Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance

{kind=link}
Abstract
:1. Introduction
2. Vessel Formation in Malignant Tumors
- (a)
- (b)
- Co-option of existing vessels [17].
- (c)
- Vasculogenesis (de novo vessel formation) through incorporation of circulating endothelial precursor cells [17].
- (d)
- Intussusception (splitting of the lumen of a vessel into two).
- (e)
- Formation of pseudo-vascular channels lined by tumor cells rather than endothelial cells (“vascular mimicry”).
- (f)
- Microvessel formation by a subset of bone marrow-derived myeloid cells infiltrating the tumor [17].
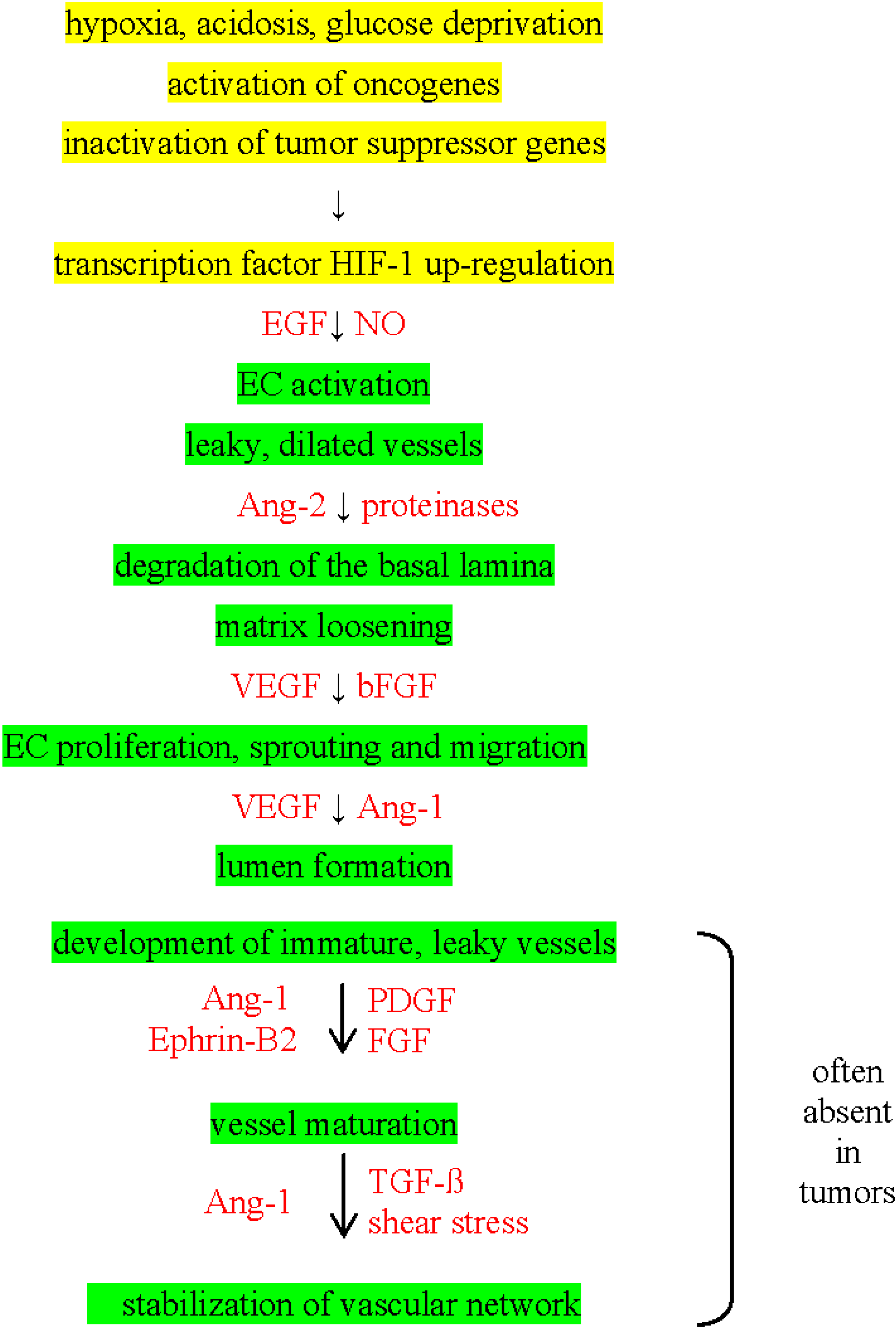
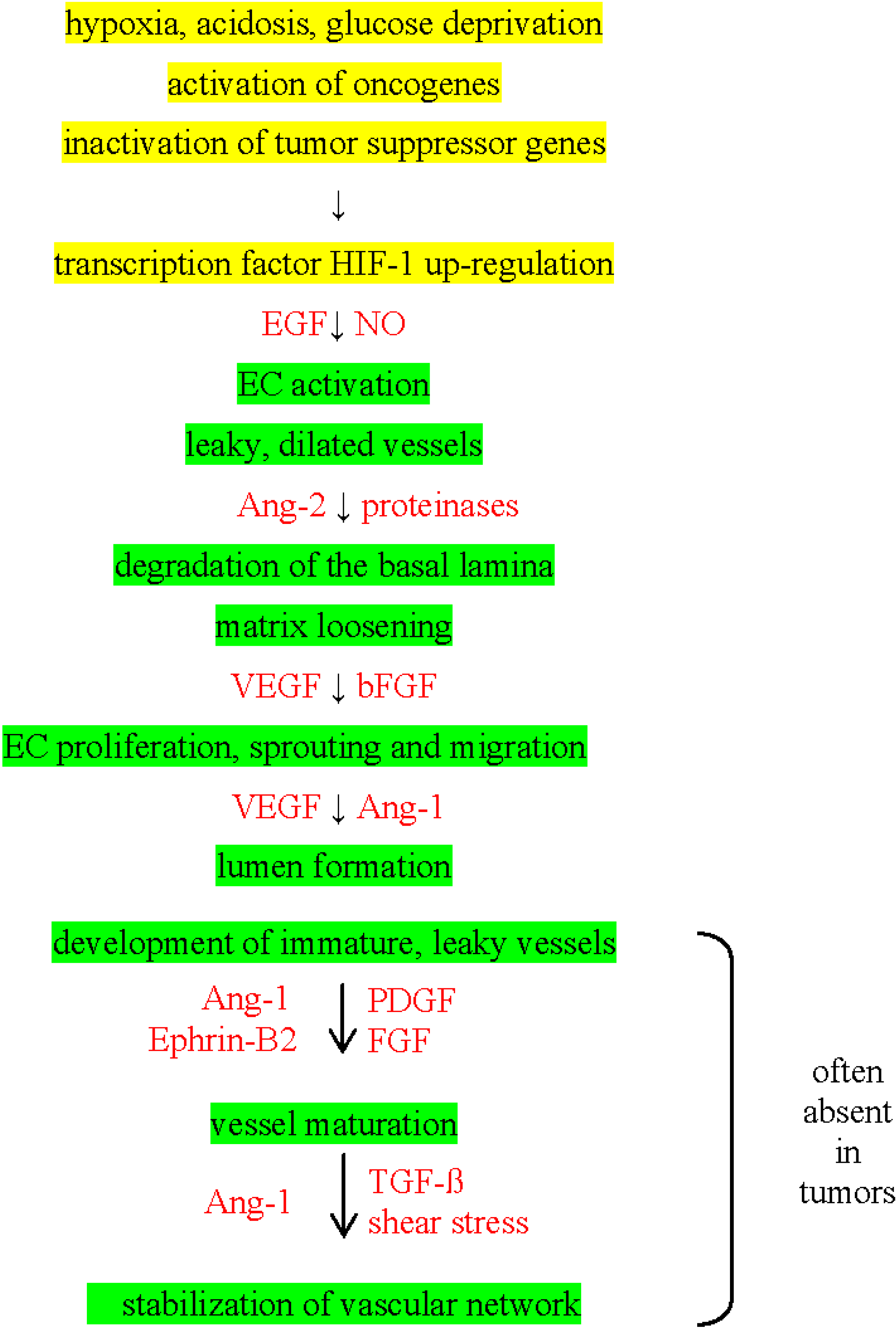
3. Angiogenic Switch in Tumors
4. Tumor Microcirculation
5. Tumor Blood Flow Rates
- (a)
- (b)
- Tumors can have flow rates which are similar to those measured in organs with a high metabolic rate such as liver, heart or brain.
- (c)
- Some tumors exhibit flow rates which are even lower than those of tissues with a low metabolic rate such as skin, resting muscle or adipose tissue.
