruption of STAT3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Investig. 2004, 114, 720–728. [Google Scholar] [CrossRef]
- Pedranzini, L.; Leitch, A.; Bromberg, J. STAT3 is required for the development of skin cancer. J. Clin. Investig. 2004, 114, 619–622. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a pten-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef]
- Seidel, H.M.; Milocco, L.H.; Lamb, P.; Darnell, J.E., Jr.; Stein, R.B.; Rosen, J. Spacing of palindromic half sites as a determinant of selective stat (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. USA 1995, 92, 3041–3045. [Google Scholar] [CrossRef]
- Yang, E.; Lerner, L.; Besser, D.; Darnell, J.E. Independent and cooperative activation of chromosomal c-fos promoter by STAT3. J. Biol. Chem. 2003, 278, 15794–15799. [Google Scholar] [CrossRef]
- Niu, G.; Briggs, J.; Deng, J.; Ma, Y.; Lee, H.; Kortylewski, M.; Kujawski, M.; Kay, H.; Cress, W.D.; Jove, R.; et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res. 2008, 6, 1099–1105. [Google Scholar] [CrossRef]
- Kiuchi, N.; Nakajima, K.; Ichiba, M.; Fukada, T.; Narimatsu, M.; Mizuno, K.; Hibi, M.; Hirano, T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med. 1999, 189, 63–73. [Google Scholar] [CrossRef]
- Foshay, K.M.; Gallicano, G.I. Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev. 2008, 17, 269–278. [Google Scholar] [CrossRef]
- Okumura, F.; Okumura, A.J.; Matsumoto, M.; Nakayama, K.I.; Hatakeyama, S. TRIM8 regulates nanog via Hsp90beta-mediated nuclear translocation of STAT3 in embryonic stem cells. Biochim. Biophys. Acta 2011, 1813, 1784–1792. [Google Scholar] [CrossRef]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.W.; Ren, L.L.; Wang, Y.C.; Su, W.Y.; Wang, J.L.; Cui, Y.; Wang, Z.H.; et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of STAT3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, S.; Shen, M.; Liu, J.; Wang, M.; Li, C.; Wang, Y.; Deng, A.; Mei, Q. STAT3 is involved in esophageal carcinogenesis through regulation of Oct-1. Carcinogenesis 2013, 34, 678–688. [Google Scholar] [CrossRef]
- Choi, H.J.; Han, J.S. Overexpression of phospholipase D enhances Bcl-2 expression by activating STAT3 through independent activation of ERK and p38MAPK in HeLa cells. Biochim. Biophys. Acta 2012, 1823, 1082–1091. [Google Scholar] [CrossRef]
- Becker, T.M.; Boyd, S.C.; Mijatov, B.; Gowrishankar, K.; Snoyman, S.; Pupo, G.M.; Scolyer, R.A.; Mann, G.J.; Kefford, R.F.; Zhang, X.D.; et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene 2013, 33, 1158–1166. [Google Scholar]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of STAT3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of STAT3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and c-Jun suppresses fas transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Li, S.; Patterson, C.; Runge, M.S. Thrombin regulates vascular smooth muscle cell growth and heat shock proteins via the JAK-STAT pathway. J. Biol. Chem. 2001, 276, 18915–18924. [Google Scholar] [CrossRef]
- Chen, X.S.; Zhang, Y.; Wang, J.S.; Li, X.Y.; Cheng, X.K.; Zhang, Y.; Wu, N.H.; Shen, Y.F. Diverse effects of STAT1 on the regulation of Hsp90alpha gene under heat shock. J. Cell. Biochem. 2007, 102, 1059–1066. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef]
- Schaefer, A.; Unterberger, C.; Frankenberger, M.; Lohrum, M.; Staples, K.J.; Werner, T.; Stunnenberg, H.; Ziegler-Heitbrock, L. Mechanism of interferon-gamma mediated down-regulation of interleukin-10 gene expression. Mol. Immunol. 2009, 46, 1351–1359. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by STAT3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Kinjyo, I.; Inoue, H.; Hamano, S.; Fukuyama, S.; Yoshimura, T.; Koga, K.; Takaki, H.; Himeno, K.; Takaesu, G.; Kobayashi, T.; et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J. Exp. Med. 2006, 203, 1021–1031. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol. Cancer Res. 2010, 8, 232–245. [Google Scholar] [CrossRef]