- (d)
- Blood flow in human tumors can be higher or lower than that of the tissue of origin, depending on the functional state of the latter tissue (e.g., average blood flow in breast cancers is substantially higher than that of postmenopausal breast and significantly lower than flow data obtained in the lactating, parenchymal breast).
- (e)
- The average perfusion rate of carcinomas does not deviate substantially from that of tissue sarcomas.
- (f)
- Metastatic lesions exhibit a blood supply which is comparable to that of the primary tumor [11].
- (g)
- In some tumor entities, blood flow in the periphery is distinctly higher than in the center whereas in others, blood flow is significantly higher at the tumor center compared to the tumor edge.
- (h)
- Flow data from multiple sites of measurement show marked heterogeneity within individual tumors. In cervical cancer, the intra-tumor heterogeneity was similar to the inter-tumor heterogeneity [50].
- (i)
- (j)
- (k)
- Tumor blood flow is not regulated according to the metabolic demand as is the case in normal tissues.
6. Arterio-Venous Shunt Perfusion in Tumors
7. Tumor Hypoxia and HIF
8. Therapeutic Interventions to Overcome Hypoxia-Related Radioresistance
9. Conclusions
Abbreviations
| Akt | protein kinase B (PKB) |
| Ang | Angiopoietin |
| EC | endothelial cell |
| EGF | epidermal growth factor |
| EGFR | EGF receptor |
| bFGF | basic fibroblast growth factor |
| ERK | extracellular signal-regulated kinase |
| FIH-1 | factor inhibiting HIF-1 |
| FDG PET | fluorine-18-fluorodeoxyglucose positron emission tomography |
| HIF | hypoxia-inducible factor |
| HSP | heat shock protein |
| IL | Interleukin |
| IFN | Interferon |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor kappa-B |
| PDGF | platelet-derived growth factor |
| PDGFR | PDGF receptor |
| PGE2 | prostaglandin E2 |
| PI3K | phosphatidylinositol 3-kinase |
| RACK-1 | receptor of activated protein kinase C |
| Raf | rapidly growing fibrosarcoma protein |
| Ras | rat sarcoma protein |
| TGF-β | transforming growth factor beta |
| TMZ | temozolomide |
| TNF | tumor necrosis factor |
| uPA | urokinase-type plasminogen activator |
| VEGF | vascular endothelial growth factor |
| VEGFR | VEGF receptor |
Acknowledgment
Conflicts of Interest
References
- Risau, W.; Sariola, H.; Zerwes, H.G.; Sasse, J.; Ekblom, P.; Kemler, R.; Doetschman, T. Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development 1988, 102, 471–478. [Google Scholar]
- Patel-Hett, S.; D’amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int. J. Dev. Biol. 2011, 55, 353–363. [Google Scholar]
- Folkman, J.; Bach, M.; Rowe, J.W.; Davidoff, F.; Lambert, P.; Hirsch, C.; Goldberg, A.; Hiatt, H.H.; Glass, J.; Henshaw, E. Tumor angiogenesis - Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef]
- Arnold, F.; West, D.C. Angiogenesis in wound healing. Pharmacol. Ther. 1991, 52, 407–422. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Redmer, D.A. Angiogenesis in the placenta. Biol. Reprod. 2001, 64, 1033–1040. [Google Scholar] [CrossRef]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Jung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; Keshet, E. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Sutherland, R.M. Cell and environment interactions in tumor microregions: The multicell spheroid model. Science 1988, 240, 177–184. [Google Scholar]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Vaupel, P.; Höckel, M. Blood supply, oxygenation status and metabolic micromilieu of breast cancers: Characterization and therapeutic relevance. Int. J. Oncol. 2000, 17, 869–879. [Google Scholar]
- Vaupel, P. Blood flow and metabolic microenvironment of brain tumors. J. Neurooncol. 1994, 22, 261–267. [Google Scholar] [CrossRef]
- Vaupel, P. Physiological properties of malignant tumours. NMR Biomed. 1992, 5, 220–225. [Google Scholar] [CrossRef]
- Reinhold, H.S.; van den Berg-Blok, A. Vascularization of experimental tumours. Ciba Found. Symp. 1983, 100, 100–119. [Google Scholar]
- Reinhold, H.S.; van den Berg-Blok, A. Circulation physiology of tumors. In Rodent Tumor Models in Experimental Cancer Therapy; Kallmann, R.F., Ed.; Pergamon Press: New York, NY, USA, 1987. [Google Scholar]
- Vaupel, P. Abnormal microvasculature and defective microcirculatory function in solid tumors. In Vascular-Targeted Therapies in Oncology; Siemann, D.W., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006; pp. 9–29. [Google Scholar]
- Shchors, K.; Evan, G. Tumor angiogenesis: Cause or consequence of cancer? Cancer Res. 2007, 67, 7059–7061. [Google Scholar] [CrossRef]
- Sonveaux, P. Provascular strategy: Targeting functional adaptations of mature blood vessels in tumors to selectively influence the tumor vascular reactivity and improve cancer treatment. Radiother. Oncol. 2008, 86, 300–313. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomaki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim. Biophys. Acta 2008, 1784, 150–158. [Google Scholar] [CrossRef]
- Eibl, G.; Bruemmer, D.; Okada, Y.; Duffy, J.P.; Law, R.E.; Reber, H.A.; Hines, O.J. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003, 306, 887–897. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; Lecouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Salven, P.; Lymboussaki, A.; Heikkila, P.; Jaaskela-Saari, H.; Enholm, B.; Aase, K.; von Euler, G.; Eriksson, U.; Alitalo, K.; Joensuu, H. Vascular endothelial growth factors VEGF-b and VEGF-c are expressed in human tumors. Am. J. Pathol. 1998, 153, 103–108. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFa and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Hoffmann, J.; Junker, H.; Schmieder, A.; Venz, S.; Brandt, R.; Multhoff, G.; Falk, W.; Radons, J. EGCG downregulates IL-1R expression and suppresses IL-1-induced tumorigenic factors in human pancreatic adenocarcinoma cells. Biochem. Pharmacol. 2011, 82, 1153–1162. [Google Scholar] [CrossRef]
- Hardtner, C.; Multhoff, G.; Falk, W.; Radons, J. (−)-epigallocatechin-3-gallate, a green tea-derived catechin, synergizes with celecoxib to inhibit IL-1-induced tumorigenic mediators by human pancreatic adenocarcinoma cells colo35. Eur. J. Pharmacol. 2012, 684, 36–43. [Google Scholar] [CrossRef]
- Honicke, A.S.; Ender, S.A.; Radons, J. Combined administration of EGCG and IL-1 receptor antagonist efficiently downregulates IL-1-induced tumorigenic factors in U-2 OS human osteosarcoma cells. Int. J. Oncol. 2012, 41, 753–758. [Google Scholar]
- Radons, J.; Falk, W.; Schubert, T.E. Interleukin-10 does not affect IL-1-induced interleukin-6 and metalloproteinase production in human chondrosarcoma cells, SW1353. Int. J. Mol. Med. 2006, 17, 377–383. [Google Scholar]
- Murray, B.; Wilson, D.J. A study of metabolites as intermediate effectors in angiogenesis. Angiogenesis 2001, 4, 71–77. [Google Scholar] [CrossRef]
- Sonveaux, P.; Brouet, A.; Havaux, X.; Gregoire, V.; Dessy, C.; Balligand, J.L.; Feron, O. Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: Implications for tumor radiotherapy. Cancer Res. 2003, 63, 1012–1019. [Google Scholar]
- Maddirela, D.R.; Kesanakurti, D.; Gujrati, M.; Rao, J.S. Mmp-2 suppression abrogates irradiation-induced microtubule formation in endothelial cells by inhibiting alpha-v-beta3-mediated SDF-1/CXCR4 signaling. Int. J. Oncol. 2013, 42, 1279–1288. [Google Scholar]
- Asuthkar, S.; Velpula, K.K.; Nalla, A.K.; Gogineni, V.R.; Gondi, C.S.; Rao, J.S. Irradiation-induced angiogenesis is associated with an MMP-9-MIR-494-Syndecan-1 regulatory loop in medulloblastoma cells. Oncogene 2013. [Google Scholar] [CrossRef]
- Multhoff, G.; Vaupel, P. Radiation-induced changes in microcirculation and interstitial fluid pressure affecting the delivery of macromolecules and nanotherapeutics to tumors. Front. Oncol. 2012. [Google Scholar] [CrossRef]