- Itoh, M.; Murata, T.; Suzuki, T.; Shindoh, M.; Nakajima, K.; Imai, K.; Yoshida, K. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene 2006, 25, 1195–1204. [Google Scholar]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. STAT3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef]
- Liu, M.; Wilson, N.O.; Hibbert, J.M.; Stiles, J.K. Stat3 regulates MMP3 in heme-induced endothelial cell apoptosis. PLoS One 2013, 8, e71366. [Google Scholar]
- Song, Y.; Qian, L.; Song, S.; Chen, L.; Zhang, Y.; Yuan, G.; Zhang, H.; Xia, Q.; Hu, M.; Yu, M.; et al. Fra-1 and STAT3 synergistically regulate activation of human MMP-9 gene. Mol. Immun. 2008, 45, 137–143. [Google Scholar] [CrossRef]
- Snyder, M.; Huang, X.Y.; Zhang, J.J. Signal transducers and activators of transcription 3 (STAT3) directly regulates cytokine-induced fascin expression and is required for breast cancer cell migration. J. Biol. Chem. 2011, 286, 38886–38893. [Google Scholar] [CrossRef]
- Wu, Y.; Diab, I.; Zhang, X.; Izmailova, E.S.; Zehner, Z.E. STAT3 enhances vimentin gene expression by binding to the antisilencer element and interacting with the repressor protein, ZBP-89. Oncogene 2004, 23, 168–178. [Google Scholar] [CrossRef]
- Schiavone, D.; Dewilde, S.; Vallania, F.; Turkson, J.; di Cunto, F.; Poli, V. The Rhou/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem. J. 2009, 421, 283–292. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Timmer, H.; Luttickhuizen, D.; Vellenga, E.; Kruijer, W. c-Jun and c-Fos cooperate with STAT3 in IL-6-induced transactivation of the IL-6 respone element (IRE). Cytokine 2001, 14, 78–87. [Google Scholar] [CrossRef]
- Jung, M.; Weigert, A.; Tausendschon, M.; Mora, J.; Oren, B.; Sola, A.; Hotter, G.; Muta, T.; Brune, B. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol. Cell. Biol. 2012, 32, 3938–3948. [Google Scholar] [CrossRef]
- Bousquet, C.; Zatelli, M.C.; Melmed, S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 2000, 106, 1417–1425. [Google Scholar] [CrossRef]
- Hagihara, K.; Nishikawa, T.; Sugamata, Y.; Song, J.; Isobe, T.; Taga, T.; Yoshizaki, K. Essential role of STAT3 in cytokine-driven NF-κb-mediated serum amyloid a gene expression. Genes Cells: Devoted Mol. Cell. Mech. 2005, 10, 1051–1063. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive STAT3 activity up-regulates vegf expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Huang, Y.H.; Wu, M.P.; Pan, S.C.; Su, W.C.; Chen, Y.W.; Wu, L.W. STAT1 activation by venous malformations mutant Tie2-R849W antagonizes VEGF-A-mediated angiogenic response partly via reduced bFGF production. Angiogenesis 2013, 16, 207–222. [Google Scholar] [CrossRef]
- Hung, W.; Elliott, B. Co-operative effect of c-Src tyrosine kinase and STAT3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 2001, 276, 12395–12403. [Google Scholar] [CrossRef]
- Nakagawa, K.; Takasawa, S.; Nata, K.; Yamauchi, A.; Itaya-Hironaka, A.; Ota, H.; Yoshimoto, K.; Sakuramoto-Tsuchida, S.; Miyaoka, T.; Takeda, M.; et al. Prevention of Reg I-induced beta-cell apoptosis by IL-6/dexamethasone through activation of HGF gene regulation. Biochim. Biophys. Acta 2013, 1833, 2988–2995. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting STAT3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- Przanowski, P.; Dabrowski, M.; Ellert-Miklaszewska, A.; Kloss, M.; Mieczkowski, J.; Kaza, B.; Ronowicz, A.; Hu, F.; Piotrowski, A.; Kettenmann, H.; et al. The signal transducers STAT1 and STAT3 and their novel target JMJD3 drive the expression of inflammatory genes in microglia. J. Mol. Med. 2013, 92, 239–254. [Google Scholar]