- Vaupel, P. Pathophysiology of solid tumors. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies; Molls, M., Vaupel, P., Nieder, C., Anscher, M.S., Eds.; Springer: Berlin, Heidelberg, Germany; New York, NY, USA, 2009; pp. 51–92. [Google Scholar]
- Butler, T.P.; Grantham, F.H.; Gullino, P.M. Bulk transfer of fluid in the interstitial compartment of mammary tumors. Cancer Res. 1975, 35, 3084–3088. [Google Scholar]
- Sevick, E.M.; Jain, R.K. Viscous resistance to blood flow in solid tumors: Effect of hematocrit on intratumor blood viscosity. Cancer Res. 1989, 49, 3513–3519. [Google Scholar]
- Gillies, R.J.; Schornack, P.A.; Secomb, T.W.; Raghunand, N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1999, 1, 197–207. [Google Scholar]
- Matsumoto, S.; Yasui, H.; Mitchell, J.B.; Krishna, M.C. Imaging cycling tumor hypoxia. Cancer Res. 2010, 70, 10019–10023. [Google Scholar] [CrossRef]
- Cardenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008, 68, 5812–5819. [Google Scholar]
- Vaupel, P.; Mayer, A. Imaging tumor hypoxia: Blood-borne delivery of imaging agents is fundamentally different in hypoxia subtypes. J. Innov. Opt. Health Sci. 2014. [Google Scholar] [CrossRef]
- Vaupel, P.; Kelleher, D.K.; Höckel, M. Oxygen status of malignant tumors: Pathogenesis of hypoxia and significance for tumor therapy. Semin. Oncol. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A.; Briest, S.; Höckel, M. Oxygenation gain factor: A novel parameter characterizing the association between hemoglobin level and the oxygenation status of breast cancers. Cancer Res. 2003, 63, 7634–7637. [Google Scholar]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Lyng, H.; Vorren, A.O.; Sundfor, K.; Taksdal, I.; Lien, H.H.; Kaalhus, O.; Rofstad, E.K. Intra- and inter-tumor heterogeneity in blood perfusion of human cervical cancer before treatment and after radiotherapy. Int. J. Cancer 2001, 96, 182–190. [Google Scholar] [CrossRef]
- Haider, M.A.; Milosevic, M.; Fyles, A.; Sitartchouk, I.; Yeung, I.; Henderson, E.; Lockwood, G.; Lee, T.Y.; Roberts, T.P. Assessment of the tumor microenvironment in cervix cancer using dynamic contrast enhanced CT, interstitial fluid pressure and oxygen measurements. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1100–1107. [Google Scholar] [CrossRef]
- Höckel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schäffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar]
- Hill, S.A.; Pigott, K.H.; Saunders, M.I.; Powell, M.E.; Arnold, S.; Obeid, A.; Ward, G.; Leahy, M.; Hoskin, P.J.; Chaplin, D.J. Microregional blood flow in murine and human tumours assessed using laser Doppler microprobes. Br. J. Cancer 1996, 27, S260–S263. [Google Scholar]
- Pigott, K.H.; Hill, S.A.; Chaplin, D.J.; Saunders, M.I. Microregional fluctuations in perfusion within human tumours detected using laser Doppler flowmetry. Radiother. Oncol. 1996, 40, 45–50. [Google Scholar] [CrossRef]
- Wilson, C.B.; Lammertsma, A.A.; Mckenzie, C.G.; Sikora, K.; Jones, T. Measurements of blood flow and exchanging water space in breast tumors using positron emission tomography: A rapid and noninvasive dynamic method. Cancer Res. 1992, 52, 1592–1597. [Google Scholar]
- Feldmann, H.J.; Molls, M.; Vaupel, P. Blood flow and oxygenation status of human tumors. Clinical investigations. Strahlenther. Onkol. 1999, 175, 1–9. [Google Scholar]
- Vaupel, P.; Manz, R.; Müller-Klieser, W.; Grunewald, W.A. Intracapillary HbO2 saturations within tissue-isolated malignant-tumors during hyperoxia. Pflügers Arch. Eur. J. Physiol. 1978, 373, R39. [Google Scholar] [CrossRef]
- Weiss, L.; Hultborn, R.; Tveit, E. Blood-flow characteristics in induced rat mammary neoplasia. Microvasc. Res. 1979, 17, S119. [Google Scholar]
- Endrich, B.; Hammersen, F.; Götz, A.; Messmer, K. Microcirculatory blood flow, capillary morphology and local oxygen pressure of the hamster amelanotic melanoma a-Mel-3. J. Natl. Cancer Inst. 1982, 68, 475–485. [Google Scholar]
- Wheeler, R.H.; Ziessman, H.A.; Medvec, B.R.; Juni, J.E.; Thrall, J.H.; Keyes, J.W.; Pitt, S.R.; Baker, S.R. Tumor blood flow and systemic shunting in patients receiving intraarterial chemotherapy for head and neck cancer. Cancer Res. 1986, 46, 4200–4204. [Google Scholar]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumour hypoxia using p02 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer. 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Harada, H. How can we overcome tumor hypoxia in radiation therapy? J. Radiat. Res. 2011, 52, 545–556. [Google Scholar] [CrossRef]
- Harada, H.; Itasaka, S.; Kizaka-Kondoh, S.; Shibuya, K.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J. Biol. Chem. 2009, 284, 5332–5342. [Google Scholar]
- Schilling, D.; Duwel, M.; Molls, M.; Multhoff, G. Radiosensitization of wildtype p53 cancer cells by the MDM2-inhibitor PXN727 is associated with altered heat shock protein 70 (Hsp70) levels. Cell Stress Chaperones. 2013, 18, 183–191. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. Rack1 competes with hsp90 for binding to HIF-1 alpha and is required for O2-independent and Hsp90 inhibitor-induced degradation of HIF-1 alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Yoshimura, M.; Itasaka, S.; Harada, H.; Hiraoka, M. Microenvironment and radiation therapy. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 610–616. [Google Scholar] [CrossRef]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through s-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Yasui, H.; Ogura, A.; Asanuma, T.; Matsuda, A.; Kashiwakura, I.; Kuwabara, M.; Inanami, O. Inhibition of HIF-1alpha by the anticancer drug Tas106 enhances x-ray-induced apoptosis in vitro and in vivo. Br. J. Cancer 2008, 99, 1442–1452. [Google Scholar] [CrossRef]
- Schwartz, D.L.; Powis, G.; Thitai-Kumar, A.; He, Y.; Bankson, J.; Williams, R.; Lemos, R.; Oh, J.; Volgin, A.; Soghomonyan, S.; et al. The selective hypoxia inducible factor-1 inhibitor px-478 provides in vivo in vivo radiosensitization through tumor stromal effects. Mol. Cancer Ther. 2009, 8, 947–958. [Google Scholar] [CrossRef]
- Harada, H.; Itasaka, S.; Zhu, Y.; Zeng, L.; Xie, X.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. Treatment regimen determines whether an HIF-1 inhibitor enhances or inhibits the effect of radiation therapy. Br. J. Cancer 2009, 100, 747–757. [Google Scholar] [CrossRef]
- Li, G.; Xie, B.; Li, X.; Chen, Y.; Wang, Q.; Xu, Y.; Xu-Welliver, M.; Zou, L. Down-regulation of survivin and hypoxia-inducible factor-1 alpha by beta-elemene enhances the radiosensitivity of lung adenocarcinoma xenograft. Cancer Biother. Radiopharm. 2012, 27, 56–64. [Google Scholar] [CrossRef]
- Okamoto, K.; Ito, D.; Miyazaki, K.; Watanabe, S.; Tohyama, O.; Yokoi, A.; Ozawa, Y.; Asano, M.; Kawamura, T.; Yamane, Y.; et al. Microregional antitumor activity of a small-molecule hypoxia-inducible factor 1 inhibitor. Int. J. Mol. Med. 2012, 29, 541–549. [Google Scholar]
- Quesada, J.; Amato, R. The molecular biology of soft-tissue sarcomas and current trends in therapy. Sarcoma 2012, 2012, 849456. [Google Scholar]
- Wehland, M.; Bauer, J.; Magnusson, N.E.; Infanger, M.; Grimm, D. Biomarkers for anti-angiogenic therapy in cancer. Int. J. Mol. Sci. 2013, 14, 9338–9364. [Google Scholar] [CrossRef]
- Mauceri, H.J.; Hanna, N.N.; Beckett, M.A.; Gorski, D.H.; Staba, M.J.; Stellato, K.A.; Bigelow, K.; Heimann, R.; Gately, S.; Dhanabal, M.; et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 1998, 394, 287–291. [Google Scholar] [CrossRef]
- Itasaka, S.; Komaki, R.; Herbst, R.S.; Shibuya, K.; Shintani, T.; Hunter, N.R.; Onn, A.; Bucana, C.D.; Milas, L.; Ang, K.K.; et al. Endostatin improves radioresponse and blocks tumor revascularization after radiation therapy for A431 xenografts in mice. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 870–878. [Google Scholar]