- Hamilton, K.E.; Simmons, J.G.; Ding, S.; van Landeghem, L.; Lund, P.K. Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol. Cancer Res. 2011, 9, 1718–1731. [Google Scholar]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced s1pr1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Ahmad, R.; Rajabi, H.; Kosugi, M.; Joshi, M.D.; Alam, M.; Vasir, B.; Kawano, T.; Kharbanda, S.; Kufe, D. MUC1-C oncoprotein promotes STAT3 activation in an autoinductive regulatory loop. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef]
- Gaemers, I.C.; Vos, H.L.; Volders, H.H.; van der Valk, S.W.; Hilkens, J. A STAT-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J. Biol. Chem. 2001, 276, 6191–6199. [Google Scholar]
- Oh, H.M.; Yu, C.R.; Golestaneh, N.; Amadi-Obi, A.; Lee, Y.S.; Eseonu, A.; Mahdi, R.M.; Egwuagu, C.E. STAT3 protein promotes T-cell survival and inhibits interleukin-2 production through up-regulation of class o forkhead transcription factors. J. Biol. Chem. 2011, 286, 30888–30897. [Google Scholar]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates Foxp3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef]
- Haviland, R.; Eschrich, S.; Bloom, G.; Ma, Y.; Minton, S.; Jove, R.; Cress, W.D. Necdin, a negative growth regulator, is a novel STAT3 target gene down-regulated in human cancer. PLoS One 2011, 6, e24923. [Google Scholar]
- Bellido, T.; O’Brien, C.A.; Roberson, P.K.; Manolagas, S.C. Transcriptional activation of the p21 WAF1,CIP1,SDI1 gene by interleukin-6 type cytokines: A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J. Biol. Chem. 1998, 273, 21137–21144. [Google Scholar] [CrossRef]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar]
- Abell, K.; Bilancio, A.; Clarkson, R.W.; Tiffen, P.G.; Altaparmakov, A.I.; Burdon, T.G.; Asano, T.; Vanhaesebroeck, B.; Watson, C.J. STAT3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat. Cell Biol. 2005, 7, 392–398. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.-W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for STAT3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef]
- Kang, J.W.; Park, Y.S.; Lee, D.H.; Kim, J.H.; Kim, M.S.; Bak, Y.; Hong, J.; Yoon, D.Y. Intracellular interaction of interleukin (IL)-32alpha with protein kinase cepsilon (pkcepsilon) and STAT3 protein augments IL-6 production in THP-1 promonocytic cells. J. Biol. Chem. 2012, 287, 35556–35564. [Google Scholar]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by STAT-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Chappell, V.L.; Le, L.X.; LaGrone, L.; Mileski, W.J. STAT proteins play a role in tumor necrosis factor alpha gene expression. Shock 2000, 14, 400–402. [Google Scholar] [CrossRef]
- Kusaba, H.; Ghosh, P.; Derin, R.; Buchholz, M.; Sasaki, C.; Madara, K.; Longo, D.L. Interleukin-12-induced interferon-gamma production by human peripheral blood T cells is regulated by mammalian target of rapamycin (MTOR). J. Biol. Chem. 2005, 280, 1037–1043. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NF-κb. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef]
- Han, W.; Carpenter, R.L.; Cao, X.; Lo, H.W. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol. Carcinog. 2013, 52, 959–969. [Google Scholar] [CrossRef]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef]
- Thomas, R.M.; Sai, H.; Wells, A.D. Conserved intergenic elements and DNA methylation cooperate to regulate transcription at the IL17 locus. J. Biol. Chem. 2012, 287, 25049–25059. [Google Scholar] [CrossRef]
- Adamson, A.; Ghoreschi, K.; Rittler, M.; Chen, Q.; Sun, H.W.; Vahedi, G.; Kanno, Y.; Stetler-Stevenson, W.G.; O’Shea, J.J.; Laurence, A. Tissue inhibitor of metalloproteinase 1 is preferentially expressed in th1 and Th17 T-helper cell subsets and is a direct stat target gene. PLoS One 2013, 8, e59367. [Google Scholar] [CrossRef]
- Bugno, M.; Graeve, L.; Gatsios, P.; Koj, A.; Heinrich, P.C.; Travis, J.; Kordula, T. Identification of the interleukin-6/oncostatin m response element in the rat tissue inhibitor of metalloproteinases-1 (TIMP-1) promoter. Nucleic Acids Res. 1995, 23, 5041–5047. [Google Scholar] [CrossRef]
- Coffer, P.; Lutticken, C.; van Puijenbroek, A.; Klop-de Jonge, M.; Horn, F.; Kruijer, W. Transcriptional regulation of the junB promoter: Analysis of STAT-mediated signal transduction. Oncogene 1995, 10, 985–994. [Google Scholar]
- Barre, B.; Vigneron, A.; Coqueret, O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the CDC25a promoter. J. Biol. Chem. 2005, 280, 15673–15681. [Google Scholar] [CrossRef]
- Shaulian, E. Ap-1—the jun proteins: Oncogenes or tumor suppressors in disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar]
- Ji, J.; Wang, X.W. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin. Oncol. 2012, 39, 461–472. [Google Scholar] [CrossRef]
- Hindley, C.; Philpott, A. The cell cycle and pluripotency. Biochem. J. 2013, 451, 135–143. [Google Scholar] [CrossRef]
- Philip, B.; Ito, K.; Moreno-Sanchez, R.; Ralph, S.J. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 2013, 34, 1699–1707. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. Emt-activating transcription factors in cancer: Beyond emt and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Kang, J.; Shakya, A.; Tantin, D. Stem cells, stress, metabolism and cancer: A drama in two octs. Trends Biochem. Sci. 2009, 34, 491–499. [Google Scholar] [CrossRef]
- Obinata, D.; Takayama, K.; Urano, T.; Murata, T.; Kumagai, J.; Fujimura, T.; Ikeda, K.; Horie-Inoue, K.; Homma, Y.; Ouchi, Y.; et al. Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int. J. Cancer J. Int. Cancer 2012, 130, 1021–1028. [Google Scholar]
- Wang, M.L.; Chiou, S.H.; Wu, C.W. Targeting cancer stem cells: Emerging role of Nanog transcription factor. Onco Targets Ther. 2013, 6, 1207–1220. [Google Scholar]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Chao, J.-R.; Wang, J.-M.; Lee, S.-F.; Peng, H.-W.; Lin, Y.-H.; Chou, C.-H.; Li, J.-C.; Huang, H.-M.; Chou, C.-K.; Kuo, M.-L.; et al. Mcl-1 is an immediate-early gene activated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol. Cell. Biol. 1998, 18, 4883–4898. [Google Scholar]
- Wang, J.-M.; Chao, J.-R.; Chen, W.; Kuo, M.-L.; Yen, J.J.-Y.; Yang-Yen, H.-F. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/AKT signaling pathway through a transcription factor complex containing creb. Mol. Cell. Biol. 1999, 19, 6195–6206. [Google Scholar]
- Oskay Halacli, S.; Halacli, B.; Altundag, K. The significance of heat shock proteins in breast cancer therapy. Med. Oncol. 2013, 30. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Kitamura, H.; Kamon, H.; Sawa, S.; Park, S.J.; Katunuma, N.; Ishihara, K.; Murakami, M.; Hirano, T. IL-6-STAT3 controls intracellular MHC class II αβ dimer level through cathepsin s activity in dendritic cells. Immunity 2005, 23, 491–502. [Google Scholar] [CrossRef]
- Benkhart, E.M.; Siedlar, M.; Wedel, A.; Werner, T.; Ziegler-Heitbrock, H.W. Role of STAT3 in lipopolysaccharide-induced IL-10 gene expression. J. Immunol. 2000, 165, 1612–1617. [Google Scholar]