- Peng, F.; Xu, Z.; Wang, J.; Chen, Y.; Li, Q.; Zuo, Y.; Chen, J.; Hu, X.; Zhou, Q.; Wang, Y.; et al. Recombinant human endostatin normalizes tumor vasculature and enhances radiation response in xenografted human nasopharyngeal carcinoma models. PLoS One 2012, 7, e34646. [Google Scholar]
- Ke, Q.H.; Zhou, S.Q.; Huang, M.; Lei, Y.; Du, W.; Yang, J.Y. Early efficacy of endostar combined with chemoradiotherapy for advanced cervical cancers. Asian Pac. J. Cancer Prev. 2012, 13, 923–926. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, L.; Xu, X.; Tu, Y.; Qin, S.; Yin, Y. Antitumor activity of endostar combined with radiation against human nasopharyngeal carcinoma in mouse xenograft models. Oncol. Lett. 2012, 4, 976–980. [Google Scholar]
- Mcmeekin, D.S.; Sill, M.W.; Darcy, K.M.; Abulafia, O.; Hanjani, P.; Pearl, M.L.; Rubin, S.C.; Rose, P.G.; Small, L.; Benbrook, D.M. A phase II trial of thalidomide in patients with refractory uterine carcinosarcoma and correlation with biomarkers of angiogenesis: A gynecologic oncology group study. Gynecol. Oncol. 2012, 127, 356–361. [Google Scholar] [CrossRef]
- Knisely, J.P.; Berkey, B.; Chakravarti, A.; Yung, A.W.; Curran, W.J., Jr.; Robins, H.I.; Movsas, B.; Brachman, D.G.; Henderson, R.H.; Mehta, M.P. A phase III study of conventional radiation therapy plus thalidomide versus conventional radiation therapy for multiple brain metastases (RTOG 0118). Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 79–86. [Google Scholar]
- Viani, G.A.; Manta, G.B.; Fonseca, E.C.; de Fendi, L.I.; Afonso, S.L.; Stefano, E.J. Whole brain radiotherapy with radiosensitizer for brain metastases. J. Exp. Clin. Cancer Res. 2009, 28, 1. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Brooks, C.; Sheu, T.; Bridges, K.; Mason, K.; Kuban, D.; Mathew, P.; Meyn, R. Preclinical evaluation of sunitinib, a multi-tyrosine kinase inhibitor, as a radiosensitizer for human prostate cancer. Radiat. Oncol. 2012. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; Mcgraw, S.; Jaeckle, K.A.; Buckner, J.C. North central cancer treatment group phase I trial N057k of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 468–475. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Shih, K.C.; Shepard, G.C.; Tillinghast, G.W.; Brinker, B.T.; Spigel, D.R. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clin. Adv. Hematol. Oncol. 2012, 10, 240–246. [Google Scholar]
- Zaidi, S.; Mclaughlin, M.; Bhide, S.A.; Eccles, S.A.; Workman, P.; Nutting, C.M.; Huddart, R.A.; Harrington, K.J. The HSP90 inhibitor NVP-AUY922 radiosensitizes by abrogation of homologous recombination resulting in mitotic entry with unresolved DNA damage. PLoS One 2012, 7, e35436. [Google Scholar]
- Gandhi, N.; Wild, A.T.; Chettiar, S.T.; Aziz, K.; Kato, Y.; Gajula, R.P.; Williams, R.D.; Cades, J.A.; Annadanam, A.; Song, D.; et al. Novel hsp90 inhibitor nvp-auy922 radiosensitizes prostate cancer cells. Cancer Biol. Ther. 2013, 14, 347–356. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Multhoff, G.; Radons, J.; Vaupel, P. Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance. Cancers 2014, 6, 813-828. https://doi.org/10.3390/cancers6020813
Multhoff G, Radons J, Vaupel P. Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance. Cancers. 2014; 6(2):813-828. https://doi.org/10.3390/cancers6020813
Chicago/Turabian StyleMulthoff, Gabriele, Jürgen Radons, and Peter Vaupel. 2014. "Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance" Cancers 6, no. 2: 813-828. https://doi.org/10.3390/cancers6020813
APA StyleMulthoff, G., Radons, J., & Vaupel, P. (2014). Critical Role of Aberrant Angiogenesis in the Development of Tumor Hypoxia and Associated Radioresistance. Cancers, 6(2), 813-828. https://doi.org/10.3390/cancers6020813