- Ziegler-Heitbrock, L.; Lotzerich, M.; Schaefer, A.; Werner, T.; Frankenberger, M.; Benkhart, E. IFN-α induces the human IL-10 gene by recruiting both ifn regulatory factor 1 and STAT3. J. Immunol. 2003, 171, 285–290. [Google Scholar]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Cheng, J.; Fan, X.M. Role of cyclooxygenase-2 in gastric cancer development and progression. World J. Gastroenterol. 2013, 19, 7361–7368. [Google Scholar] [CrossRef]
- Zugowski, C.; Lieder, F.; Muller, A.; Gasch, J.; Corvinus, F.M.; Moriggl, R.; Friedrich, K. STAT3 controls matrix metalloproteinase-1 expression in colon carcinoma cells by both direct and AP-1-mediated interaction with the MMP-1 promoter. Biol. Chem. 2011, 392, 449–459. [Google Scholar]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by STAT3-c. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Jakoncic, J.; Zhang, J.J.; Huang, X.Y. Migrastatin analogues target fascin to block tumour metastasis. Nature 2010, 464, 1062–1066. [Google Scholar] [CrossRef]
- Chuang, Y.Y.; Valster, A.; Coniglio, S.J.; Backer, J.M.; Symons, M. The atypical Rho family GTPase Wrch-1 regulates focal adhesion formation and cell migration. J. Cell Sci. 2007, 120, 1927–1934. [Google Scholar] [CrossRef]
- Ory, S.; Brazier, H.; Blangy, A. Identification of a bipartite focal adhesion localization signal in Rhou/Wrch-1, a Rho family GTPase that regulates cell adhesion and migration. Biol. Cell 2007, 99, 701–716. [Google Scholar] [CrossRef]
- Kobayashi, H.; Boelte, K.C.; Lin, P.C. Endothelial cell adhesion molecules and cancer progression. Curr. Med. Chem. 2007, 14, 377–386. [Google Scholar] [CrossRef]
- Veitonmaki, N.; Hansson, M.; Zhan, F.; Sundberg, A.; Lofstedt, T.; Ljungars, A.; Li, Z.C.; Martinsson-Niskanen, T.; Zeng, M.; Yang, Y.; et al. A human icam-1 antibody isolated by a function-first approach has potent macrophage-dependent antimyeloma activity in vivo. Cancer Cell 2013, 23, 502–515. [Google Scholar] [CrossRef]
- Zhu, X.W.; Gong, J.P. Expression and role of icam-1 in the occurrence and development of hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2013, 14, 1579–1583. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta 2012, 1826, 129–169. [Google Scholar]
- Sherbet, G.V. Hormonal influences on cancer progression and prognosis. Vitam. Horm. 2005, 71, 147–200. [Google Scholar] [CrossRef]
- Malle, E.; Sodin-Semrl, S.; Kovacevic, A. Serum amyloid A: An acute-phase protein involved in tumour pathogenesis. Cell. Mol. Life Sci. 2009, 66, 9–26. [Google Scholar] [CrossRef]
- Deng, J.; Liang, H.; Zhang, R.; Sun, D.; Pan, Y.; Liu, Y.; Zhang, L.; Hao, X. STAT3 is associated with lymph node metastasis in gastric cancer. Tumour Biol. 2013, 34, 2791–2800. [Google Scholar] [CrossRef]
- Huang, X.; Meng, B.; Iqbal, J.; Ding, B.B.; Perry, A.M.; Cao, W.; Smith, L.M.; Bi, C.; Jiang, C.; Greiner, T.C.; et al. Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B-cell lymphoma treated with R-chop. J. Clin. Oncol. 2013, 31, 4520–4528. [Google Scholar] [CrossRef]
- Xu, Y.H.; Lu, S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur. J. Surg. Oncol. 2014, 40, 311–317. [Google Scholar] [CrossRef]
- Zetter, B.R. The scientific contributions of M. Judah Folkman to cancer research. Nat. Rev. Cancer 2008, 8, 647–654. [Google Scholar] [CrossRef]
- Megeney, L.A.; Perry, R.L.; LeCouter, J.E.; Rudnicki, M.A. bFGF and LIF signaling activates STAT3 in proliferating myoblasts. Dev. Genet. 1996, 19, 139–145. [Google Scholar] [CrossRef]
- Cheung, M.; Testa, J.R. Diverse mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets 2013, 13, 234–244. [Google Scholar] [CrossRef]
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Shirogane, T.; Fukada, T.; Muller, J.M.; Shima, D.T.; Hibi, M.; Hirano, T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity 1999, 11, 709–719. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Takedatsu, H.; Cario, E.; de Jong, Y.P.; Ooi, C.J.; Xavier, R.J.; Terhorst, C.; Podolsky, D.K.; Bhan, A.K. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 2002, 122, 134–144. [Google Scholar] [CrossRef]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J.H. Foxo family members in cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef]
- Douglass, S.; Ali, S.; Meeson, A.P.; Browell, D.; Kirby, J.A. The role of FOXP3 in the development and metastatic spread of breast cancer. Cancer Metastasis Rev. 2012, 31, 843–854. [Google Scholar] [CrossRef]
- Asai, T.; Liu, Y.; Nimer, S.D. Necdin, a p53 target gene, in stem cells. Oncotarget 2013, 4, 806–807. [Google Scholar]
- Hasegawa, K.; Yoshikawa, K. Necdin regulates p53 acetylation via sirtuin1 to modulate DNA damage response in cortical neurons. J. Neurosci. 2008, 28, 8772–8784. [Google Scholar] [CrossRef]
- Chapman, R.S.; Lourenco, P.C.; Tonner, E.; Flint, D.J.; Selbert, S.; Takeda, K.; Akira, S.; Clarke, A.R.; Watson, C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of STAT3. Genes Dev. 1999, 13, 2604–2616. [Google Scholar] [CrossRef]
- Allin, K.H.; Nordestgaard, B.G. Elevated c-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit. Rev. Clin. Lab. Sci. 2011, 48, 155–170. [Google Scholar] [CrossRef]
- Kolev, M.; Towner, L.; Donev, R. Complement in cancer and cancer immunotherapy. Arch. Immunol. Ther. Exp. 2011, 59, 407–419. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. JAK-STAT 2013, 2, e23353. [Google Scholar] [CrossRef]
- Ernst, M.; Najdovska, M.; Grail, D.; Lundgren-May, T.; Buchert, M.; Tye, H.; Matthews, V.B.; Armes, J.; Bhathal, P.S.; Hughes, N.R.; et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Investig. 2008, 118, 1727–1738. [Google Scholar]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector t cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Qi, W.; Huang, X.; Wang, J. Correlation between Th17 cells and tumor microenvironment. Cell. Immunol. 2013, 285, 18–22. [Google Scholar] [CrossRef]
- Ye, J.; Livergood, R.S.; Peng, G. The role and regulation of human Th17 cells in tumor immunity. Am. J. Pathol. 2013, 182, 10–20. [Google Scholar] [CrossRef]
- Wurtz, S.O.; Schrohl, A.S.; Sorensen, N.M.; Lademann, U.; Christensen, I.J.; Mouridsen, H.; Brunner, N. Tissue inhibitor of metalloproteinases-1 in breast cancer. Endocr.-Relat. Cancer 2005, 12, 215–227. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Rao, C.V. Inos-selective inhibitors for cancer prevention: Promise and progress. Future Med. Chem. 2012, 4, 2193–2204. [Google Scholar] [CrossRef]
- Johnson, E.S.; Kornbluth, S. Phosphatases driving mitosis: Pushing the gas and lifting the brakes. Prog. Mol. Biol. Transl. Sci. 2012, 106, 327–341. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carpenter, R.L.; Lo, H.-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897-925. https://doi.org/10.3390/cancers6020897
Carpenter RL, Lo H-W. STAT3 Target Genes Relevant to Human Cancers. Cancers. 2014; 6(2):897-925. https://doi.org/10.3390/cancers6020897
Chicago/Turabian StyleCarpenter, Richard L., and Hui-Wen Lo. 2014. "STAT3 Target Genes Relevant to Human Cancers" Cancers 6, no. 2: 897-925. https://doi.org/10.3390/cancers6020897
APA StyleCarpenter, R. L., & Lo, H.-W. (2014). STAT3 Target Genes Relevant to Human Cancers. Cancers, 6(2), 897-925. https://doi.org/10.3390/cancers6020897